
An Inert Continuous Microreactor for the Isolation and Analysis of a Single Microbial Cell



Abstract
:
1. Introduction
2. Experimental Section
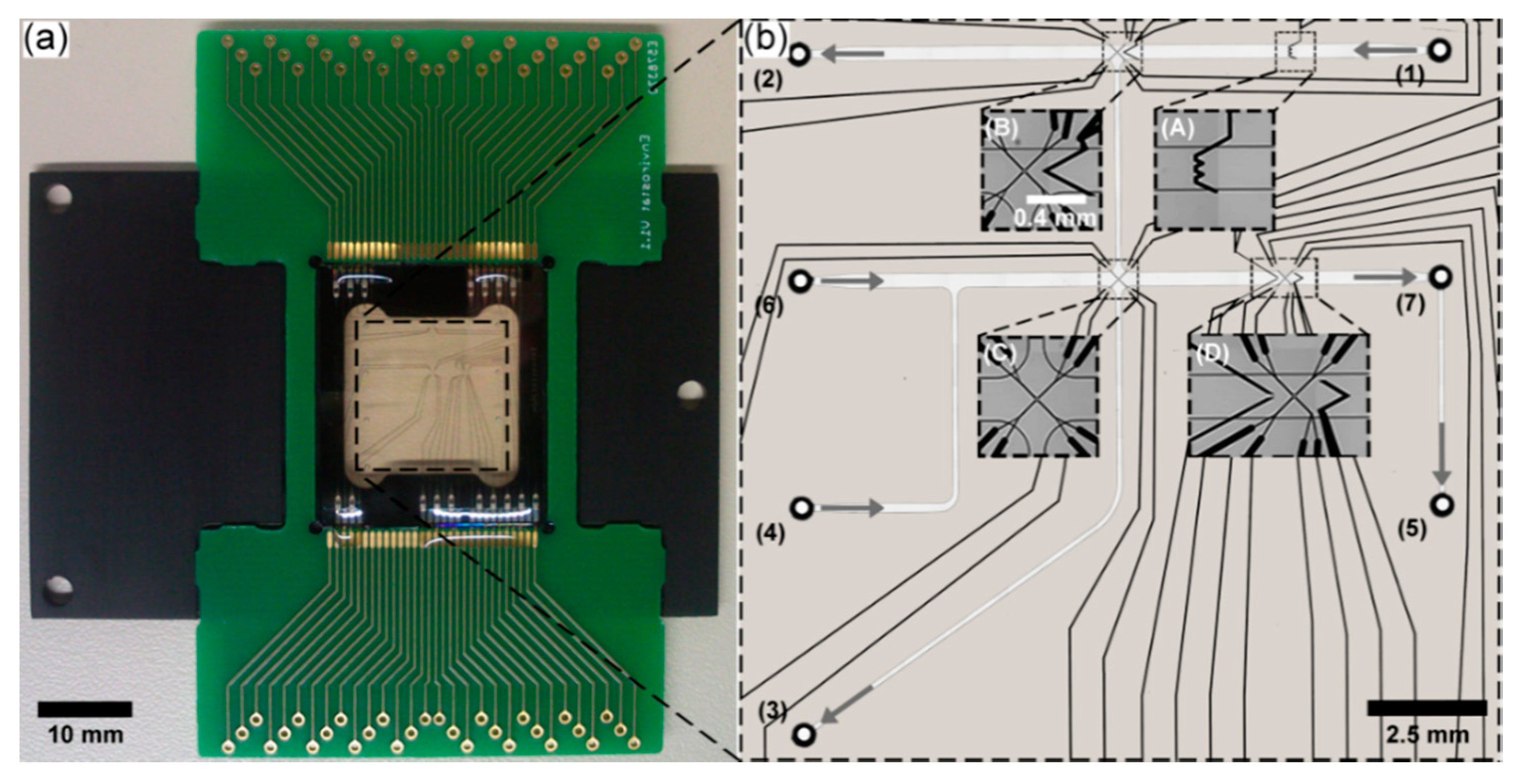
2.1. LOC Design
2.2. LOC Microfabrication
2.3. LOC Periphery
2.4. Cell Separation and Trapping with nDEP
2.5. Microscopy
2.6. Comparative Characterization of the LOC Device
2.7. Strains and Media

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain/Plasmid | Characteristic | Reference |
|---|---|---|
| C. glutamicum ATCC13032 | Wildtype strain, biotin-auxotroph | [39] |
| C. glutamicum DM1728 | pyc(P458S), hom(V59A) | [40] |
| C. glutamicum DM1730 | pyc(P458S), hom(V59A), lysC(T311I), Δpck | Evonik Industries |
| C. glutamicum DM1919 | pyc(P458S), hom(V59A), 2 copies of lysC(T311I), Δpck | Evonik Industries |
| pSenLys | Encodes LysG and its target promoter of lysE with a transcriptional fusion to eyfp, KnR | [38] |
2.8. Cultivation Conditions
3. Results and Discussion
3.1. Material Selection and Bonding Methods for an Inert nDEP-Based Single-Cell Analysis System
| Characteristics | SU-8-Glass (UV Cured Polymer) | PD 955 PY-Glass (Thermally Cured Polymer) | Glass-Glass (Direct Fusion) |
|---|---|---|---|
| Bond strength | 1–2 MPa [41] | >25 Mpa a | ~bond strength of glass (67 MPa) b |
| Biocompatibility | Compatible [42] | Compatible | Compatible |
| Solvent resistance | Resistant | Not resistant | Resistant |
| Chemically inert | Inert | Not inert | Inert |
| Bonding homogeneity | Homogeneous | Heterogeneous | Homogeneous |
3.2. Implementation of Functional Electrodes into Direct Fusion-Bonded LOCs for nDEP Cell Trapping

3.3. LOC Design and Characterization

3.4. Experimental Validation of Characteristics and Comparison of a Direct Fusion- and Adhesive-Bonded LOC Device
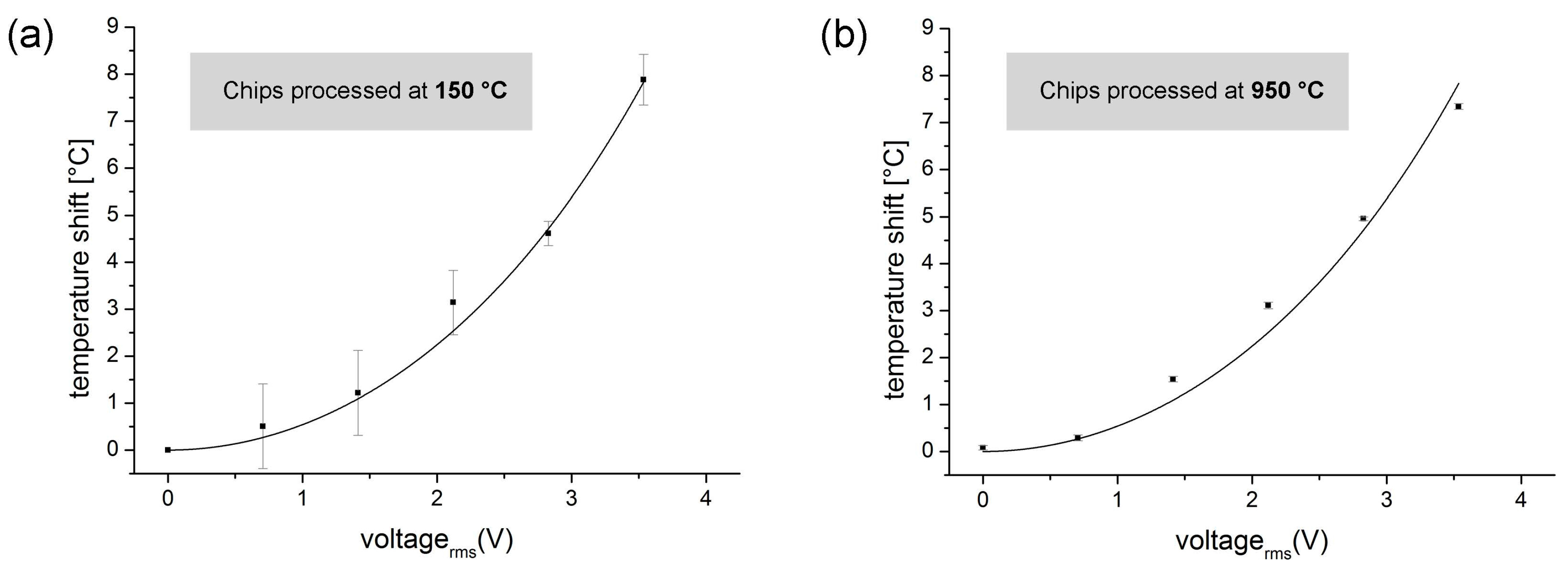
