Separation and Characterization of Prostate Cancer Cell Subtype according to Their Motility Using a Multi-Layer CiGiP Culture



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
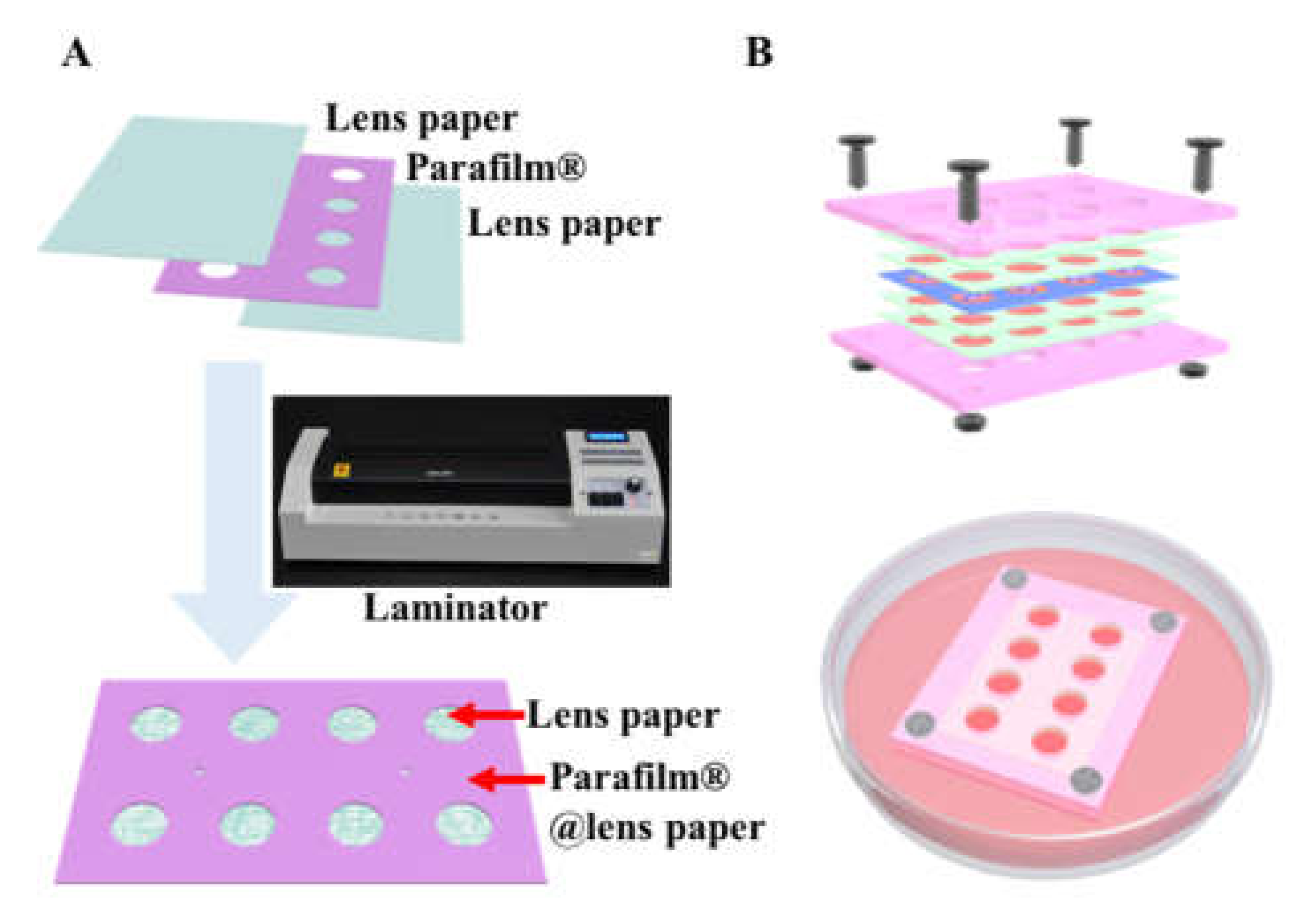
2.2. Preparation of the Multi-Layer CiGiP Platform
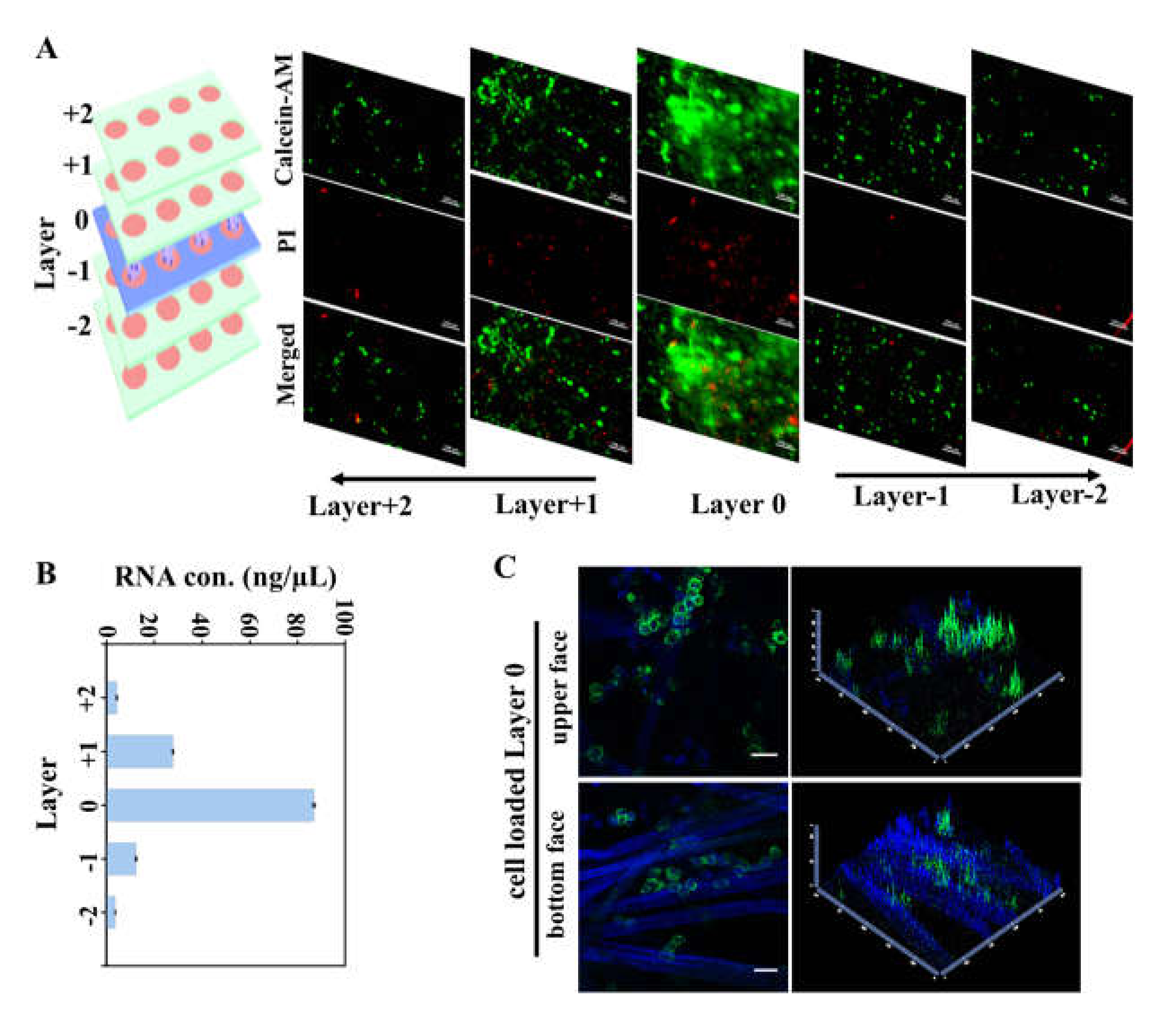
2.3. Characterization of Cell Invasion on Multi-Layer Cultures
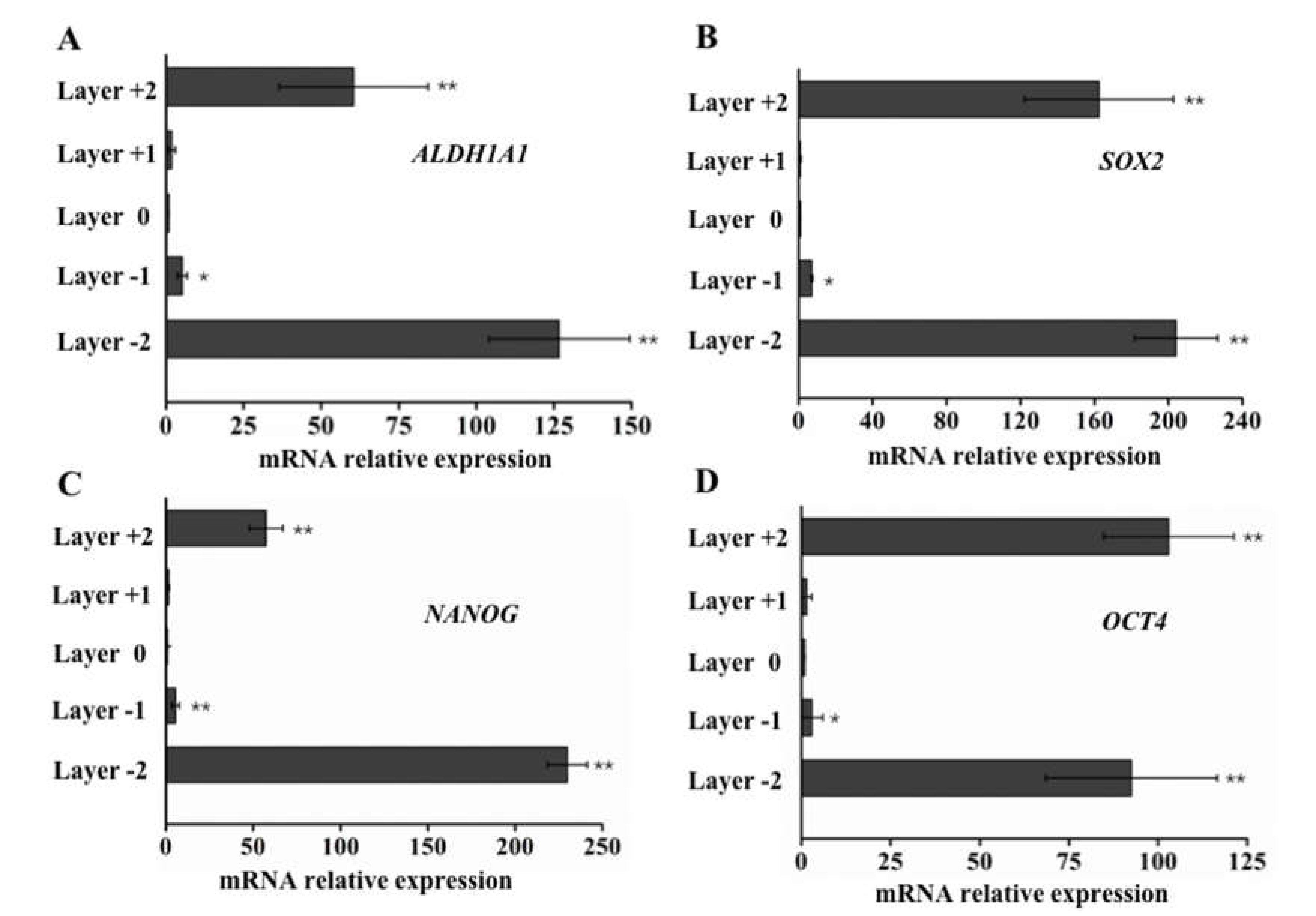
2.4. qPCR to Evaluate Gene Expression in Cells that Invade Different Paper Layers
2.5. MTT Assay to Test the Cytotoxicity Effects of Doxorubicin on Cells Migrating Different Paper Sheets
2.6. Statistic Analysis
3. Results
3.1. Hydrophobic Parafilm® Patterned Lens Paper for Assembling of Multi-Layer Paper-Based Cell Culture Platform
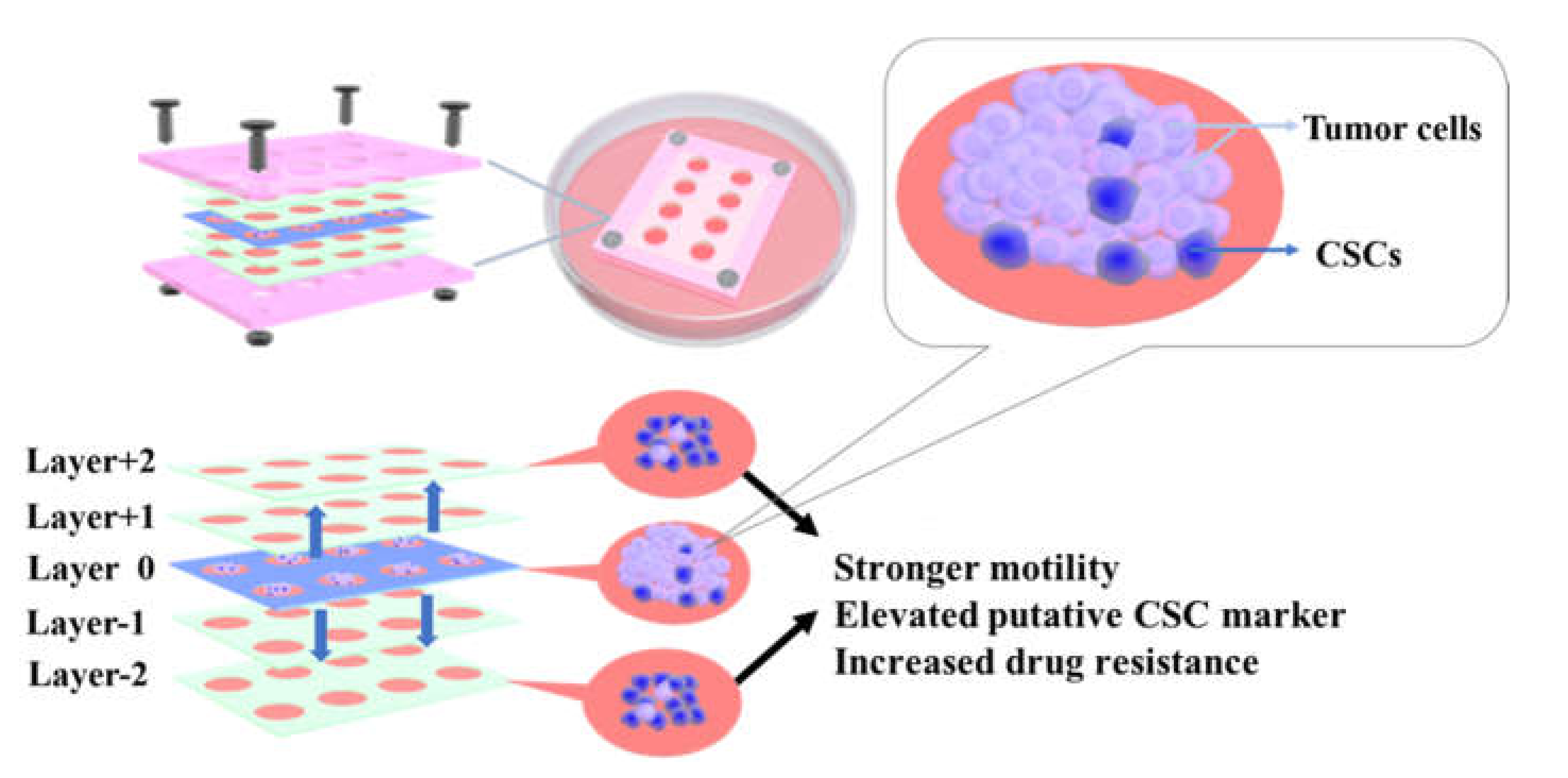
3.2. CiGiP Multi-Layer Cultures Assisted Separation and Isolation of Cells According to Their Motility
3.3. Elevated ALDH1A1, SOX2, NANOG, and OCT4 Expression in Cells with Higher Motility
3.4. Increased Resistance to Doxorubicin of Cells with Higher Motility
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sampieri, K.; Fodde, R. Cancer stem cells and metastasis. Semin. Cancer Biol. 2012, 22, 187–193. [Google Scholar] [CrossRef]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.; Nelson, W.J. Running with neighbors: Coordinating cell migration and cell-cell adhesion. Curr. Opin. Cell Biol. 2015, 36, 62–70. [Google Scholar] [CrossRef]
