8-(Pyridin-2-yl)quinolin-7-ol as a Platform for Conjugated Proton Cranes: A DFT Structural Design



Abstract
:1. Introduction
2. Theoretical Simulations
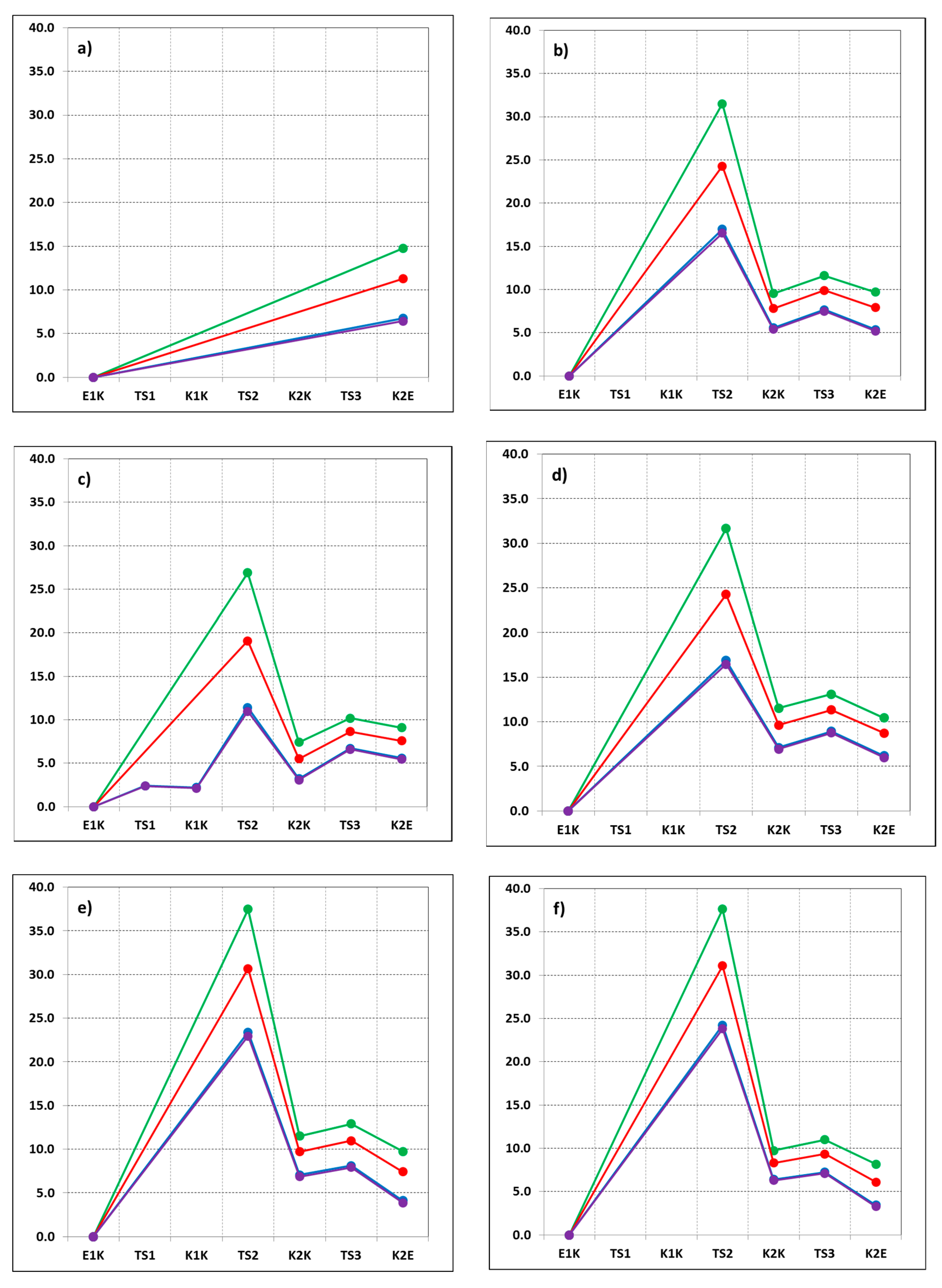
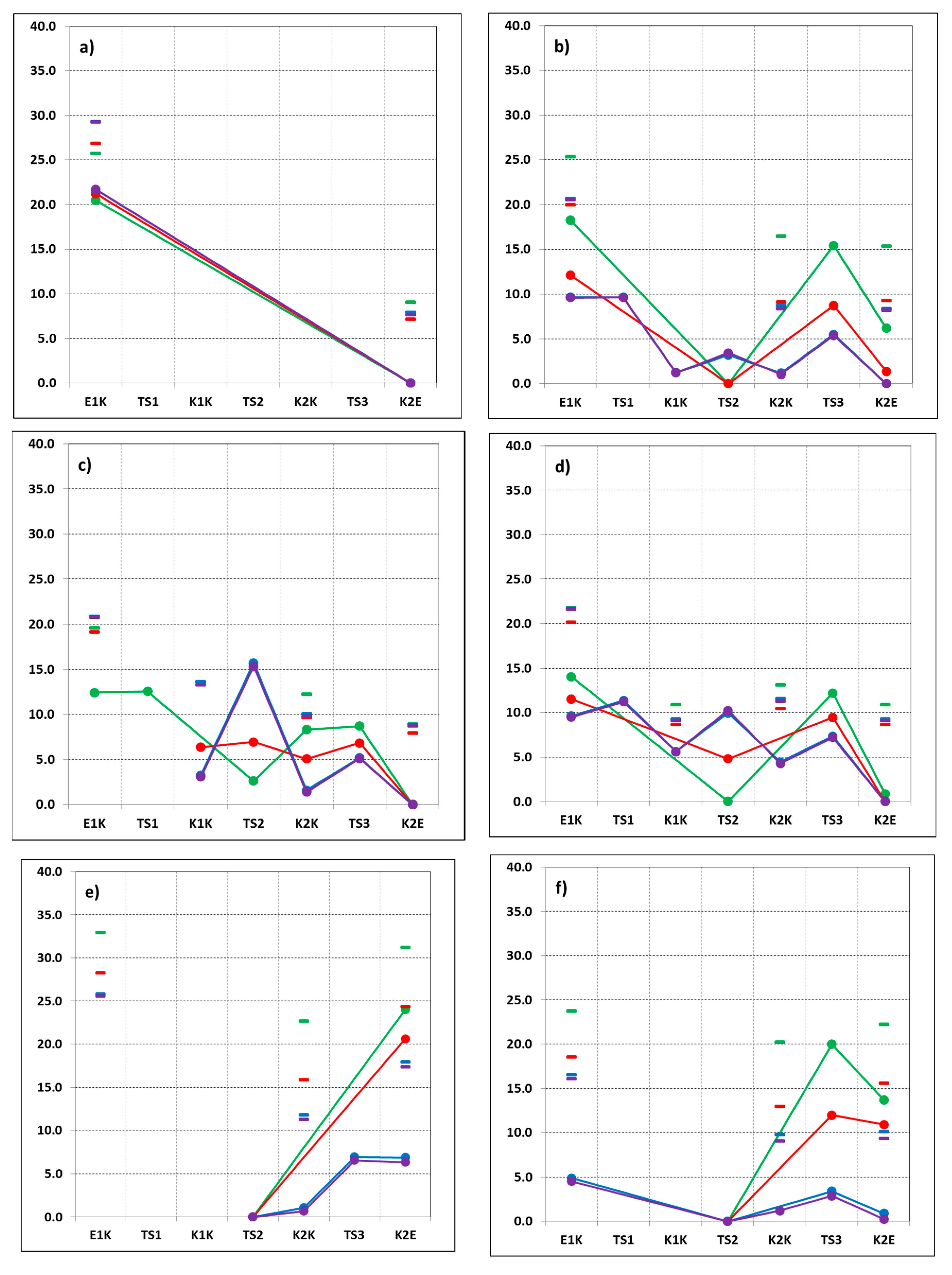
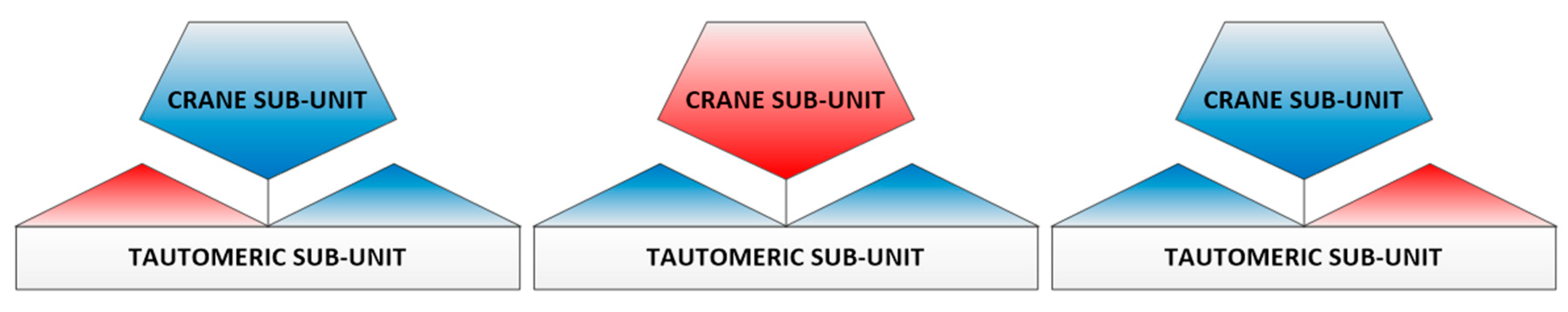
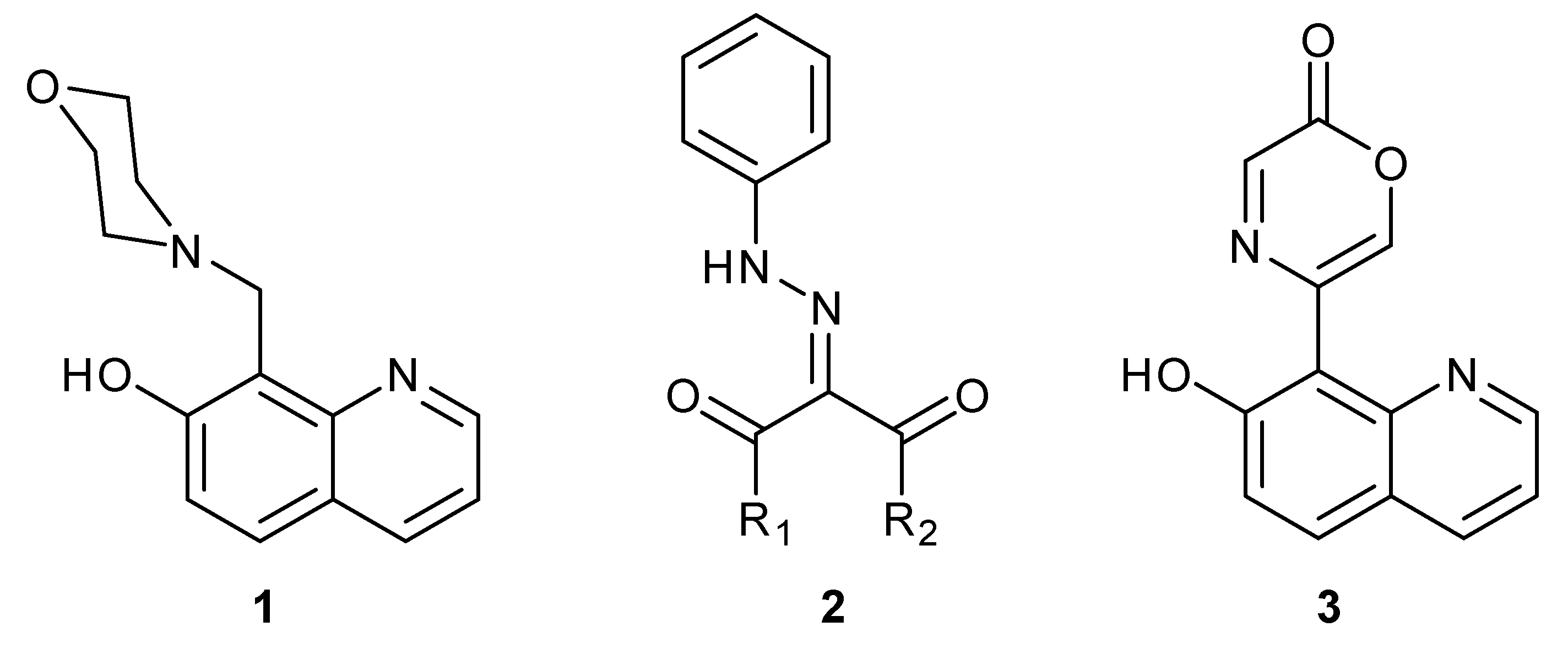
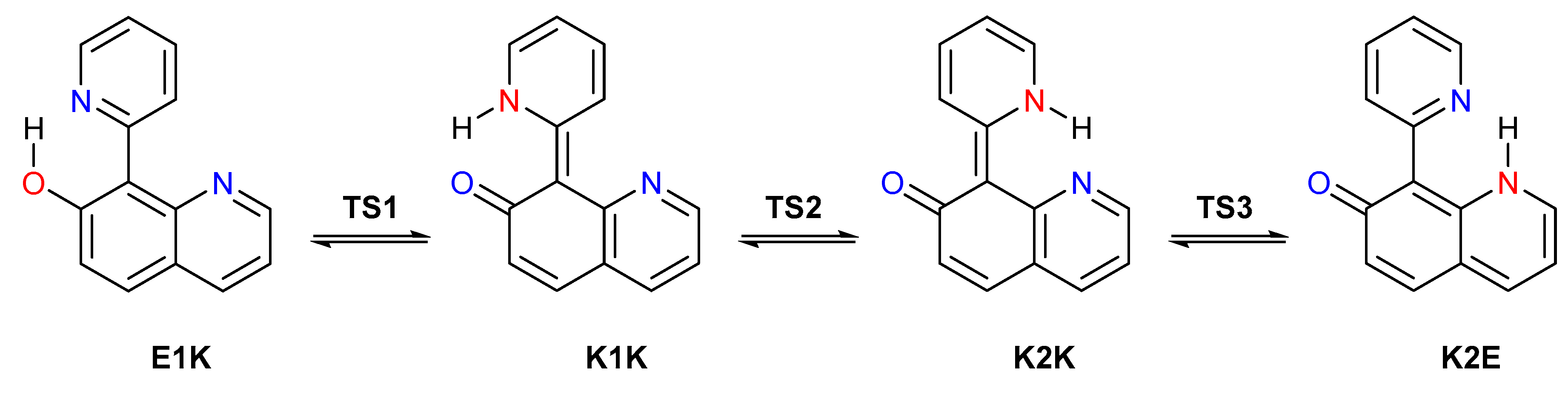
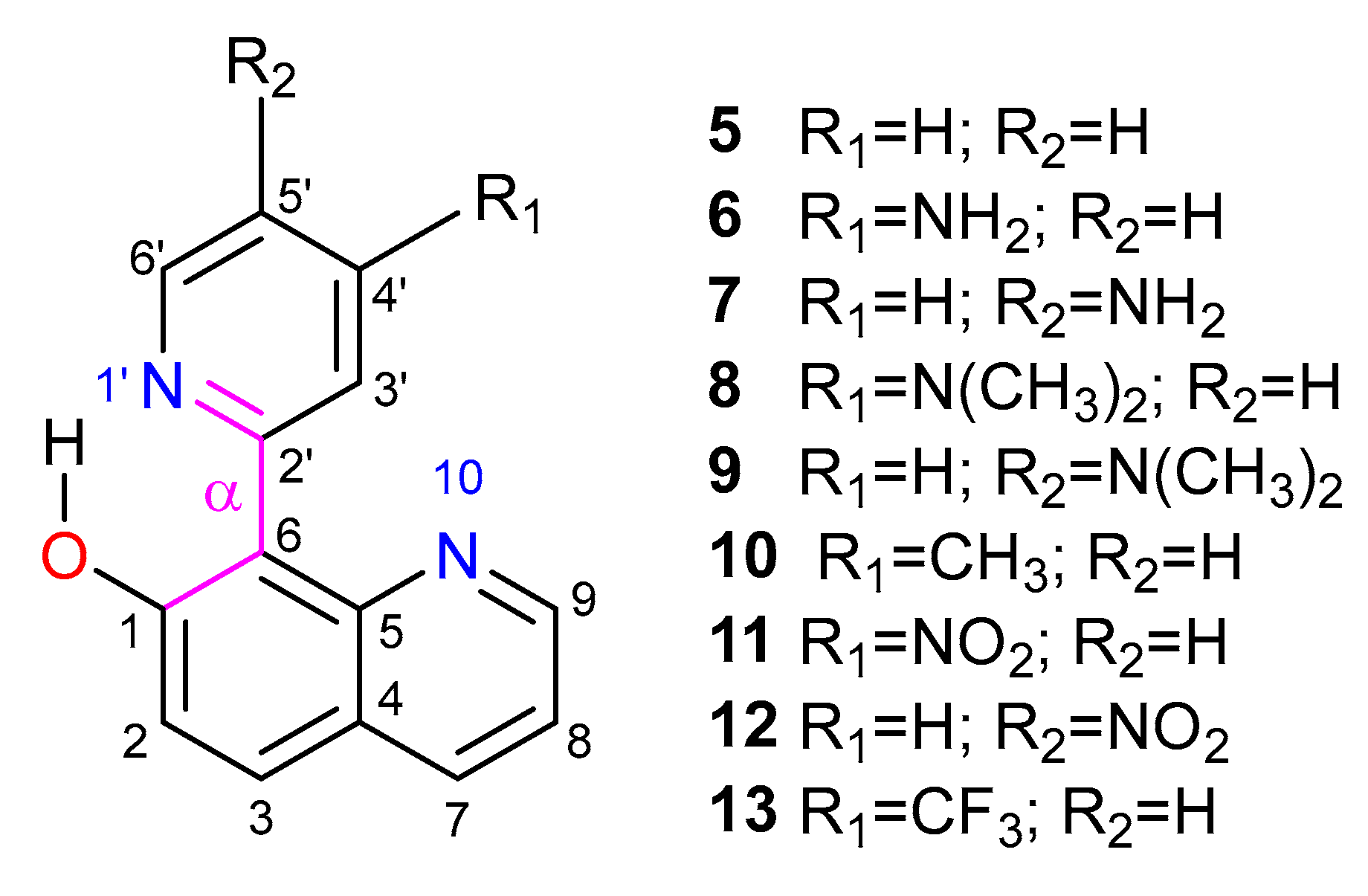
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hynes, J.T.; Klinman, J.P.; Limbach, H.-H.; Schowen, R.L. (Eds.) Hydrogen-Transfer Reactions; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; ISBN 978-3-527-61154-6. [Google Scholar]
- Antonov, L. (Ed.) Tautomerism: Methods and Theories; Wiley-VCH: Weinheim, Germany, 2014; ISBN 978-3-527-33294-6. [Google Scholar]
- Stasyuk, A.J.; Cywiński, P.J.; Gryko, D.T. Excited-state intramolecular proton transfer in 2′-(2′-hydroxyphenyl)imidazo[1,2- a ]pyridines. J. Photochem. Photobiol. C Photochem. Rev. 2016, 28, 116–137. [Google Scholar] [CrossRef]
- Serdiuk, I.E.; Roshal, A.D. Exploring double proton transfer: A review on photochemical features of compounds with two proton-transfer sites. Dyes Pigments 2017, 138, 223–244. [Google Scholar] [CrossRef]
- Demchenko, A.P.; Tang, K.-C.; Chou, P.-T. Excited-state proton coupled charge transfer modulated by molecular structure and media polarization. Chem. Soc. Rev. 2013, 42, 1379–1408. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.E.; Park, S.Y. Advanced Organic Optoelectronic Materials: Harnessing Excited-State Intramolecular Proton Transfer (ESIPT) Process. Adv. Mater. 2011, 23, 3615–3642. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, M.; Taniguchi, M. Single Molecule Electronics and Devices. Sensors 2012, 12, 7259–7298. [Google Scholar] [CrossRef]
- Sakai, K.; Tsuzuki, T.; Itoh, Y.; Ichikawa, M.; Taniguchi, Y. Using proton-transfer laser dyes for organic laser diodes. Appl. Phys. Lett. 2005, 86, 081103. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-Y.; Hsieh, C.-C.; Cheng, Y.-M.; Lai, C.-H.; Chou, P.-T. Extensive spectral tuning of the proton transfer emission from 550 to 675 nm via a rational derivatization of 10-hydroxybenzo[h]quinoline. Chem. Commun. 2006, 4395. [Google Scholar] [CrossRef]
- Smith, T.P.; Zaklika, K.A.; Thakur, K.; Walker, G.C.; Tominaga, K.; Barbara, P.F. Ultrafast studies on proton transfer in photostabilizers. J. Photochem. Photobiol. Chem. 1992, 65, 165–175. [Google Scholar] [CrossRef]
- Irie, M.; Fukaminato, T.; Matsuda, K.; Kobatake, S. Photochromism of Diarylethene Molecules and Crystals: Memories, Switches, and Actuators. Chem. Rev. 2014, 114, 12174–12277. [Google Scholar] [CrossRef]