3.4.1. Quantitative Measurements of Joule Heating in a Direct Fusion- and Adhesive-Bonded LOC Device

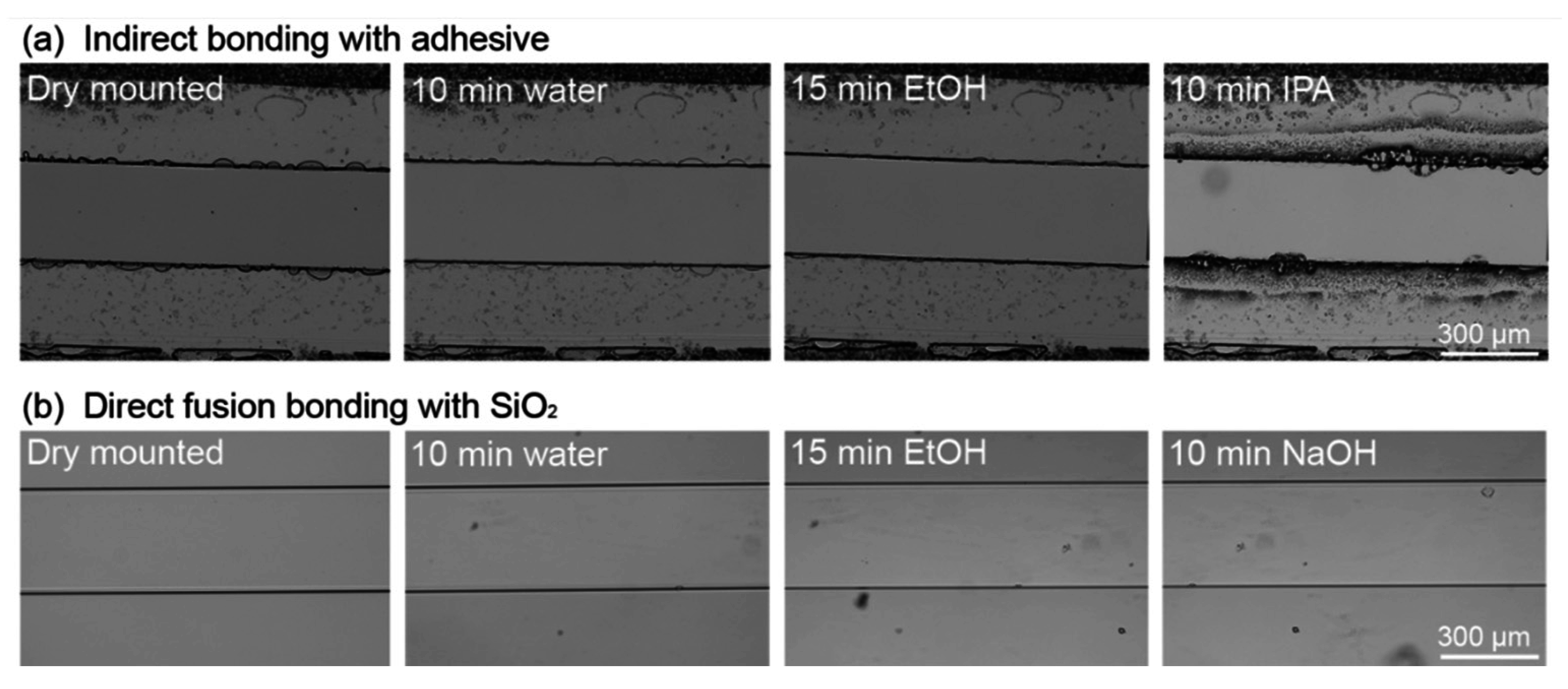
3.4.2. Solvent Resistance of a Direct Fusion- and Adhesive-Bonded LOC Device
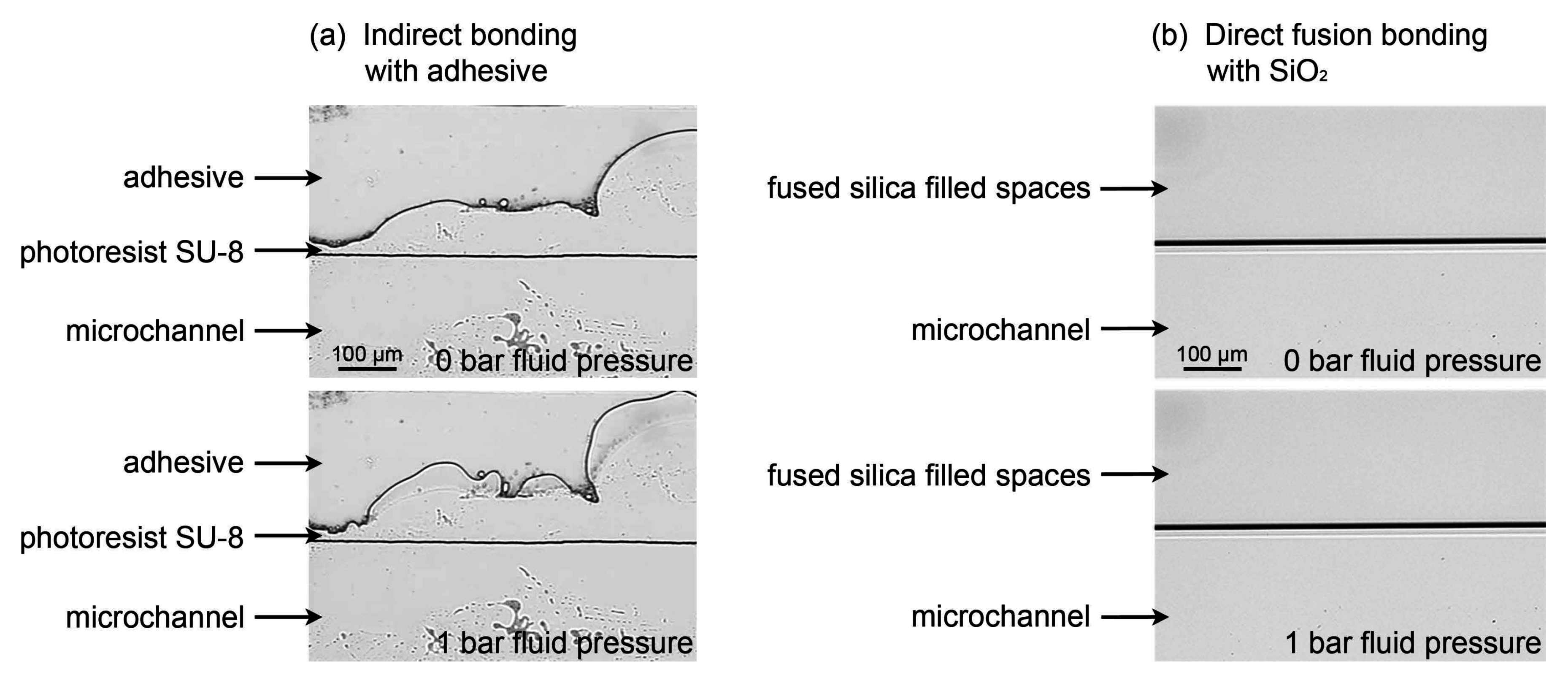
3.4.3. Pressure Resistance of a Direct Fusion- and Adhesive-Bonded LOC Device


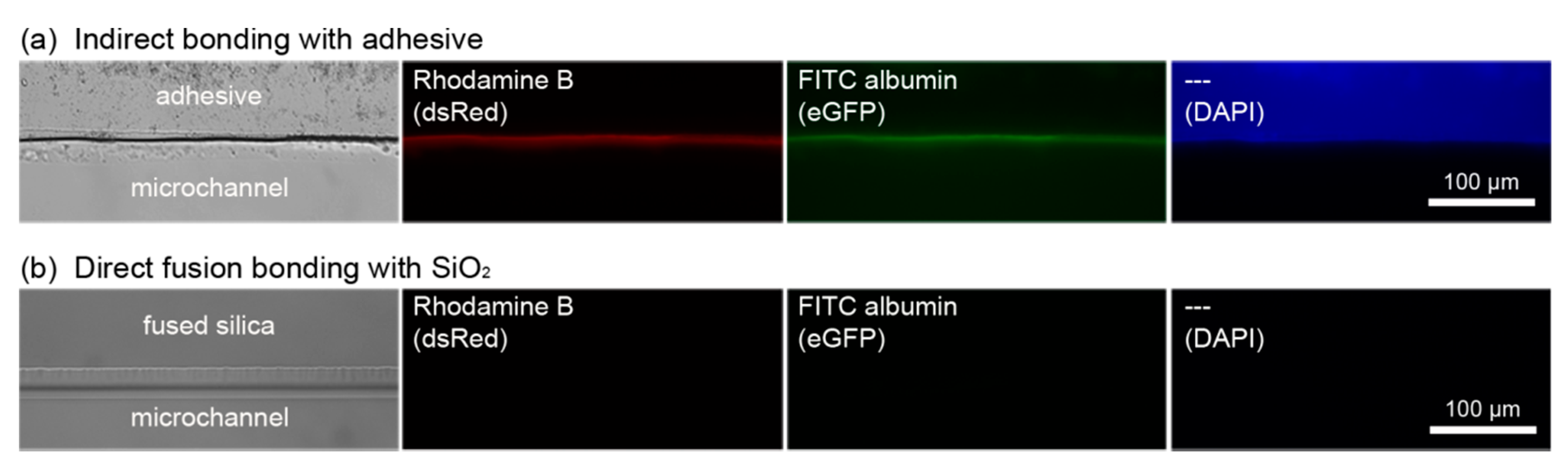
3.4.4. Adsorption of (Bio-)Molecules

3.5. Application of the LOC Device for Analyses of Single-Cell Growth and Productivity
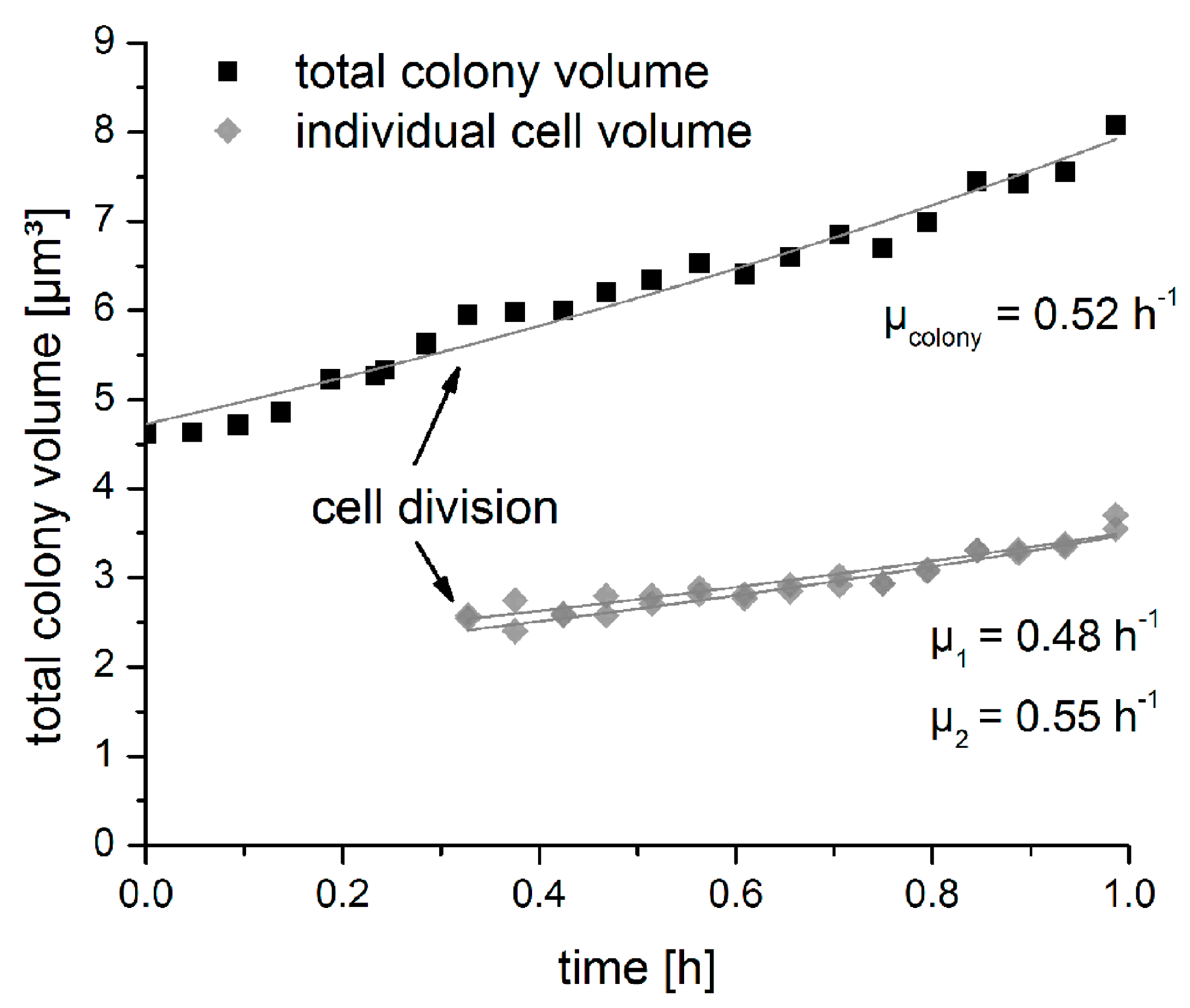
3.5.1. Growth of an Isolated Single Bacterial Cell

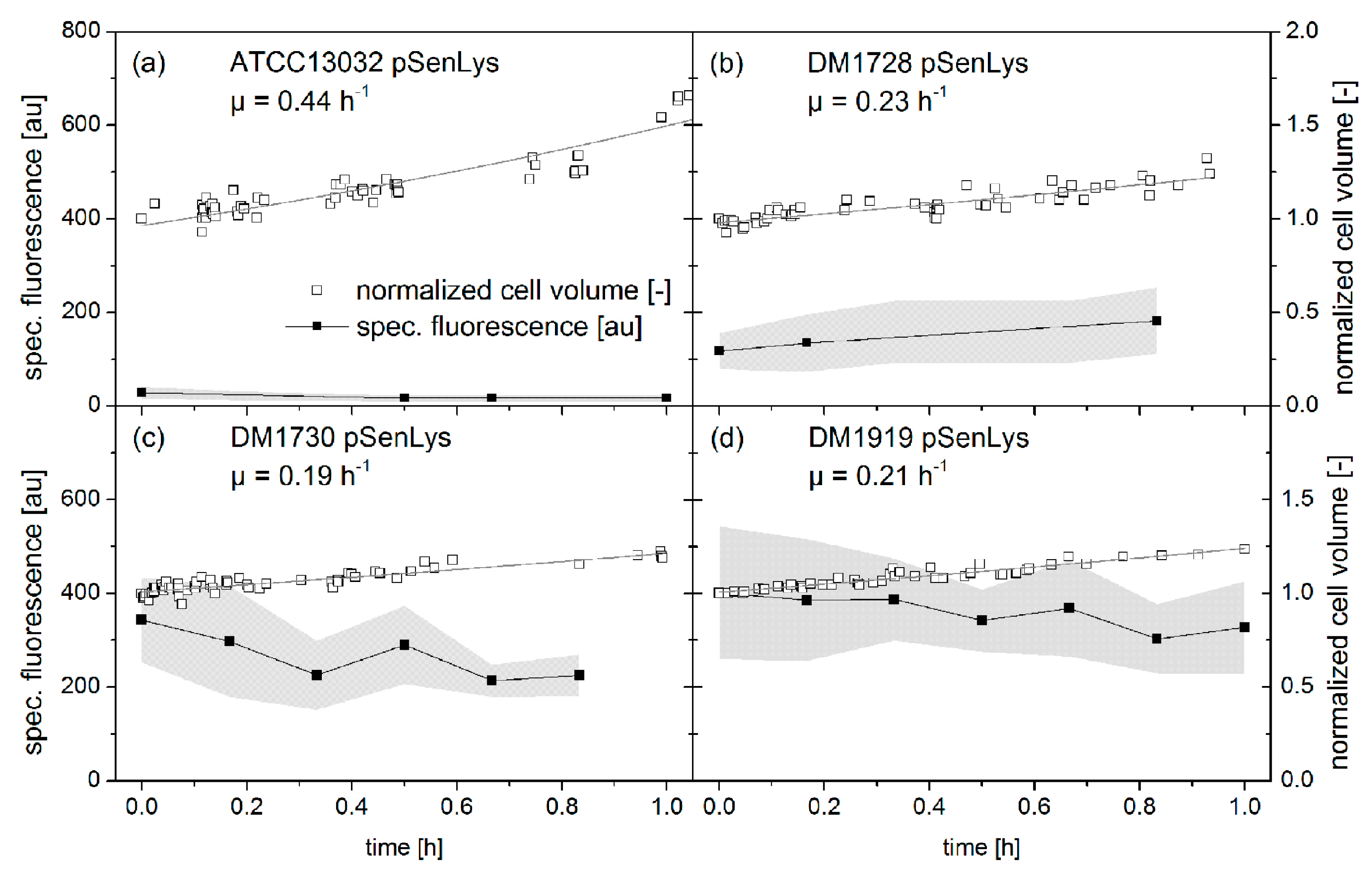
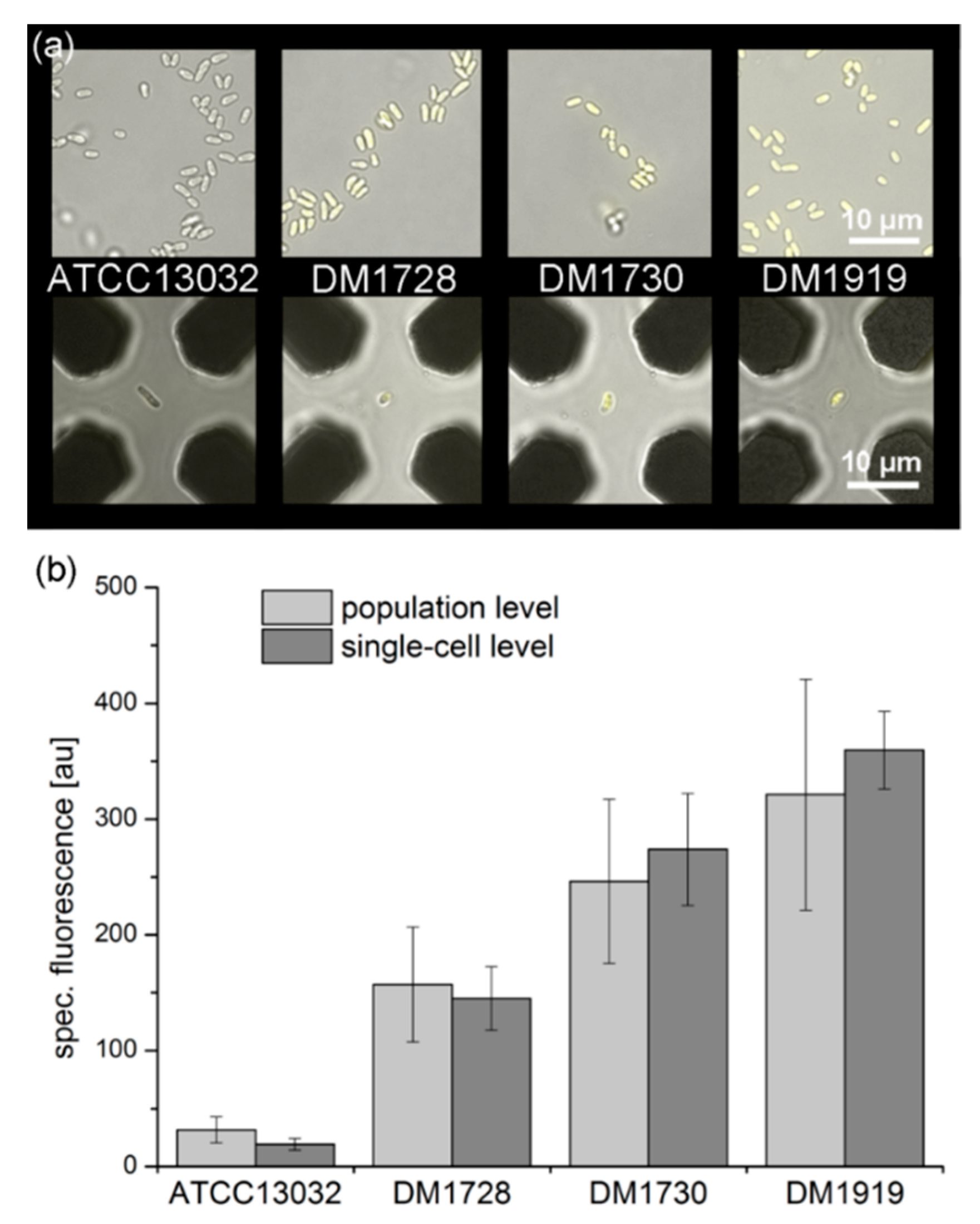
3.5.2. Qualitative eYFP-Reported l-Lysine Production at a Single-Cell Level


4. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
References
- Schmid, A.; Kortmann, H.; Dittrich, P.S.; Blank, L.M. Chemical and biological single cell analysis. Curr. Opin. Biotechnol. 2010, 21, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Arriaga, E.A. Determining biological noise via single cell analysis. Anal. Bioanal. Chem. 2009, 393, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Love, K.R.; Bagh, S.; Choi, J.; Love, J.C. Microtools for single-cell analysis in biopharmaceutical development and manufacturing. Trends Biotechnol. 2013, 31, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Dusny, C.; Schmid, A. Challenging biological limits with microfluidic single cell analysis. Microb. Biotechnol. 2015, 8, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Dusny, C.; Schmid, A. Microfluidic single-cell analysis links boundary environments and individual microbial phenotypes. Environ. Microbiol. 2015, 17, 1839–1856. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Tago, K.; Senoo, K. Single-cell analysis and isolation for microbiology and biotechnology: Methods and applications. Appl. Microbiol. Biotechnol. 