- Jung, A.; Brabletz, T.; Kirchner, T. The Migrating Cancer Stem Cells Model—A Conceptual Explanation of Malignant Tumour Progression. In Cancer Stem Cells; Springer: Berlin/Heidelberg, Germany, 2007; pp. 109–124. [Google Scholar]
- Robinson, B.D.; Sica, G.L.; Liu, Y.F.; Rohan, T.E.; Gertler, F.B.; Condeelis, J.S.; Jones, J.G. Tumor microenvironment of metastasis in human breast carcinoma: A potential prognostic marker linked to hematogenous dissemination. Clin. Cancer Res. 2009, 15, 2433–2441. [Google Scholar] [CrossRef] [PubMed]
- Ehsan, S.M.; Welch-Reardon, K.M.; Waterman, M.L.; Hughes, C.C.; George, S.C. A three-dimensional in vitro model of tumor cell intravasation. Integr. Biol. (Camb.) 2014, 6, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Kalchman, J.; Fujioka, S.; Chung, S.; Kikkawa, Y.; Mitaka, T.; Kamm, R.D.; Tanishita, K.; Sudo, R. A three-dimensional microfluidic tumor cell migration assay to screen the effect of anti-migratory drugs and interstitial flow. Microfluid. Nanofluid. 2012, 14, 969–981. [Google Scholar] [CrossRef] [Green Version]
- Justus, C.R.; Leffler, N.; Ruiz-Echevarria, M.; Yang, L.V. In vitro cell migration and invasion assays. J. Vis. Exp. 2014, 88, e51046. [Google Scholar]