- Nedeltcheva-Antonova, D.; Antonov, L. Controlled Tautomerism: Is It Possible? In Tautomerism: Concepts and Applications in Science and Technology; Antonov, L., Ed.; Wiley-VCH: Weinheim, Germany, 2016; pp. 273–294. ISBN 978-3-527-69571-3. [Google Scholar]
- Kassem, S.; van Leeuwen, T.; Lubbe, A.S.; Wilson, M.R.; Feringa, B.L.; Leigh, D.A. Artificial molecular motors. Chem. Soc. Rev. 2017, 46, 2592–2621. [Google Scholar] [CrossRef] [Green Version]
- Kassem, S.; Lee, A.T.L.; Leigh, D.A.; Markevicius, A.; Solà, J. Pick-up, transport and release of a molecular cargo using a small-molecule robotic arm. Nat. Chem. 2015, 8, 138–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalink, C.J.; van Ingen, W.M.; Huizer, A.H.; Varma, C.A.G.O. Prospects for using photoinduced intramolecular proton transfer to study the dynamics of conformational changes in flexible molecular chains. J. Chem. Soc. Faraday Trans. 1991, 87, 1103. [Google Scholar] [CrossRef]
- Jalink, C.J.; Huizer, A.H.; Varma, C.A.G.O. Rate-limiting action of a proton crane in long-range intramolecular proton transfer. J. Chem. Soc. Faraday Trans. 1992, 88, 2655. [Google Scholar] [CrossRef]
- de Bekker, E.J.A.; Geerlings, J.D.; Varma, C.A.G.O. Mechanism of a Photoinduced Solvent-Assisted Transfer of a Proton to a Specified Remote Target. J. Phys. Chem. A 2000, 104, 5916–5927. [Google Scholar] [CrossRef]
- de Bekker, E.J.A.; Pugzlys, A.; Varma, C.A.G.O. Elementary Processes in Photoinduced Proton Transfers in 2-Hydroxy-1-( N -morpholinomethyl)naphthalene and 7-Hydroxy-8-( N -morpholinomethyl)quinoline in Liquid Solutions. J. Phys. Chem. A 2001, 105, 399–409. [Google Scholar] [CrossRef]
- van der Loop, T.H.; Ruesink, F.; Amirjalayer, S.; Sanders, H.J.; Buma, W.J.; Woutersen, S. Unraveling the Mechanism of a Reversible Photoactivated Molecular Proton Crane. J. Phys. Chem. B 2014, 118, 12965–12971. [Google Scholar] [CrossRef]
- Wu, K.-C.; Cheng, Y.-M.; Lin, Y.-S.; Yeh, Y.-S.; Pu, S.-C.; Hu, Y.-H.; Yu, J.-K.; Che, P.-T. Competitive intramolecular hydrogen bonding formation and excited-state proton transfer reaction in 1-[(diethylamino)-methyl]-2-hydroxy-3-naphthaldehyde. Chem. Phys. Lett. 2004, 384, 203–209. [Google Scholar] [CrossRef]
- Wu, K.-C.; Lin, Y.-S.; Yeh, Y.-S.; Chen, C.-Y.; Ahmed, M.O.; Chou, P.-T.; Hon, Y.-S. Design and synthesis of intramolecular hydrogen bonding systems. Their application in metal cation sensing based on excited-state proton transfer reaction. Tetrahedron 2004, 60, 11861–11868. [Google Scholar] [CrossRef]
- Antonov, L.; Deneva, V.; Simeonov, S.; Kurteva, V.; Nedeltcheva, D.; Wirz, J. Exploiting Tautomerism for Switching and Signaling. Angew. Chem. Int. Ed. 2009, 48, 7875–7878. [Google Scholar] [CrossRef]
- Antonov, L.M.; Kurteva, V.B.; Simeonov, S.P.; Deneva, V.V.; Crochet, A.; Fromm, K.M. Tautocrowns: A concept for a sensing molecule with an active side-arm. Tetrahedron 2010, 66, 4292–4297. [Google Scholar] [CrossRef] [Green Version]