2010, 86, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Brehm-Stecher, B.F.; Johnson, E.A. Single-cell microbiology: Tools, technologies, and applications. Microbiol. Mol. Biol. Rev. 2004, 68, 538–559. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Huang, H.B.; Chen, L.G.; Li, X.P.; Ge, Z.B.; Chen, T.; Yang, Z.; Sun, L.N. Dielectrophoresis for bioparticle manipulation. International Journal of Molecular Sciences 2014, 15, 18281–18309. [Google Scholar] [CrossRef] [PubMed]
- Voldman, J. Electrical forces for microscale cell manipulation. Annu. Rev. Biomed. Eng. 2006, 8, 425–454. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, S.B.; Cao, W.B.; Li, W.H.; Wen, W.J.; Alici, G. Improved concentration and separation of particles in a 3D dielectrophoretic chip integrating focusing, aligning and trapping. Microfluid. Nanofluidics 2013, 14, 527–539. [Google Scholar] [CrossRef]
- Dusny, C.; Fritzsch, F.S.O.; Frick, O.; Schmid, A. Isolated microbial single cells and resulting micropopulations grow faster in controlled environments. Appl. Environ. Microbiol. 2012, 78, 7132–7136. [Google Scholar] [CrossRef] [PubMed]
- Fritzsch, F.S.; Rosenthal, K.; Kampert, A.; Howitz, S.; Dusny, C.; Blank, L.M.; Schmid, A. Picoliter nDEP traps enable time-resolved contactless single bacterial cell analysis in controlled microenvironments. Lab Chip 2013, 13, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Kortmann, H.; Chasanis, P.; Blank, L.M.; Franzke, J.; Kenig, E.; Schmid, A. The Envirostat - A new bioreactor concept. Lab Chip 2009, 9, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Dusny, C.; Gruenberger, A.; Probst, C.; Wiechert, W.; Kohlheyer, D.; Schmid, A. Technical bias of microcultivation environments on single-cell physiology. Lab Chip 2015, 15, 1822–1834. [Google Scholar] [CrossRef] [PubMed]
