
Applicability of Artificial Vascularized Liver Tissue to Proteomic Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Two-Dimensional Cell Culture
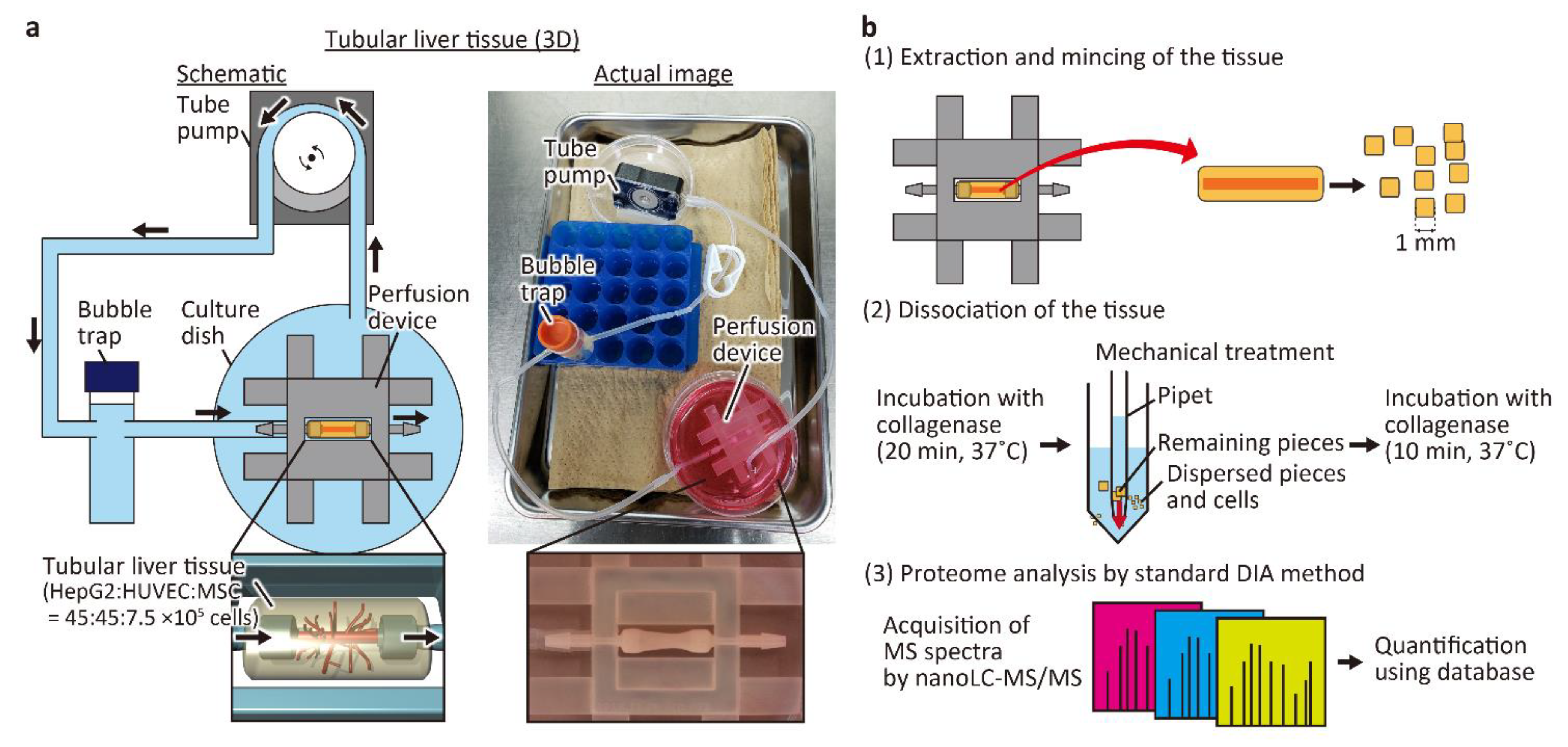
2.2. Preparation of the Tubular Liver Tissue
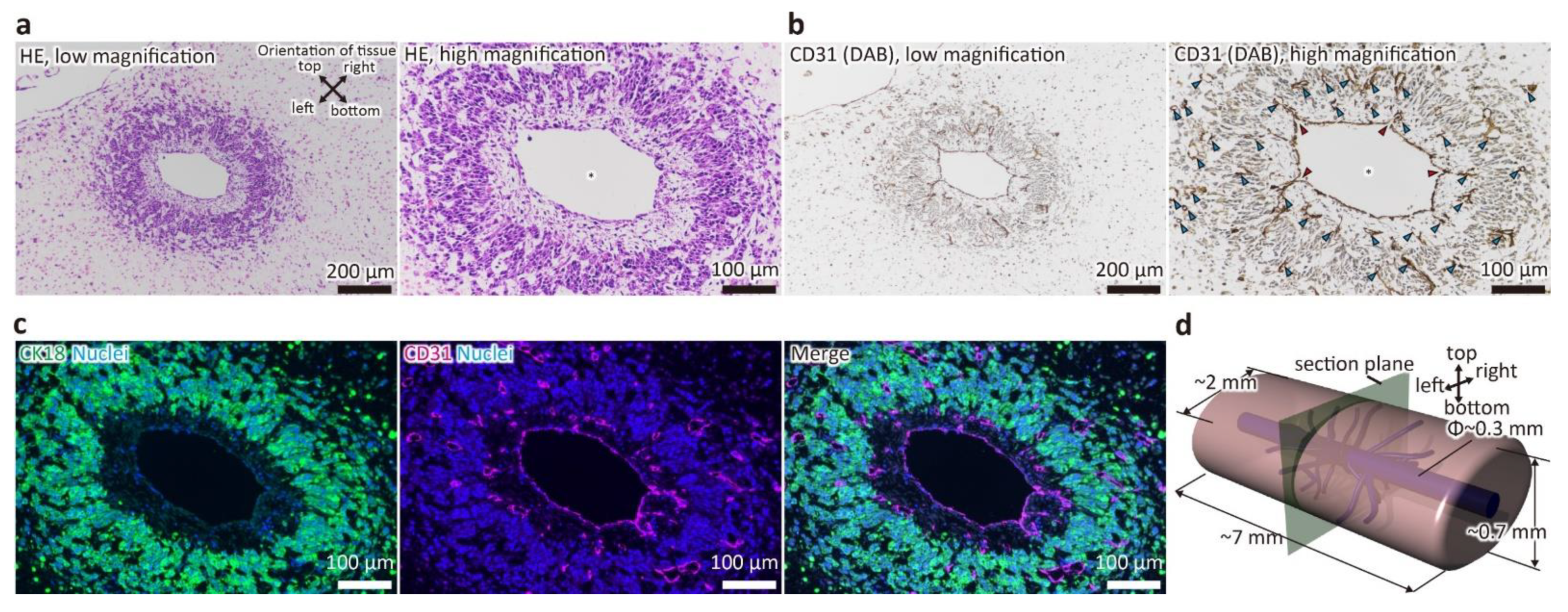
2.3. Histological Analysis
2.4. Cell Collection and Proteomic Analysis
3. Results and Discussion
3.1. Evaluation of Tissue Morphology
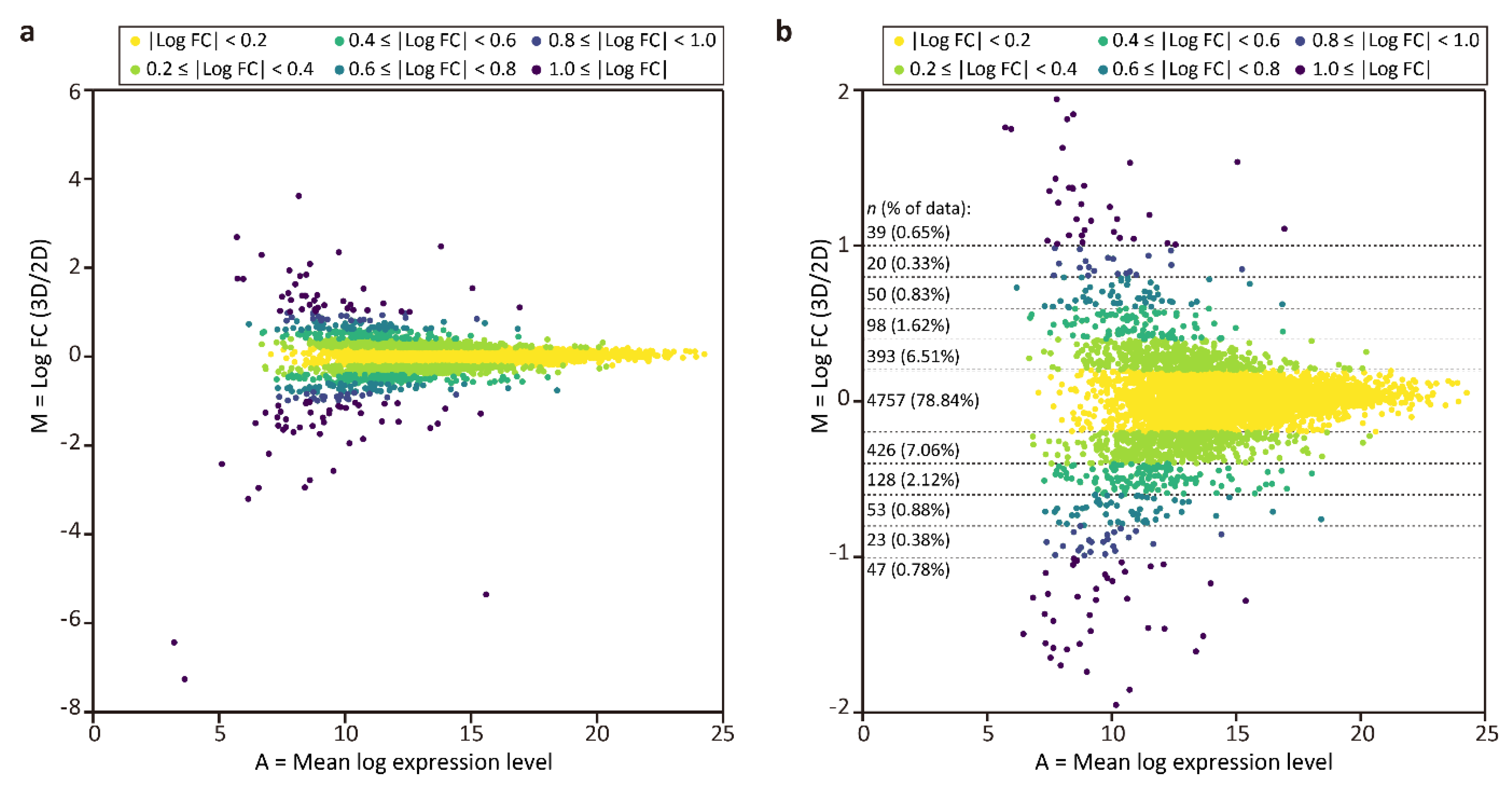
3.2. Evaluation of the Proteomic Analysis Process
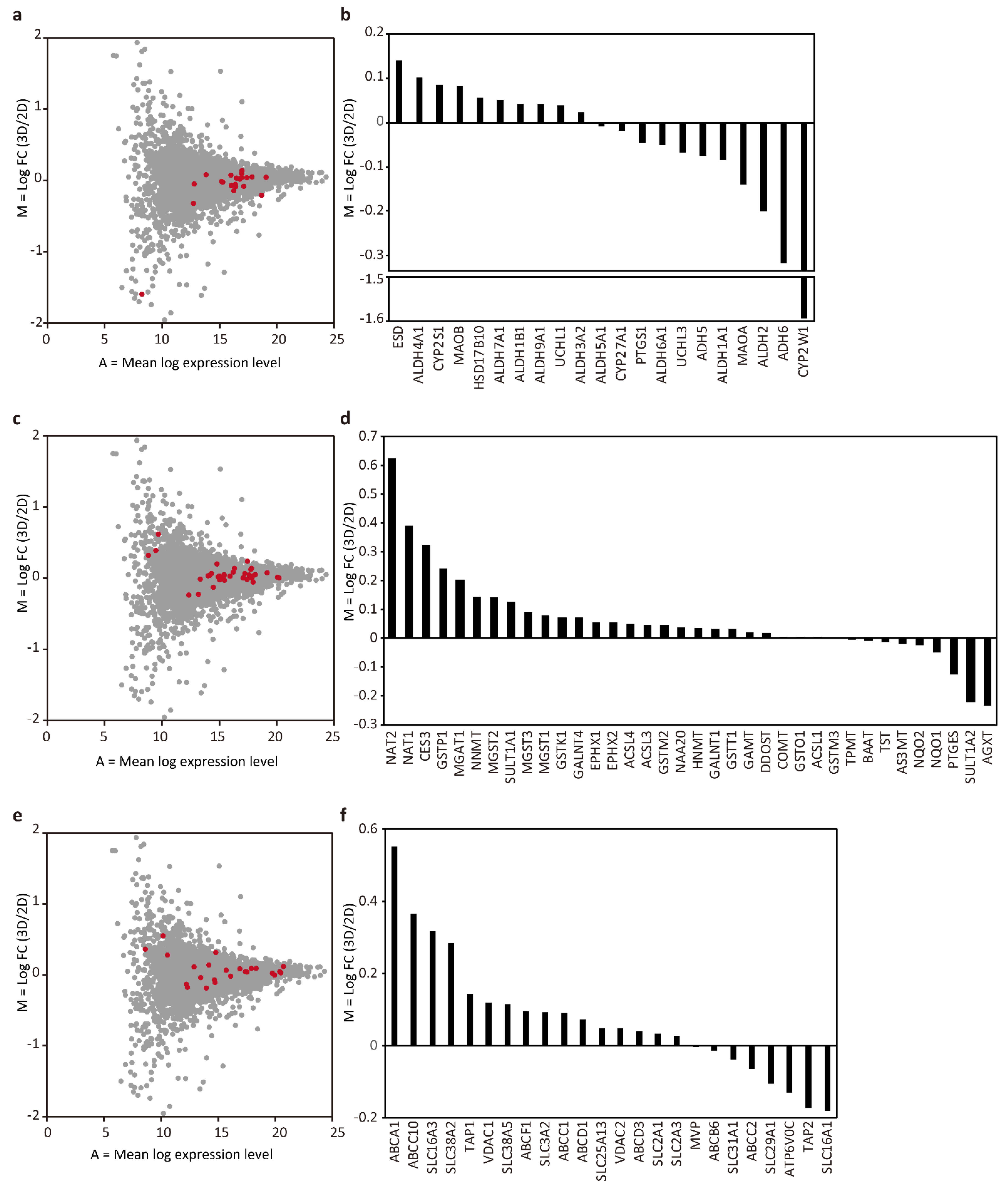
3.3. Proteomic Comparison between the Tubular Liver Tissue and 2D Culture
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jin, Y.; Kim, J.; Lee, J.S.; Min, S.; Kim, S.; Ahn, D.-H.; Kim, Y.-G.; Cho, S.-W. Vascularized Liver Organoids Generated Using Induced Hepatic Tissue and Dynamic Liver-Specific Microenvironment as a Drug Testing Platform. Adv. Funct. Mater. 2018, 28, 1801954. [Google Scholar] [CrossRef]