- Nedeltcheva, D.; Kurteva, V.; Antonov, L. Gas-phase study of molecular switches based on tautomeric proton transfer. Eur. J. Mass Spectrom. 2011, 17, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Courtot, P.; Pichon, R.; Le Saint, J. Determination du site de chelation chez les arylhydrazones de tricetones et D’ α-dicetones substituees. Tetrahedron Lett. 1976, 17, 1177–1180. [Google Scholar] [CrossRef]
- Courtot, P.; Pichon, R.; Le Saint, J. Photochromisme par isomerisation syn-anti de phenylhydrazones-2- de tricetones-1,2,3 et de dicetones-1,2 substituees. Tetrahedron Lett. 1976, 17, 1181–1184. [Google Scholar] [CrossRef]
- Courtot, P.; Pichon, R.; Le Saint, J. Échanges inter et intra-molćulaires du proton entre deux atomes d’azote de cycles chélatés hydrazone-imine et azo-énamine. Tetrahedron Lett. 1979, 20, 1591–1594. [Google Scholar] [CrossRef]
- Pichon, R.; Le Saint, J.; Courtot, P. Photoisomerisation d’arylhydrazones-2 de dicetones-1,2 substituees en 2. Tetrahedron 1981, 37, 1517–1524. [Google Scholar] [CrossRef]
- Yoder, C.H.; Barth, R.C.; Richter, W.M.; Snavely, F.A. Nuclear magnetic resonance study of some nitrogen-15 substituted azo heterocycles. J. Org. Chem. 1972, 37, 4121–4123. [Google Scholar] [CrossRef]
- Shawali, A.S.; Harb, N.M.S.; Badahdah, K.O. A study of tautomerism in diazonium coupling products of 4-hydroxycoumarin. J. Heterocycl. Chem. 1985, 22, 1397–1403. [Google Scholar] [CrossRef]
- Yordanov, D.; Deneva, V.; Georgiev, A.; Vassilev, N.; Vala, M.; Zhivkov, I.; Antonov, L. 4-OH coumarin based rotary switches: Tautomeric state and effect of the stator. Dyes Pigments 2020, 108861. [Google Scholar] [CrossRef]
- Su, X.; Aprahamian, I. Hydrazone-based switches, metallo-assemblies and sensors. Chem. Soc. Rev. 2014, 43, 1963. [Google Scholar] [CrossRef] [Green Version]
- Tatum, L.A.; Su, X.; Aprahamian, I. Simple Hydrazone Building Blocks for Complicated Functional Materials. Acc. Chem. Res. 2014, 47, 2141–2149. [Google Scholar] [CrossRef]
- Foy, J.T.; Ray, D.; Aprahamian, I. Regulating signal enhancement with coordination-coupled deprotonation of a hydrazone switch. Chem. Sci. 2015, 6, 209–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.D.; Moran, M.J.; Aprahamian, I. New molecular switch architectures. Proc. Natl. Acad. Sci. USA 2018, 115, 9414–9422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobolewski, A.L. Reversible molecular switch driven by excited-state hydrogen transfer. Phys. Chem. Chem. Phys. 2008, 10, 1243. [Google Scholar] [CrossRef] [PubMed]
- Lapinski, L.; Nowak, M.J.; Nowacki, J.; Rode, M.F.; Sobolewski, A.L. A Bistable Molecular Switch Driven by Photoinduced Hydrogen-Atom Transfer. ChemPhysChem 2009, 10, 2290–2295. [Google Scholar] [CrossRef]