- Chonan, Y.; Taki, S.; Sampetrean, O.; Saya, H.; Sudo, R. Endothelium-induced three-dimensional invasion of heterogeneous glioma initiating cells in a microfluidic coculture platform. Integr. Biol. (Camb.) 2017, 9, 762–773. [Google Scholar] [CrossRef]
- Cui, X.; Guo, W.; Sun, Y.; Sun, B.; Hu, S.; Sun, D.; Lam, R.H.W. A microfluidic device for isolation and characterization of transendothelial migrating cancer cells. Biomicrofluidics 2017, 11, 014105. [Google Scholar] [CrossRef] [Green Version]
- Doyle, A.D.; Petrie, R.J.; Kutys, M.L.; Yamada, K.M. Dimensions in cell migration. Curr. Opin. Cell Biol. 2013, 25, 642–649. [Google Scholar] [CrossRef] [Green Version]
- Derda, R.; Laromaine, A.; Mammoto, A.; Tang, S.K.; Mammoto, T.; Ingber, D.E.; Whitesides, G.M. Paper-supported 3D cell culture for tissue-based bioassays. Proc. Natl. Acad. Sci. USA 2009, 106, 18457–18462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.B. Three-dimensional tissue culture models in cancer biology. Semin. Cancer Biol. 2005, 15, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Cukierman, E. Modeling Tissue Morphogenesis and Cancer in 3D. Cell 2007, 130, 601–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, V.L.; Zheng, Y.; Choi, N.W.; Verbridge, S.S.; Sutermaster, B.A.; Bonassar, L.J.; Fischbach, C.; Stroock, A.D. Dense type I collagen matrices that support cellular remodeling and microfabrication for studies of tumor angiogenesis and