- Waldbaur, A.; Rapp, H.; Lange, K.; Rapp, B.E. Let there be chip-towards rapid prototyping of microfluidic devices: One-step manufacturing processes. Anal. Methods 2011, 3, 2681–2716. [Google Scholar] [CrossRef]
- Duffy, D.C.; McDonald, J.C.; Schueller, O.J.; Whitesides, G.M. Rapid prototyping of microfluidic systems in poly(dimethylsiloxane). Anal. Chem. 1998, 70, 4974–4984. [Google Scholar] [CrossRef] [PubMed]
- Scharnweber, T.; Truckenmueller, R.; Schneider, A.M.; Welle, A.; Reinhardt, M.; Giselbrecht, S. Rapid prototyping of microstructures in polydimethylsiloxane (PDMS) by direct UV-lithography. Lab Chip 2011, 11, 1368–1371. [Google Scholar] [CrossRef] [PubMed]
- Kuncova-Kallio, J.; Kallio, P.J. In PDMS and its suitability for analytical microfluidic devices. In Conference proceedings: 28th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, New York, NY, USA, 30 August–3 September 2006; 2006; pp. 2486–2489. [Google Scholar]
- Li, N.; Schwartz, M.; Ionescu-Zanetti, C. PDMS compound adsorption in context. J. Biomol. Screen. 2009, 14, 194–202. [Google Scholar] [PubMed]
- Velve-Casquillas, G.; le Berre, M.; Piel, M.; Tran, P.T. Microfluidic tools for cell biological research. Nano today 2010, 5, 28–47. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Saurabh, S.; Bruchez, M.P.; Schwartz, R.; Leduc, P. Molecular crowding shapes gene expression in synthetic cellular nanosystems. Nat. Nanotechnol. 2013, 8, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Bodas, D.; Khan-Malek, C. Hydrophilization and hydrophobic recovery of PDMS by oxygen plasma and chemical treatment - An SEM investigation. Sens. Actuators B Chem. 2007, 123, 368–373. [Google Scholar] [CrossRef]
- Wang, J.D.; Douville, N.J.; Takayama, S.; ElSayed, M. Quantitative analysis of molecular absorption into PDMS microfluidic channels. Ann. Biomed. Eng. 2012, 40, 1862–1873. [Google Scholar] [CrossRef] [PubMed]