- Rode, M.F.; Sobolewski, A.L. Effect of Chemical Substituents on the Energetical Landscape of a Molecular Photoswitch: An Ab Initio Study. J. Phys. Chem. A 2010, 114, 11879–11889. [Google Scholar] [CrossRef]
- Vetokhina, V.; Nowacki, J.; Pietrzak, M.; Rode, M.F.; Sobolewski, A.L.; Waluk, J.; Herbich, J. 7-Hydroxyquinoline-8-carbaldehydes. 1. Ground- and Excited-State Long-Range Prototropic Tautomerization. J. Phys. Chem. A 2013, 117, 9127–9146. [Google Scholar] [CrossRef]
- Csehi, A.; Illés, L.; Halász, G.J.; Vibók, Á. The effect of chemical substituents on the functionality of a molecular switch system: A theoretical study of several quinoline compounds. Phys. Chem. Chem. Phys. 2013, 15, 18048. [Google Scholar] [CrossRef]
- Csehi, A.; Woywod, C.; Halász, G.; Vibók, Á. Ab initio studies of two pyrimidine derivatives as possible photo-switch systems. Open Phys. 2013, 11. [Google Scholar] [CrossRef] [Green Version]
- Csehi, A.; Halász, G.J.; Vibók, Á. Molecular switch properties of 7-hydroxyquinoline compounds. Int. J. Quantum Chem. 2014, 114, 1135–1145. [Google Scholar] [CrossRef]
- Ortiz-Sánchez, J.M.; Gelabert, R.; Moreno, M.; Lluch, J.M.; Anglada, J.M.; Bofill, J.M. Bipyridyl Derivatives as Photomemory Devices: A Comparative Electronic-Structure Study. Chem.-Eur. J. 2010, 16, 6693–6703. [Google Scholar] [CrossRef]
- Kaczmarek, L.S. Bipyridines. Part XVIII. On the Synthesis of [2,2’-Bipyridine]-3-ol and Other Novel 2,2’-Bipyridine Derivatives from [2,2’-Bipyridine]-3,3’-diamine. Bull. Pol. Acad. Sci. Tech. Sci. 1985, 33, 401–409. [Google Scholar]
- Kaczmarek, Ł; Balicki, R.; Lipkowski, J.; Borowicz, P.; Grabowska, A. Structure and photophysics of deazabipyridyls. Excited internally hydrogen-bonded systems with one proton transfer reaction site. J. Chem. Soc. Perkin Trans. 2 1994, 1603. [Google Scholar] [CrossRef]
- Borowicz, P.; Grabowska, A.; Leś, A.; Kaczmarek, Ł.; Zagrodzki, B. New phototautomerizing systems: Non-symmetric derivatives of [2,2′-bipyridyl]-3,3′-diol. Chem. Phys. Lett. 1998, 291, 351–359. [Google Scholar] [CrossRef]
- Kaczmarek, Ł.; Zagrodzki, B.; Kamieński, B.; Pietrzak, M.; Schilf, W.; Leś, A. Synthesis and NMR study of new derivatives of [2,2′-bipyridyl]-3,3′-diol and [2,2′-bipyridyl]-3-ol. J. Mol. Struct. 2000, 553, 61–72. [Google Scholar] [CrossRef]
- Böhnke, H.; Bahrenburg, J.; Ma, X.; Röttger, K.; Näther, C.; Rode, M.F.; Sobolewski, A.L.; Temps, F. Ultrafast dynamics of the ESIPT photoswitch N-(3-pyridinyl)-2-pyridinecarboxamide. Phys. Chem. Chem. Phys. 2018, 20, 2646–2655. [Google Scholar] [CrossRef] [PubMed]
- Antonov, L. Favipiravir tautomerism: A theoretical insight. Theor. Chem. Acc. 2020, 139, 145. [Google Scholar] [CrossRef]