vasculogenesis in vitro. Biomaterials 2010, 31, 8596–8607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, C.C.; Boyce, M.W.; Lockett, M.R. Paper-based Invasion Assays for Quantifying Cellular Movement in Three-dimensional Tissue-like Structures. Curr. Protoc. Chem. Biol. 2017, 9, 75–95. [Google Scholar] [PubMed]
- Mosadegh, B.; Lockett, M.R.; Minn, K.T.; Simon, K.A.; Gilbert, K.; Hillier, S.; Newsome, D.; Li, H.; Hall, A.B.; Boucher, D.M.; et al. A paper-based invasion assay: Assessing chemotaxis of cancer cells in gradients of oxygen. Biomaterials 2015, 52, 262–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Zhang, Q.; Chen, B.; Liang, G.-T.; Li, W.-X.; Zhou, X.-M.; Liu, D.-Y. Study on Microenvironment Acidification by Microfluidic Chip with Multilayer-paper Supported Breast Cancer Tissue. Chin. J. Anal. Chem. 2013, 41, 822–827. [Google Scholar] [CrossRef]
- Simon, K.A.; Mosadegh, B.; Minn, K.T.; Lockett, M.R.; Mohammady, M.R.; Boucher, D.M.; Hall, A.B.; Hillier, S.M.; Udagawa, T.; Eustace, B.K.; et al. Metabolic response of lung cancer cells to radiation in a paper-based 3D cell culture system. Biomaterials 2016, 95, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Yu, S.J.; Yang, K.; Jin, Y.; Cho, A.N.; Kim, J.; Lee, B.; Yang, H.S.; Im, S.G.; Cho, S.W. Paper-based bioactive scaffolds for stem cell-mediated bone tissue engineering. Biomaterials 2014, 35, 9811–9823. [Google Scholar] [CrossRef]