- Saheli, M.; Sepantafar, M.; Pournasr, B.; Farzaneh, Z.; Vosough, M.; Piryaei, A.; Baharvand, H. Three-dimensional liver-derived extracellular matrix hydrogel promotes liver organoids function. J. Cell. Biochem. 2018, 119, 4320–4333. [Google Scholar] [CrossRef]
- Bell, C.C.; Dankers, A.C.A.; Lauschke, V.M.; Sison-Young, R.; Jenkins, R.; Rowe, C.; Goldring, C.E.; Park, K.; Regan, S.L.; Walker, T.; et al. Comparison of Hepatic 2D Sandwich Cultures and 3D Spheroids for Long-term Toxicity Applications: A Multicenter Study. Toxicol. Sci. 2018, 162, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Luckert, C.; Schulz, C.; Lehmann, N.; Thomas, M.; Hofmann, U.; Hammad, S.; Hengstler, J.G.; Braeuning, A.; Lampen, A.; Hessel, S. Comparative analysis of 3D culture methods on human HepG2 cells. Arch. Toxicol. 2017, 91, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Mizukami, Y.; Tanaka, Y.; Nishikawa, T.; Ogino, Y.; Nishikawa, M.; Konishi, S.; Kusamori, K.; Mizuno, N.; Takakura, Y.; et al. Optimization of Albumin Secretion