- Antonov, L. Tautomerism in Azo and Azomethyne Dyes: When and If Theory Meets Experiment. Molecules 2019, 24, 2252. [Google Scholar] [CrossRef] [Green Version]
- Deneva, V.; Vassilev, N.G.; Hristova, S.; Yordanov, D.; Hayashi, Y.; Kawauchi, S.; Fennel, F.; Völzer, T.; Lochbrunner, S.; Antonov, L. Chercher de l’eau: The switching mechanism of the rotary switch ethyl-2-(2-(quinolin-8-yl)hydrazono)-2-(pyridin-2-yl)acetate. Comput. Mater. Sci. 2020, 177, 109570. [Google Scholar] [CrossRef]
- Manolova, Y.; Marciniak, H.; Tschierlei, S.; Fennel, F.; Kamounah, F.S.; Lochbrunner, S.; Antonov, L. Solvent control of intramolecular proton transfer: Is 4-hydroxy-3-(piperidin-1-ylmethyl)-1-naphthaldehyde a proton crane? Phys. Chem. Chem. Phys. 2017, 19, 7316–7325. [Google Scholar] [CrossRef]
- Marciniak, H.; Hristova, S.; Deneva, V.; Kamounah, F.S.; Hansen, P.E.; Lochbrunner, S.; Antonov, L. Dynamics of excited state proton transfer in nitro substituted 10-hydroxybenzo[h]quinolines. Phys. Chem. Chem. Phys. 2017, 19, 26621–26629. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Kawauchi, S.; Antonov, L. Description of the Tautomerism in Some Azonaphthols. J. Phys. Org. Chem. 2013, 26, 643–652. [Google Scholar] [CrossRef]
- Antonov, L.; Kurteva, V.; Crochet, A.; Mirolo, L.; Fromm, K.M.; Angelova, S. Tautomerism in 1-phenylazo-4-naphthols: Experimental results vs quantum-chemical predictions. Dyes Pigments 2012, 92, 714–723. [Google Scholar] [CrossRef] [Green Version]
- Angelova, S.; Paskaleva, V.; Kochev, N.; Antonov, L. DFT study of hydrazone-based molecular switches: The effect of different stators on the on/off state distribution. Mol. Phys. 2019, 117, 1604–1612. [Google Scholar] [CrossRef]
- Fang, H.; Kim, Y. Hydrogen-bonded channel-dependent mechanism of long-range proton transfer in the excited-state tautomerization of 7-hydroxyquinoline: A theoretical study. Theor. Chem. Acc. 2017, 136, 28. [Google Scholar] [CrossRef]
- Mori, Y. Reaction pathway and H/D kinetic isotope effects of the triple proton transfer in a 7-hydroxyquinoline-methanol complex in the ground state: A computational approach. J. Phys. Org. Chem. 2018, 31, e3747. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Improta, R. UV-Visible Absorption and Emission Energies in Condensed Phase by PCM/TD-DFT Methods. In Computational Strategies for Spectroscopy; Barone, V., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 37–75. ISBN 978-1-118-00872-0. [Google Scholar]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Antonov, L.; Kawauchi, S.; Okuno, Y. Prediction of the color of dyes by using time-dependent density functional theory. Bulg. Chem. Commun. 2014, 46, 228–237. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2005; ISBN 978-0-521-83128-4. [Google Scholar]