- Iliescu, C.; Taylor, H.; Avram, M.; Miao, J.; Franssila, S. A practical guide for the fabrication of microfluidic devices using glass and silicon. Biomicrofluidics 2012, 6, 016505. [Google Scholar] [CrossRef] [PubMed]
- Mazurczyk, R.; Mansfield, C.D.; Lygan, M. Glass microstructure capping and bonding techniques. Methods Mol. Biol. 2013, 949, 141–151. [Google Scholar] [PubMed]
- Becker, H.; Locascio, L.E. Polymer microfluidic devices. Talanta 2002, 56, 267–287. [Google Scholar] [CrossRef]
- Temiz, Y.; Lovchik, R.D.; Kaigala, G.V.; Delamarche, E. Lab-on-a-chip devices: How to close and plug the lab? Microelectron. Eng. 2015, 132, 156–175. [Google Scholar] [CrossRef]
- Niklaus, F.; Stemme, G.; Lu, J.Q.; Gutmann, R.J. Adhesive wafer bonding. J. Appl. Phys. 2006, 99, 031101. [Google Scholar] [CrossRef]
- Schmidt, M.A. Wafer-to-wafer bonding for microstructure formation. Proc. IEEE 1998, 86, 1575–1585. [Google Scholar] [CrossRef]
- Puigcorbe, J.; Vogel, D.; Michel, B.; Vila, A.; Gracia, I.; Cane, C.; Morante, J.R. High temperature degradation of Pt/Ti electrodes in micro-hotplate gas sensors. J. Micromech. Microeng. 2003, 13, 119–124. [Google Scholar] [CrossRef]
- Rosenthal, A.; Taff, B.M.; Voldman, J. Quantitative modeling of dielectrophoretic traps. Lab Chip 2006, 6, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Schnelle, T.; Hagedorn, R.; Fuhr, G.; Fiedler, S.; Mueller, T. Three-dimensional electric field traps for manipulation of cells—calculation and experimental verification. Biochim. Biophys. Acta 1993, 1157, 127–140. [Google Scholar] [CrossRef]
- Fuhr, G.; Arnold, W.M.; Hagedorn, R.; Muller, T.; Benecke, W.; Wagner, B.; Zimmermann, U. Levitation, holding, and rotation of cells within traps made by high-frequency fields. Biochim. Biophys. Acta 1992, 1108, 215–223. [Google Scholar] [CrossRef]
