
Refined Method for Droplet Microfluidics-Enabled Detection of Plasmodium falciparum Encoded Topoisomerase I in Blood from Malaria Patients

Abstract
:1. Introduction
2. Experimental Section
2.1. Cloning and Purification of Topoisomerase I
2.2. Blood Sampling
2.3. Enzyme Extraction Using Droplet Microfluidics
2.4. Lysis of Blood Using Bead Beating
2.5. Substrate Preparation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | 5ʹ Modification |
|---|---|---|
| Primer 1 | ATTTTTCTAAGTCTTTTAGATCGAACGACTCAGAATGATGCATGTATACTAAACTCACAAATTAGAGC | |
| Primer 2 | TTTTTTTTTTTTTTTTTTTTTTTTTGCTTTCTCATAGCTCACGCTG | |
| Am-Cl | [AmC6F]ACTACCATTCTGAGTCGTTCGATCTAAAAGACTTAGA | Amine |
| Biotin-Cl | Biotin-ACTACCATTCTGAGTCGTTCGATCTAAAAGACTTAGA | Biotin |
| Cl | ACTACCATTCTGAGTCGTTCGATCTAAAAGACTTAGA | |
| NCl | ATTTTTCTAAGTCTTTTAGATCGAACGACTCAGAATG |
2.6. Polyacrylamide Gel-Enabled Detection of pfTopoI Activity
2.7. Fluorescence Microscopy-Enabled Detection of pfTopoI Activity
2.8. Radioactivity-Enabled Detection of pfTopoI Activity Using Magnetic Beads
3. Results
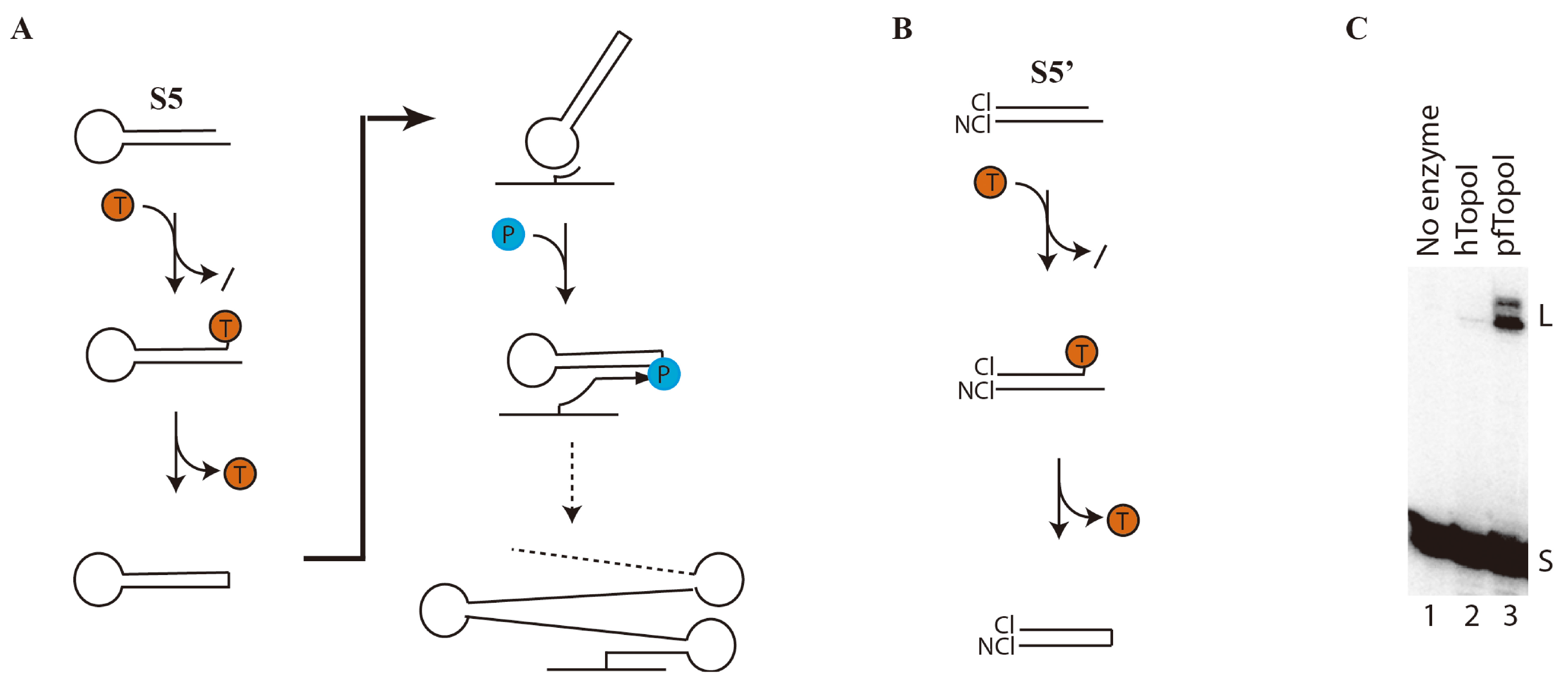