and Metabolic Activity of Cytochrome P450 1A1 of Human Hepatoblastoma HepG2 Cells in Multicellular Spheroids by Controlling Spheroid Size. Biol. Pharm. Bull. Pharm. Bull. 2017, 40, 334–338. [Google Scholar] [CrossRef] [Green Version]
- Takebe, T.; Zhang, R.R.; Koike, H.; Kimura, M.; Yoshizawa, E.; Enomura, M.; Koike, N.; Sekine, K.; Taniguchi, H. Generation of a vascularized and functional human liver from an iPSC-derived organ bud transplant. Nat. Protoc. 2014, 9, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Ramaiahgari, S.C.; den Braver, M.W.; Herpers, B.; Terpstra, V.; Commandeur, J.N.M.; van de Water, B.; Price, L.S. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Arch. Toxicol. 2014, 88, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Mori, N.; Akagi, Y.; Imai, Y.; Takayama, Y.; Kida, Y.S. Fabrication of Perfusable Vascular Channels and Capillaries in 3D Liver-like Tissue. Sci. Rep. 2020, 10, 5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, N.; Kida, Y.S. Expression of genes involved in drug metabolism differs between perfusable 3D liver tissue and conventional 2D-cultured hepatocellular carcinoma cells. FEBS Open Bio 2020. [Google Scholar] [CrossRef]
- Krasny, L.; Huang, P.H. Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Mol. Omi. 2021. [Google Scholar] [CrossRef] [PubMed]
- Walgren, J. Application of proteomic technologies in the drug development process. Toxicol. Lett. 2004, 149, 377–385. [Google Scholar] [CrossRef]
- Hewick, R.M.; Lu, Z.; Wang, J.H. Proteomics in Drug Discovery. In Advances in Protein Chemistry; Elsevier Inc.: Amsterdam, The Netherlands, 2003; Volume 65, pp. 309–342. [Google Scholar]
- Ghazalpour, A.; Bennett, B.; Petyuk, V.A.; Orozco, L.; Hagopian, R.; Mungrue, I.N.; Farber, C.R.; Sinsheimer, J.; Kang, H.M.; Furlotte, N.; et al. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallström, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Qiu, Z.; Ouyang, W.; Miao, J.; Xiong, P.; Mao, D.; Feng, K.; Li, M.; Luo, M.; Xiao, H.; et al. Hepatic transcriptome and proteome analyses provide new insights into the regulator mechanism of dietary avicularin in diabetic mice. Food Res. Int. 2019, 125, 108570. [Google Scholar] [CrossRef] [PubMed]
- Moritz, A.; Li, Y.; Guo, A.; Villen, J.; Wang, Y.; MacNeill, J.; Kornhauser, J.; Sprott, K.; Zhou, J.; Possemato, A.; et al. Akt-RSK-S6 Kinase Signaling Networks Activated by Oncogenic Receptor Tyrosine Kinases. Sci. Signal. 2010, 3, ra64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, Y.; Watanabe, E.; Umeyama, T.; Nakajima, D.; Hattori, M.; Honda, K.; Ohara, O. Optimization of Data-Independent Acquisition Mass Spectrometry for Deep and Highly Sensitive Proteomic Analysis. Int. J. Mol. Sci. 2019, 20, 5932. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Fresno, C.; Fernandez, E.A. RDAVIDWebService: A versatile R interface to DAVID. Bioinformatics 2013, 29, 2810–2811. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 25 March 2021).