- Sapp, M.C.; Fares, H.J.; Estrada, A.C.; Grande-Allen, K.J. Multilayer three-dimensional filter paper constructs for the culture and analysis of aortic valvular interstitial cells. Acta Biomater. 2015, 13, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Justice, B.A.; Badr, N.A.; Felder, R.A. 3D cell culture opens new dimensions in cell-based assays. Drug Discov. Today 2009, 14, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.; Gao, B.; Yong, K.W.; Li, Y.; Shi, M.; Zhao, X.; Li, Z.; Zhang, X.; Pingguan-Murphy, B.; Yang, H.; et al. Paper-based cell culture platform and its emerging biomedical applications. Mater. Today 2017, 20, 32–44. [Google Scholar] [CrossRef]
- Ma, I.; Allan, A.L. The Role of Human Aldehyde Dehydrogenase in Normal and Cancer Stem Cells. Stem Cell Rev. 2011, 7, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Su, Y.; Mei, Y.; Leng, Q.; Leng, B.; Liu, Z.; Stass, S.A.; Jiang, F. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab. Investig. 2010, 90, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodda, D.J.; Chew, J.L.; Lim, L.H.; Loh, Y.H.; Wang, B.; Ng, H.H.; Robson, P. Transcriptional regulation of nanog by OCT4 and SOX2. J. Biol. Chem. 2005, 280, 24731–24737. [Google Scholar] [CrossRef]