- Seger-Sauli, U.; Panayiotou, M.; Schnydrig, S.; Jordan, M.; Renaud, P. Temperature measurements in microfluidic systems: Heat dissipation of negative dielectrophoresis barriers. Electrophoresis 2005, 26, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, M.S.; Mueller, T.; Schnelle, T. Thermometry in dielectrophoresis chips for contact-free cell handling. J. Phys. D Appl. Phys. 2007, 40, 95–105. [Google Scholar] [CrossRef]
- Schnelle, T.; Mueller, T.; Fuhr, G. Trapping in AC octode field cages. J. Electrost. 2000, 50, 17–29. [Google Scholar] [CrossRef]
- Binder, S.; Schendzielorz, G.; Stabler, N.; Krumbach, K.; Hoffmann, K.; Bott, M.; Eggeling, L. A high-throughput approach to identify genomic variants of bacterial metabolite producers at the single-cell level. Genome Biol. 2012, 13, R40. [Google Scholar] [CrossRef] [PubMed]
- Keilhauer, C.; Eggeling, L.; Sahm, H. Isoleucine synthesis in Corynebacterium glutamicum - Molecular analysis of the Ilvb-Ilvn-Ilvc operon. J. Bacteriol. 1993, 175, 5595–5603. [Google Scholar] [PubMed]
- Abe, S.; Takayama, K.I.; Kinoshit, S. Taxonomical studies on glutamic acid-producing bacteria. J. Gen. Appl. Microbiol. 1967, 13, 279–301. [Google Scholar] [CrossRef]
- Blombach, B.; Hans, S.; Bathe, B.; Eikmanns, B.J. Acetohydroxyacid synthase, a novel target for improvement of l-lysine production by Corynebacterium glutamicum. Appl. Environ. Microbiol. 2009, 75, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Wang, W.; Xie, Q. Review article: Fabrication of nanofluidic devices. Biomicrofluidics 2013, 7, 26501. [Google Scholar] [CrossRef] [PubMed]
- Conradie, E.H.; Moore, D.F. SU-8 thick photoresist processing as a functional material for MEMS applications. J. Micromech. Microeng. 2002, 12, 368–374. [Google Scholar] [CrossRef]
- Tsao, C.W.; DeVoe, D.L. Bonding of thermoplastic polymer microfluidics. Microfluid. Nanofluidics 2009, 6, 1–16. [Google Scholar] [CrossRef]