- Konijnenberg, J.; Ekelmans, G.B.; Huizer, A.H.; Varma, C.A.G.O. Mechanism and solvent dependence of the solvent-catalysed pseudo-intramolecular proton transfer of 7-hydroxyquinoline in the first electronically excited singlet state and in the ground state of its tautomer. J. Chem. Soc. Faraday Trans 2 1989, 85, 39–51. [Google Scholar] [CrossRef]
- Al-Lawatia, N.; Husband, J.; Steinbrecher, T.; Abou-Zied, O.K. Tautomerism in 7-Hydroxyquinoline: A Combined Experimental and Theoretical Study in Water. J. Phys. Chem. A 2011, 115, 4195–4201. [Google Scholar] [CrossRef]
- Miura, M.; Harada, J.; Ogawa, K. Temperature-Induced Reversal of Proton Tautomerism: Role of Hydrogen Bonding and Aggregation in 7-Hydroxyquinolines. J. Phys. Chem. A 2007, 111, 9854–9858. [Google Scholar] [CrossRef]
- Lee, S.-I.; Jang, D.-J. Proton Transfers of Aqueous 7-Hydroxyquinoline in the First Excited Singlet, Lowest Triplet, and Ground States. J. Phys. Chem. 1995, 99, 7537–7541. [Google Scholar] [CrossRef]
- Lavin, A.; Collins, S. The ground-state stabilization of the keto tautomer of 7-hydroxyquinoline in methanol/argon matrices at 10 K. Chem. Phys. Lett. 1993, 204, 96–100. [Google Scholar] [CrossRef]
- Itoh, M.; Adachi, T. Transient absorption and two-step laser excitation fluorescence studies of the excited-state proton transfer and relaxation in the methanol solution of 7-hydroxyflavone. J. Am. Chem. Soc. 1984, 106, 4320–4324. [Google Scholar] [CrossRef]
- Kang, W.-K.; Cho, S.-J.; Lee, M.; Kim, D.-H.; Ryoo, R.; Jung, K.-H.; Jang, D.-J. Excited State Intramolecular Proton Transfer and Physical Properties of 7-Hydroxyquinoline. Bull. Korean Chem. Soc. 1992, 13, 140–145. [Google Scholar]
- Park, S.-Y.; Jang, D.-J. Accumulated Proton-Donating Ability of Solvent Molecules in Proton Transfer. J. Am. Chem. Soc. 2010, 132, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.F.; Philp, J.; Smith, B.E. Prototropic equilibria of electronically excited molecules. Part II. 3-, 6-, and 7-Hydroxyquinoline. J. Chem. Soc. Inorg. Phys. Theor. 1968, 3051. [Google Scholar] [CrossRef]
- Bardez, E. Excited-State Proton Transfer in Bifunctional Compounds. Isr. J. Chem. 1999, 39, 319–332. [Google Scholar] [CrossRef]
- Kwon, O.-H.; Lee, Y.-S.; Yoo, B.K.; Jang, D.-J. Excited-State Triple Proton Transfer of 7-Hydroxyquinoline along a Hydrogen-Bonded Alcohol Chain: Vibrationally Assisted Proton Tunneling. Angew. Chem. Int. Ed. 2006, 45, 415–419. [Google Scholar] [CrossRef]
- Park, S.-Y.; Kim, B.; Lee, Y.-S.; Kwon, O.-H.; Jang, D.-J. Triple proton transfer of excited 7-hydroxyquinoline along a hydrogen-bonded water chain in ethers: Secondary solvent effect on the reaction rate. Photochem. Photobiol. Sci. 2009, 8, 1611–1617. [Google Scholar] [CrossRef]