- Tay, Y.; Zhang, J.; Thomson, A.M.; Lim, B.; Rigoutsos, I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 2008, 455, 1124–1128. [Google Scholar] [CrossRef]
- Jeter, C.R.; Liu, B.; Liu, X.; Chen, X.; Liu, C.; Calhoun-Davis, T.; Repass, J.; Zaehres, H.; Shen, J.J.; Tang, D.G. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene 2011, 30, 3833–3845. [Google Scholar] [CrossRef] [Green Version]
- Linn, D.E.; Yang, X.; Sun, F.; Xie, Y.; Chen, H.; Jiang, R.; Chen, H.; Chumsri, S.; Burger, A.M.; Qiu, Y. A Role for OCT4 in Tumor Initiation of Drug-Resistant Prostate Cancer Cells. Genes Cancer 2010, 1, 908–916. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.; Li, X.; Xu, Y.; Zhang, S.; Mou, W.; Liu, Y.; Liu, Y.; Lv, D.; Liu, C.H.; Tan, X.; et al. SOX2 promotes tumorigenesis and increases the anti-apoptotic property of human prostate cancer cel. J. Mol. Cell Biol. 2011, 3, 230–238. [Google Scholar] [CrossRef]
- Postovit, L.M.; Costa, F.F.; Bischof, J.M.; Seftor, E.A.; Wen, B.; Seftor, R.E.; Feinberg, A.P.; Soares, M.B.; Hendrix, M.J. The commonality of plasticity underlying multipotent tumor cells and embryonic stem cells. J. Cell. Biochem. 2007, 101, 908–917. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence (5′—3′) |
|---|---|---|
| OCT4 | F | AACCGAGTGAGAGGCAACCT |
| R | ACAGAACCACACTCGGACCA | |
| SOX 2 | F | TGACCAGCTCGCAGACCTAC |
| R | TCGGACTTGACCACCGAAC | |
| NANOG | F | CGCGTTGTGATCTCCTTCTG |