3.1. Specific and Efficient DNA Substrate for pfTopoI

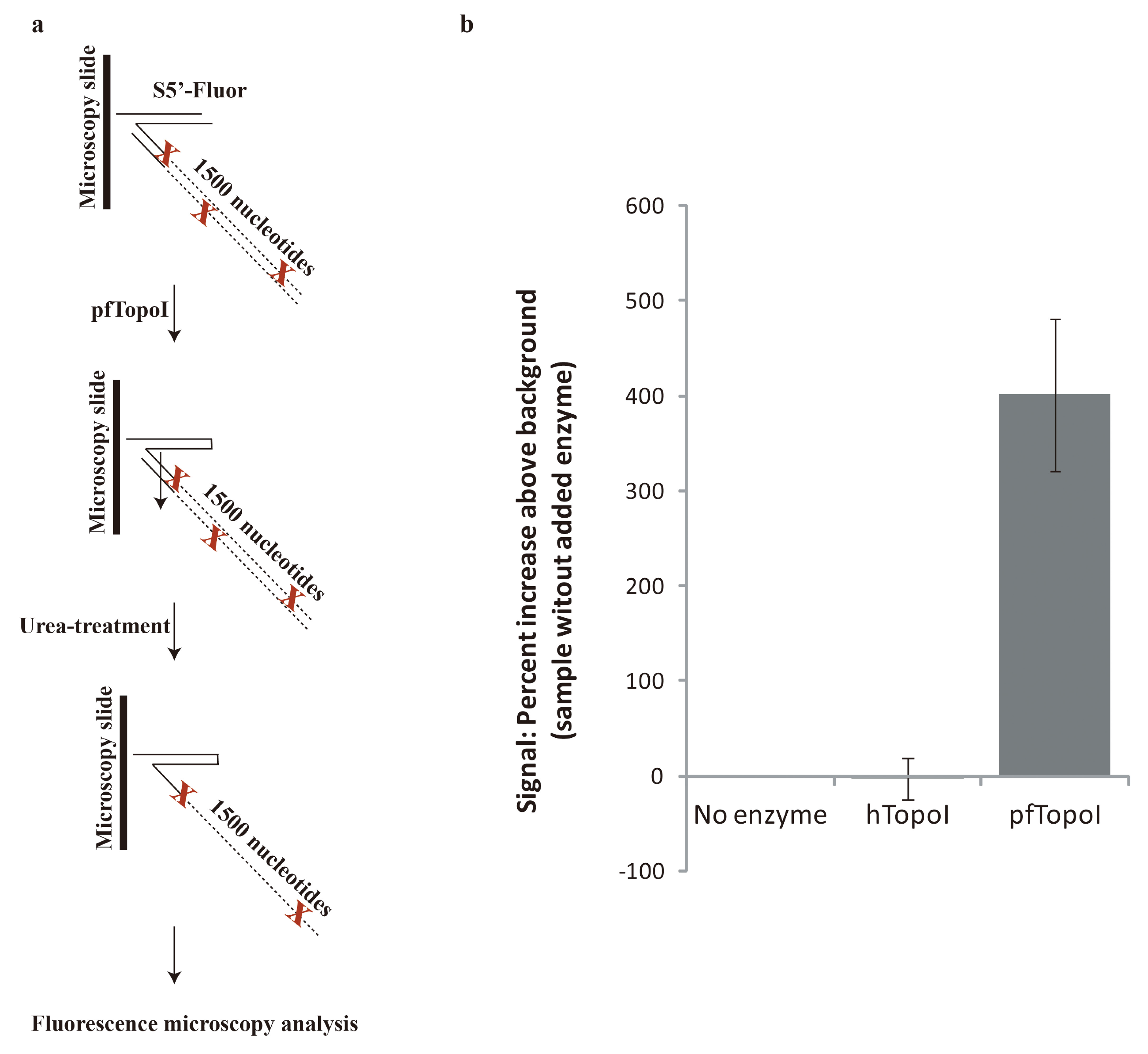
3.2. Towards Development of a User-Friendly Readout


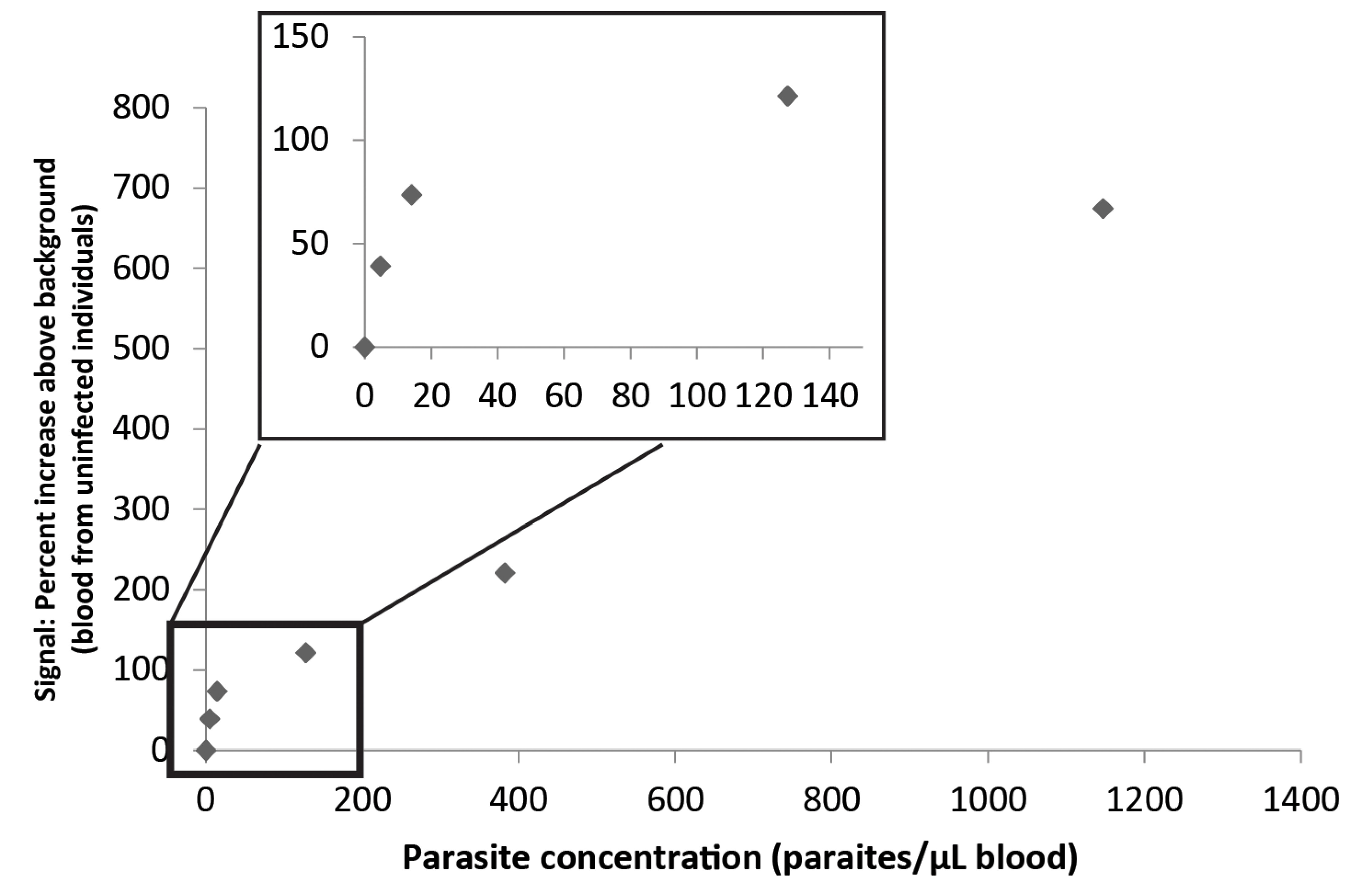
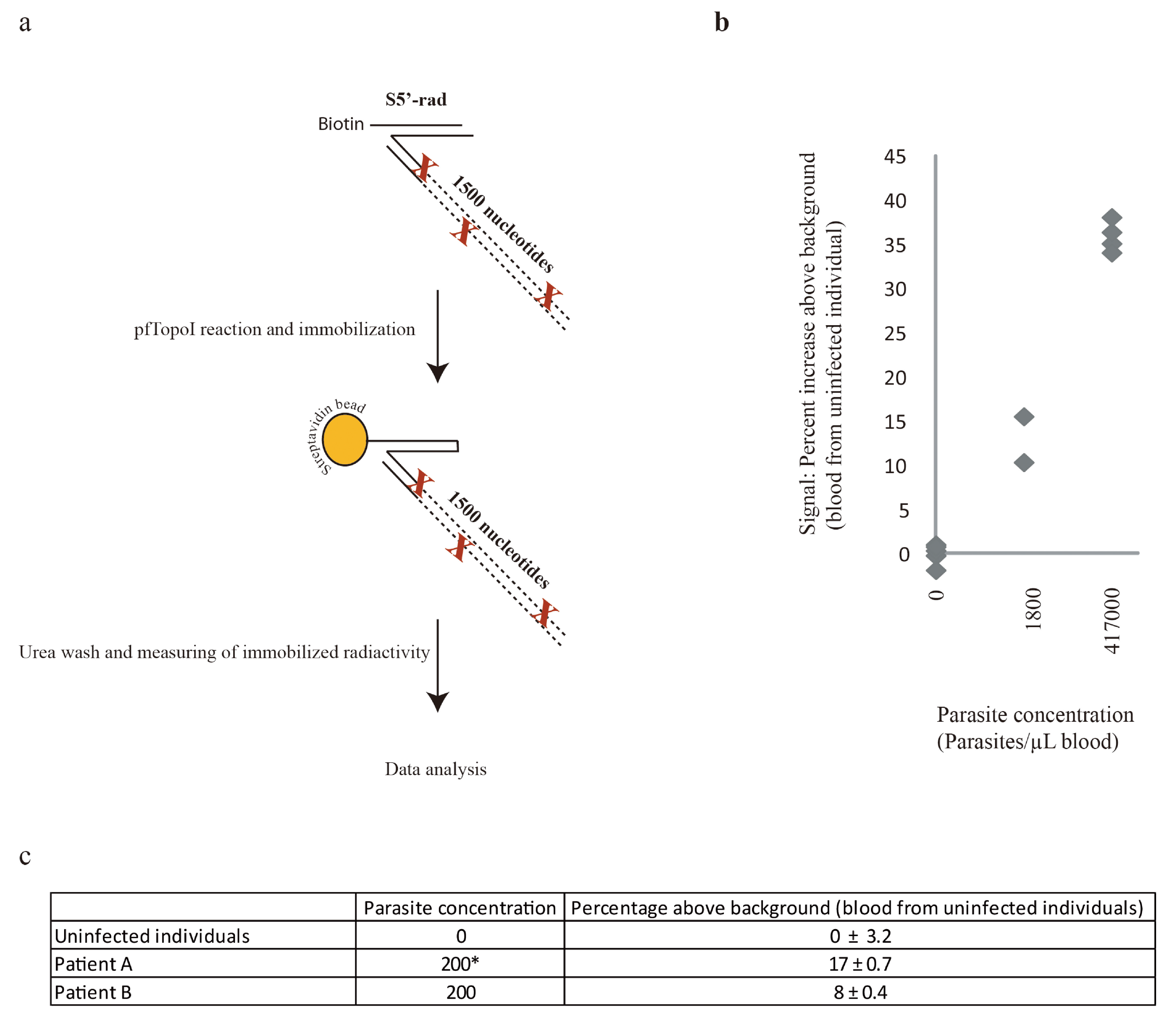
3.3. Combination of the Enzyme-Based Malaria Detection with a Droplet Microfluidics Platform

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| P. falciparum | Plasmodium falciparum |
| pfTopoI | P. falciparum topoisomerae I |
| hTopoI | human topoisomerase I |
| REEAD | rolling circle enhanced enzyme activity detection |
| PCR | polymerase chain reaction |
References
- Tangpukdee, N.; Duangdee, C.; Wilairatana, P.; Krudsood, S. Malaria diagnosis: A brief review. Korean J. Parasitol. 2009, 47, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.; Nielsen, C.J.; Labouriau, R.; Roy, A.; Tesauro, C.; Jensen, P.W.; Harmsen, C.; Kristoffersen, E.L.; Chiu, Y.L.; Frohlich, R.; et al. Droplet microfluidics platform for highly sensitive and quantitative detection of malaria-causing plasmodium parasites based on enzyme activity measurement. ACS Nano 2012, 6, 10676–10683. [Google Scholar] [CrossRef] [PubMed]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Ann. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Lisby, M.; Olesen, J.R.; Skouboe, C.; Krogh, B.O.; Straub, T.; Boege, F.; Velmurugan, S.; Martensen, P.M.; Andersen, A.H.; Jayaram, M.; et al. Residues within the N-terminal domain of human topoisomerase I play a direct role in relaxation. J. Biol. Chem. 2001, 276, 20220–20227. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Xia, Y.; Whitesides, G.M. Soft lithography for micro- and nanoscale patterning. Nat. Protoc. 2010, 5, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.P.; Grigsby, C.L.; Zhao, F.; Leong, K.W. Tuning physical properties of nanocomplexes through microfluidics-assisted confinement. Nano Lett. 2011, 11, 2178–2182. [Google Scholar] [CrossRef] [PubMed]
- Juul, S.; Ho, Y.P.; Koch, J.; Andersen, F.F.; Stougaard, M.; Leong, K.W.; Knudsen, B.R. Detection of single enzymatic events in rare or single cells using microfluidics. ACS Nano 2011, 5, 8305–8310. [Google Scholar] [CrossRef] [PubMed]
- Abate, A.R.; Weitz, D.A. Syringe-vacuum microfluidics: A portable technique to create monodisperse emulsions. Biomicrofluidics 2011, 5, 14107. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, C.; Juul, S.; Arno, B.; Nielsen, C.J.; Fiorani, P.; Frohlich, R.F.; Andersen, F.F.; Desideri, A.; Stougaard, M.; Petersen, E.; et al. Specific detection of topoisomerase I from the malaria causing P. falciparum parasite using isothermal rolling circle amplification. In Proceedings of 2012 Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), San Diego, CA, USA, 28 August–1 September 2012; pp. 2416–2419.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hede, M.S.; Okorie, P.N.; Fruekilde, S.K.; Fjelstrup, S.; Thomsen, J.; Franch, O.; Tesauro, C.; Bugge, M.T.; Christiansen, M.; Picot, S.; et al. Refined Method for Droplet Microfluidics-Enabled Detection of Plasmodium falciparum Encoded Topoisomerase I in Blood from Malaria Patients. Micromachines 2015, 6, 1505-1513. https://doi.org/10.3390/mi6101432
Hede MS, Okorie PN, Fruekilde SK, Fjelstrup S, Thomsen J, Franch O, Tesauro C, Bugge MT, Christiansen M, Picot S, et al. Refined Method for Droplet Microfluidics-Enabled Detection of Plasmodium falciparum Encoded Topoisomerase I in Blood from Malaria Patients. Micromachines. 2015; 6(10):1505-1513. https://doi.org/10.3390/mi6101432
Chicago/Turabian StyleHede, Marianne Smedegaard, Patricia Nkem Okorie, Signe Kirk Fruekilde, Søren Fjelstrup, Jonas Thomsen, Oskar Franch, Cinzia Tesauro, Magnus Tobias Bugge, Mette Christiansen, Stéphane Picot, and et al. 2015. "Refined Method for Droplet Microfluidics-Enabled Detection of Plasmodium falciparum Encoded Topoisomerase I in Blood from Malaria Patients" Micromachines 6, no. 10: 1505-1513. https://doi.org/10.3390/mi6101432
APA StyleHede, M. S., Okorie, P. N., Fruekilde, S. K., Fjelstrup, S., Thomsen, J., Franch, O., Tesauro, C., Bugge, M. T., Christiansen, M., Picot, S., Lötsch, F., Mombo-Ngoma, G., Mischlinger, J., Adegnika, A. A., Pedersen, F. S., Ho, Y. -P., Petersen, E., Stougaard, M., Ramharter, M., & Knudsen, B. R. (2015). Refined Method for Droplet Microfluidics-Enabled Detection of Plasmodium falciparum Encoded Topoisomerase I in Blood from Malaria Patients. Micromachines, 6(10), 1505-1513. https://doi.org/10.3390/mi6101432