Luminescent materials, in the form of powders (phosphors) and films, have been extensively studied in recent decades [
35,
36] because their great importance for a wide variety of applications such as: Lighting, image displays, signaling, lasers, medical applications, etc. [
37,
38]. They have been synthesized through a variety of physical and chemical techniques, including: Hidrothermal/Solvothermal [
39,
40,
41], solid-state reaction [
42], sol-gel [
43,
44], laser ablation [
45,
46], sputtering [
47], Pechini Method [
48], plasma electrolitic oxidation [
49], conventional melt-quenching method [
50,
51], combustion synthesis [
52], solvent evaporation method [
53,
54,
55], and co-precipitation process [
56]. Among these techniques, spray pyrolysis began to be used for this purpose in the mid-1980s, and it is still used today [
57,
58]—proving to be a practical, low cost, easy to extrapolate for large area deposition technique. In this review, an account is made on diverse luminescent materials synthesized by this technique. These materials in general involve one or more luminescent centers incorporated as dopants in a host lattice. A great variety of host lattices have been used for the synthesis (by means of spray pyrolysis) of phosphors and luminescent films, among them stand out metal oxides such as: ZrO
2, Al
2O
3, HfO
2, Y
2O
3, ZnO, In
2O
3, ZnSiO
3, CdO, (Y, Gd)BO
3, Gd
2O
3, LaPO
4, BaMgAl
10O
17, and some sulfur based compounds such as: ZnS, CaSO
4, CdS, and others. The luminescent active centers have been mainly RE (Rare Earth) and some transition metal ions. In some cases, luminescence emission has been observed to be generated by mechanisms that involve structural defects and intrinsic states in the host lattices as well. This review focuses mainly on the work done on host lattices such as: ZrO
2, Al
2O
3, HfO
2, Y
2O
3, ZnO, and ZnS with different dopants.
4.1. ZrO2
Virtually before 1999, ZrO
2 had not been used as a host lattice to produce phosphor materials synthesized by spray pyrolysis technique. In that year, some results were reported about photoluminescence (PL) and thermoluminescence (TL) properties of ZrO
2:Tb
3+ films deposited by a pneumatic spray pyrolysis (PSP) system [
59]. These films excited by 275 nm exhibited four peaks at 487, 542, 582, and 619 nm—typical of electronic transitions in the Tb
3+ ions. The TL glow curve displayed two peaks at 112 °C and 270 °C for the ZrO
2:Tb
3+ films exposed to 260 nm UV radiations. In addition, the TL response was linear in the range of 40 to 240 mJ·cm
−2 spectral UV irradiance. These results exhibited that ZrO
2:Tb
3+ films had appropriate characteristics for their use as a UV dosimeter as well as PL phosphor. In a later investigation (2001) on this material [
60], a deeper analysis was made on the thermoluminescence mechanisms. Two important parameters in TL studies such as activation energy (E) and the frequency factor (S) were investigated. In this contribution, the Lushchik and Chen methods were used to determine the kinetic parameters which showed second order kinetics for both the first and second glow TL peaks.
Furthermore, in 2001, PL and cathodoluminescence (CL) feature of ZrO
2:Tb
3+ films, deposited by the PSP, technique was reported [
61]. In this case different deposition parameters, such as substrate temperatures, doping concentrations, and the flow of the precursor solution, were studied. Substrate temperatures higher than 400 °C rendered a polycrystalline material with metastable tetragonal or cubic phases. With increasing deposition temperatures, the PL and CL emission intensities (excited with 250 nm light) also increased. The PL and CL emission spectra showed the characteristic peaks associated with the electronic transitions of Tb
3+ ions. Concentration quenching for the PL and CL emissions occurred at doping concentration greater than 1.96 and 1.17 at.%, respectively. Similar studies were conducted on ZrO
2:Eu
3+ films [
62]. Depending on the substrate temperature, these films were amorphous or polycrystalline (tetragonal-cubic phase). A strong red emission was observed which was generated by the
5D
0 →
7F
2 transition typical of the Eu
3+ ions. From those studies, it became clear that zirconia was a suitable host lattice for RE ions.
For the first time, a study on luminescent emissions from ZrO
2: Mn
2+ films deposited by the USP technique was reported in 2002 [
63]. These films were deposited at substrate temperatures ranging from 250 to 500 °C. The PL and CL (7 KeV) emission spectra showed a broad band (450–750 nm) centered at 650 nm (red), which is associated with the electronic transitions
4T
1(
4G) →
6A
1(
6S) of the Mn
2+ ions. A decrease of the luminescence, as a function of the doping concentration, substrate temperature and electron accelerating voltage was observed. The maximum emission intensity was observed for films deposited at 250 °C, EDS measurements showed that these films had a high amount of incorporated chlorine (from the precursors in the spraying solution), which acts as a co-activator for the red emission. As the deposition temperature increased, the amount of chlorine in the film (as well as the red luminescence emission intensity) decreased. The presence of chlorine was necessary for the red luminescence emission to occur. CL spectra obtained at higher electron accelerating voltages (10 KeV) from samples deposited at 500 °C showed, instead of the red emission, a wide band centered at 590 nm (yellow)—which is also characteristic of Mn
2+ ions.