- Kawamura, M.; Mashima, T.; Abe, Y.; Sasaki, K. Formation of ultra-thin continuous Pt and Al films by RF sputtering. Thin Solid Films 2000, 377, 537–542. [Google Scholar] [CrossRef]
- Lacy, F. Developing a theoretical relationship between electrical resistivity, temperature, and film thickness for conductors. Nanoscale Res. Lett. 2011, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jezequel, N.; Lagomarsino, M.C.; Heslot, F.; Thomen, P. Long-term diversity and genome adaptation of Acinetobacter baylyi in a minimal-medium chemostat. Genome Biol. Evol. 2013, 5, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, J.; Bosdriesz, E.; Nikerel, E.; Molenaar, D.; de Ridder, D.; Teusink, B.; Bruggeman, F.J. How biochemical constraints of cellular growth shape evolutionary adaptations in metabolism. Genetics 2013, 194, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Franzel, B.; Poetsch, A.; Trotschel, C.; Persicke, M.; Kalinowski, J.; Wolters, D.A. Quantitative proteomic overview on the Corynebacterium glutamicum l-lysine producing strain DM1730. J. Proteom. 2010, 73, 2336–2353. [Google Scholar] [CrossRef] [PubMed]
- Gruenberger, A.; Paczia, N.; Probst, C.; Schendzielorz, G.; Eggeling, L.; Noack, S.; Wiechert, W.; Kohlheyer, D. A disposable picolitre bioreactor for cultivation and investigation of industrially relevant bacteria on the single cell level. Lab Chip 2012, 12, 2060–2068. [Google Scholar] [CrossRef] [PubMed]
- Gruenberger, A.; van Ooyen, J.; Paczia, N.; Rohe, P.; Schiendzielorz, G.; Eggeling, L.; Wiechert, W.; Kohlheyer, D.; Noack, S. Beyond growth rate 0.6: Corynebacterium glutamicum cultivated in highly diluted environments. Biotechnol. Bioeng. 