- Hoffmann, F.; Ekimova, M.; Bekçioğlu-Neff, G.; Nibbering, E.T.J.; Sebastiani, D. Combined Experimental and Theoretical Study of the Transient IR Spectroscopy of 7-Hydroxyquinoline in the First Electronically Excited Singlet State. J. Phys. Chem. A 2016, 120, 9378–9389. [Google Scholar] [CrossRef]
- Ekimova, M.; Hoffmann, F.; Bekçioğlu-Neff, G.; Rafferty, A.; Kornilov, O.; Nibbering, E.T.J.; Sebastiani, D. Ultrafast Proton Transport between a Hydroxy Acid and a Nitrogen Base along Solvent Bridges Governed by the Hydroxide/Methoxide Transfer Mechanism. J. Am. Chem. Soc. 2019, 141, 14581–14592. [Google Scholar] [CrossRef]
- Kerdpol, K.; Daengngern, R.; Meeprasert, J.; Namuangruk, S.; Kungwan, N. Theoretical insights into photoinduced proton transfer of 7-hydroxyquinoline via intermolecular hydrogen-bonded wire of mixed methanol and water. Theor. Chem. Acc. 2016, 135, 208. [Google Scholar] [CrossRef]
- Partovi–Azar, P.; Sebastiani, D. Optimized effective potentials to increase the accuracy of approximate proton transfer energy calculations in the excited state. J. Chem. Phys. 2020, 152, 064101. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Abboud, J.L.M.; Taft, R.W. An Examination of Linear Solvation Energy Relationships. In Progress in Physical Organic Chemistry; Taft, R.W., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1981; Volume 13, pp. 485–630. ISBN 978-0-470-17192-9. [Google Scholar]
- Kamlet, M.J.; Abboud, J.L.M.; Abraham, M.H.; Taft, R.W. Linear solvation energy relationships. 23. A comprehensive collection of the solvatochromic parameters,.pi.*,.alpha., and.beta., and some methods for simplifying the generalized solvatochromic equation. J. Org. Chem. 1983, 48, 2877–2887. [Google Scholar] [CrossRef]
- Joule, J.A.; Mills, K. Heterocyclic Chemistry, 5th ed.; Wiley: Hoboken, NJ, USA, 2009; ISBN 978-1-4051-9365-8. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Environment | ΔE, kcal/mol | |||
|---|---|---|---|---|---|
| E1K | K1K | K2K | K2E | ||
| 4 | vacuum | 0.0 (20/0.08) | - | - | 15 (0.0/0.08) |
| toluene | 0.0 (21/0.13) | - | - | 11 (0.0/0.15) | |
| acetonitrile | 0.0 (22/0.22) | - | - | 6.7 (0.0/0.27) | |
| 5 | vacuum | 0.0 (18/0.26) | - (-) | 9.6 (-) | 9.7 (6.2/0.05) |
| toluene | 0.0 (12/0.38) | - (-) | 7.8 (-) | 7.9 (1.37/0.10) | |
| acetonitrile | 0.0 (9.7/0.54) | - (1.22/0.32) | 5.6 (1.18/0.52) | 5.4 (0.0/0.20) | |
| 6 | vacuum | 0.0 (12/0.23) | - (-) | 7.4 (8.3/0.23) | 9.1 (0.0/0.05) |
| toluene | 0.0 (-) | - (6.4/0.30) | 5.5 (5.1/0.34) | 7.6 (0.0/0.10) | |
| acetonitrile | 0.0 (-) | 2.2 (3.3/0.52) | 3.2 (1.6/0.44) | 5.6 (0.0/0.19) | |
| 7 | vacuum | 0.0 (14/0.30) | - (-) | 11 (-) | 10 (0.86/0.04) |
| toluene | 0.0 (11/0.46) | - (-) | 9.6 (-) | 8.7 (0.0/0.07) | |
| acetonitrile | 0.0 (9.6/0.68) | - (5.6/0.37) | 7.1 (4.4/0.55) | 6.2 (0.0/0.15) | |