| R | GTCTGGTTGCTCCAGGTTGA | |
| ALDH1A1 | F | CACAGGATCAACAGAGGTTGG |
| R | GTCCAAGTCGGCATCAGCTA | |
| GAPDH | F | CCAGGTGGTCTCCTCTGACTTCAACA |
| R | AGGGTCTCTCTCTTCCTCTTGTGCTC | |
| HIF-1α | F | GCCCCAGATTCAGGATCAGA |
| R | TGGGACTATTAGGCTCAGGTGAAC | |
| HIF-2α | F | GCCACCCAGTACCAGGACTACA |
| R | CCTCACAGTCATATCTGGTCAGTTCG |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.-X.; Zhou, Y.; Fu, J.-J.; Lu, Z.; Yu, L. Separation and Characterization of Prostate Cancer Cell Subtype according to Their Motility Using a Multi-Layer CiGiP Culture. Micromachines 2018, 9, 660. https://doi.org/10.3390/mi9120660
Wang L-X, Zhou Y, Fu J-J, Lu Z, Yu L. Separation and Characterization of Prostate Cancer Cell Subtype according to Their Motility Using a Multi-Layer CiGiP Culture. Micromachines. 2018; 9(12):660. https://doi.org/10.3390/mi9120660
Chicago/Turabian StyleWang, Lin-Xiang, Ying Zhou, Jing-Jing Fu, Zhisong Lu, and Ling Yu. 2018. "Separation and Characterization of Prostate Cancer Cell Subtype according to Their Motility Using a Multi-Layer CiGiP Culture" Micromachines 9, no. 12: 660. https://doi.org/10.3390/mi9120660
APA StyleWang, L. -X., Zhou, Y., Fu, J. -J., Lu, Z., & Yu, L. (2018). Separation and Characterization of Prostate Cancer Cell Subtype according to Their Motility Using a Multi-Layer CiGiP Culture. Micromachines, 9(12), 660. https://doi.org/10.3390/mi9120660