2013, 110, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Unthan, S.; Gruenberger, A.; van Ooyen, J.; Gaetgens, J.; Heinrich, J.; Paczia, N.; Wiechert, W.; Kohlheyer, D.; Noack, S. Beyond growth rate 0.6: What drives Corynebacterium glutamicum to higher growth rates in defined medium. Biotechnol. Bioeng. 2014, 111, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Binder, S.; Siedler, S.; Marienhagen, J.; Bott, M.; Eggeling, L. Recombineering in Corynebacterium glutamicum combined with optical nanosensors: A general strategy for fast producer strain generation. Nucleic Acids Res. 2013, 41, 6360–6369. [Google Scholar] [CrossRef] [PubMed]
- Dixit, R.; Cyr, R. Cell damage and reactive oxygen species production induced by fluorescence microscopy: Effect on mitosis and guidelines for non-invasive fluorescence microscopy. Plant J. 2003, 36, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Dailey, M.E. Handbook of biological confocal microscopy, 3rd ed.; Springer US: New York, NY, USA, 2006; pp. 381–403. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosenthal, K.; Falke, F.; Frick, O.; Dusny, C.; Schmid, A. An Inert Continuous Microreactor for the Isolation and Analysis of a Single Microbial Cell. Micromachines 2015, 6, 1836-1855. https://doi.org/10.3390/mi6121459
Rosenthal K, Falke F, Frick O, Dusny C, Schmid A. An Inert Continuous Microreactor for the Isolation and Analysis of a Single Microbial Cell. Micromachines. 2015; 6(12):1836-1855. https://doi.org/10.3390/mi6121459
Chicago/Turabian StyleRosenthal, Katrin, Floris Falke, Oliver Frick, Christian Dusny, and Andreas Schmid. 2015. "An Inert Continuous Microreactor for the Isolation and Analysis of a Single Microbial Cell" Micromachines 6, no. 12: 1836-1855. https://doi.org/10.3390/mi6121459
APA StyleRosenthal, K., Falke, F., Frick, O., Dusny, C., & Schmid, A. (2015). An Inert Continuous Microreactor for the Isolation and Analysis of a Single Microbial Cell. Micromachines, 6(12), 1836-1855. https://doi.org/10.3390/mi6121459