| 11 | vacuum | 0.0 (-) | - (-) | 11 (-) | 9.7 (24/0.14) |
| toluene | 0.0 (-) | - (-) | 9.7 (-) | 7.4 (21/0.13) | |
| acetonitrile | 0.0 (-) | - (-) | 7.0 (1.06/0.24) | 4.1 (6.8/0.15) | |
| 12 | vacuum | 0.0 (-) | - (-) | 9.7 (-) | 8.1 (14/0.06) |
| toluene | 0.0 (-) | - (-) | 8.3 (-) | 6.1 (11/0.17) | |
| acetonitrile | 0.0 (4.9/0.88) | - (-) | 6.4 (1.20/0.87) | 3.4 (0.88/0.85) | |
| Compound | Environment | E1K | K1K | K2K | K2E |
|---|---|---|---|---|---|
| Dipole moment, in D | |||||
| 4 | vacuum/ acetonitrile | 1.2/1.8 (1.3/1.9) | 7.9/12.8 (9.0/13.3) | ||
| 5 | 2.2/3.1 (2.3/3.3) | -/- (-/6.6) | 5.4/8.7 (-/8.2) | 6.1/10.0 (7.0/10.6) | |
| 6 | 3.8/5.4 (4.0/-) | -/8.3 (-/8.7) | 5.6/8.7 (5.3/8.1) | 4.8/8.0 (5.2/8.4) | |
| 7 | 3.2/4.3 (3.3/5.3) | -/- (-/8.8) | 6.5/10.3 (9.0/10.2) | 6.1/9.7 (6.5/10.7) | |
| 11 | 3.4/4.0 (-/-) | -/- (-/-) | 8.6/12.3 (-/11.7) | 10.7/15.5 (11.5/15.5) | |
| 12 | 6.2/7.8 (-/9.4) | -/- (-) | 6.5/8.9 (-/9.1) | 9.0/13.1 (10.4/14.7) | |
| dihedral angle α (Scheme 4), in o | |||||
| 5 | vacuum | 22 (16) | - (-) | 180 (-) | 180 (160) |
| toluene | 24 (15) | - (-) | 180 (-) | 173 (157) | |
| acetonitrile | 25 (15) | - (42) | 180 (163) | 164 (155) | |
| 6 | vacuum | 19 (13) | - (-) | 179 (169) | 176 (160) |
| toluene | 21 (-) | - (39) | 179 (167) | 174 (157) | |
| acetonitrile | 24 (-) | 2 (20) | 179 (166) | 169 (154) | |
| 7 | vacuum | 23 (15) | - (-) | 180 (-) | 176 (163) |
| toluene | 24 (15) | - (-) | 179 (-) | 168 (161) | |
| acetonitrile | 26 (15) | - (35) | 178 (169) | 162 (160) | |
| 11 | vacuum | 22 (-) | - (-) | 180 (-) | 173 (159) |
| toluene | 23 (-) | - (-) | 180 (-) | 171 (160) | |
| acetonitrile | 24 (-) | - (-) | 179 (157) | 168 (165) | |
| 12 | vacuum | 22 (-) | - (-) | 180 (-) | 179 (160) |
| toluene | 23 (-) | - (-) | 180 (-) | 176 (159) | |
| acetonitrile | 25 (17) | - (-) | 180 (180) | 169 (160) | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgiev, A.; Antonov, L. 8-(Pyridin-2-yl)quinolin-7-ol as a Platform for Conjugated Proton Cranes: A DFT Structural Design. Micromachines 2020, 11, 901. https://doi.org/10.3390/mi11100901
Georgiev A, Antonov L. 8-(Pyridin-2-yl)quinolin-7-ol as a Platform for Conjugated Proton Cranes: A DFT Structural Design. Micromachines. 2020; 11(10):901. https://doi.org/10.3390/mi11100901
Chicago/Turabian StyleGeorgiev, Anton, and Liudmil Antonov. 2020. "8-(Pyridin-2-yl)quinolin-7-ol as a Platform for Conjugated Proton Cranes: A DFT Structural Design" Micromachines 11, no. 10: 901. https://doi.org/10.3390/mi11100901
APA StyleGeorgiev, A., & Antonov, L. (2020). 8-(Pyridin-2-yl)quinolin-7-ol as a Platform for Conjugated Proton Cranes: A DFT Structural Design. Micromachines, 11(10), 901. https://doi.org/10.3390/mi11100901