- Guo, L.; Dial, S.; Shi, L.; Branham, W.; Liu, J.; Fang, J.-L.; Green, B.; Deng, H.; Kaput, J.; Ning, B. Similarities and Differences in the Expression of Drug-Metabolizing Enzymes between Human Hepatic Cell Lines and Primary Human Hepatocytes. Drug Metab. Dispos. 2011, 39, 528–538. [Google Scholar] [CrossRef] [Green Version]
- Stenstedt, K.; Hallstrom, M.; Johansson, I.; Ingelman-Sundberg, M.; Ragnhammar, P.; Edler, D. The expression of CYP2W1: A prognostic marker in colon cancer. Anticancer Res. 2012, 32, 3869–3874. [Google Scholar]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.-R.; Ueno, Y.; Zheng, Y.-W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Hurrell, T.; Segeritz, C.-P.; Vallier, L.; Lilley, K.S.; Cromarty, A.D. Proteomic Comparison of Various Hepatic Cell Cultures for Preclinical Safety Pharmacology. Toxicol. Sci. 2018, 164, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, L.; Axnick, J.; Buschmann, T.; Henning, C.; Urner, S.; Fang, S.; Nurmi, H.; Eichhorst, N.; Holtmeier, R.; Bódis, K.; et al. Mechanosensing by β1 integrin induces angiocrine signals for liver growth and survival. Nature 2018, 562, 128–132. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Culture Method | Number of Obtainable Cells | Number of Cells Used for Proteomic Analysis | Number of Identified and Quantified Proteins |
|---|---|---|---|
| Tubular liver tissue | (5.25 ± 0.39) × 105/device | 8.62 × 105 * | 6032 |
| 2D culture | ~0.5–5 × 104/cm2 † | 1.45 × 106 ‡ | 6034 |
| GO ID | Description | p Value |
|---|---|---|
| GO:0018105 | peptidyl-serine phosphorylation | 5.01 × 10−4 |
| GO:0006096 | glycolytic process | 5.32 × 10−4 |
| GO:0097421 | liver regeneration | 4.01 × 10−4 |
| GO:0045943 | positive regulation of transcription from RNA polymerase I promoter | 5.23 × 10−3 |
| GO:0033617 | mitochondrial respiratory chain complex IV assembly | 1.34 × 10−2 |
| GO:0016311 | dephosphorylation | 1.56 × 10−2 |
| GO:0071539 | protein localization to centrosome | 1.68 × 10−2 |
| GO:0006895 | Golgi to endosome transport | 1.86 × 10−2 |
| GO:0070194 | synaptonemal complex disassembly | 2.21 × 10−2 |
| GO:0022037 | metencephalon development | 2.21 × 10−2 |
| GO ID | Description | p Value |
|---|---|---|
| GO:0032418 | lysosome localization | 9.57 × 10−4 |
| GO:0048041 | focal adhesion assembly | 3.21 × 10−3 |
| GO:0001682 | tRNA 5′-leader removal | 7.95 × 10−3 |
| GO:0016236 | macroautophagy | 1.51 × 10−2 |
| GO:0034613 | cellular protein localization | 1.65 × 10−2 |
| GO:0030307 | positive regulation of cell growth | 2.11 × 10−2 |
| GO:0070584 | mitochondrion morphogenesis | 2.31 × 10−2 |
| GO:1900186 | negative regulation of clathrin-mediated endocytosis | 2.49 × 10−2 |
| GO:0090155 | negative regulation of sphingolipid biosynthetic process | 2.49 × 10−2 |
| GO:0007094 | mitotic spindle assembly checkpoint | 2.55 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, N.; Kida, Y.S. Applicability of Artificial Vascularized Liver Tissue to Proteomic Analysis. Micromachines 2021, 12, 418. https://doi.org/10.3390/mi12040418
Mori N, Kida YS. Applicability of Artificial Vascularized Liver Tissue to Proteomic Analysis. Micromachines. 2021; 12(4):418. https://doi.org/10.3390/mi12040418
Chicago/Turabian StyleMori, Nobuhito, and Yasuyuki S. Kida. 2021. "Applicability of Artificial Vascularized Liver Tissue to Proteomic Analysis" Micromachines 12, no. 4: 418. https://doi.org/10.3390/mi12040418
APA StyleMori, N., & Kida, Y. S. (2021). Applicability of Artificial Vascularized Liver Tissue to Proteomic Analysis. Micromachines, 12(4), 418. https://doi.org/10.3390/mi12040418