ZrO
2:Eu
3+ phosphors consisting of spherical, dense and sub-micrometer size particles were successfully synthesized by the USP technique in 2005 [
64]. The X-ray diffraction (XRD) measurements indicated that the crystallinity of these powders increased with increasing postdeposition annealing temperature. Several characterization techniques were used to study this material: Including PL emission spectra, and decay time measurements. The excitation spectrum showed a band centered at 248 nm corresponding to a charge transfer transition from Eu-O generated electronic states in the ZrO
2 host matrix. The emission spectra exhibit the typical (red) bands of Eu
3+ ions. The optimal concentration of Eu
3+ ions was 10 at.% and it was observed that the spherical morphology of the particles improves the intensity of the PL emission.
A research work on ZrO
2:Pr
3+ films was published in 2007 [
65]. In this case, PL and CL properties were studied as a function of growth parameters such as the substrate temperature and the Pr
3+ ions concentration. XRD studies indicated a tetragonal crystalline structure for zirconia as the substrate temperature was increased. The PL spectra exhibited bands centered at 490, 510, 566, 615, 642, 695, 718, 740 and 833 nm; associated with the electronic transitions
3P
0 →
3H
4,
3P
0 →
3H
4,
3P
1 +
1I
6 →
3H
5,
1D
2 →
3H
4,
3P
0 →
3H
6,
1D
2 →
3H
5,
1D
2 →
3H
5,
3P
0 →
3F
3,4, and
1D
2 →
3F
2 of the Pr
3+ ions. As the substrate temperature was increased, an increasing intensity of the PL emission was observed. Also, a quenching of the PL and CL emissions, with increasing doping concentration, was detected. Interestingly the CL spectra, as a function of the electron accelerating voltage, showed an evolution of the highest peak: For low electron accelerating voltages (4 kV) the red emission (615 nm) is the maximum, and for high voltages (15 kV) the most intense band is the blue (around 490 nm).
The cathodoluminescence properties of ZrO
2:Er
3+ films were reported in 2014 [
66]. These films were deposited at different temperatures from 400 °C up to 550 °C. As substrate temperatures are increased, the films showed a tetragonal phase. CL emission spectra showed bands centered at 524 (green), 544 (green) and 655 (red) nm associated with the electronic transition
2H
11/2 →
4I
11/2,
4S
3/2 →
4I
15/2, and
4F
9/2 →
4I
15/2 of Er
3+ ions. The highest emission intensity is achieved in samples deposited at 500 °C doped with 5 at.% of Er
3+ ions. Also, the CL emission intensity increases as the substrate temperature and electron accelerating voltage values increase.
Investigations on ZrO
2:Dy
3+ and ZrO
2:Dy
3++
xLi
+ films were published in 2015 [
67]. XRD measurements, as a function of the deposition temperature, indicated a meta-stable tetragonal crystalline structure of the zirconia. PL and CL features of the films were studied as a function of synthesis parameters such as the substrate temperature and the Dy
3+ and Li
+ concentrations. All luminescent emission spectra showed peaks located at 485 (blue), 584 (yellow), 670 (red) and 760 nm; which correspond to electronic transitions
4F
9/2 →
6H
15/2, 4F
9/2 →
6H
13/2,
4F
9/2 →
6H
11/2, and
4F
9/2 →
6H
9/2, of Dy
3+, respectively. The Li
+ incorporation in the ZrO
2:Dy
3+ films produced an improvement in the intensity of the luminescent emission, presumably because it acts as a charge compensator and because it contributes to improving the crystalline structure of the host lattice. The CIE color coordinates (0.3475, 0.3609) of these films were found within the warm white light emission region. These spectroscopic characteristics allowed to propose this material for application in solid-state lighting (SSL), especially for white lighting emission applications. It is observed that, as the concentration of Li
+ ions increases, they come closer to the perfect white area of the CIE color coordinates (0.3333, 0.3333).
Moreover, in 2015 a work on ZrO
2, ZrO
2:Dy
3+ and ZrO
2:Dy
3+ + Gd
3+ films was published [
68]. The synthesis and the characterization conditions were carried out as described in Reference [
67]. The relative concentrations of Dy
3+ and Gd
3+ ions were varied; the emission spectra of these films exhibited bands in the blue and yellow regions. The incorporation of Gd
3+ ions in ZrO
2:Dy
3+ films generated a remarkable increase in the intensity of the luminescent emission (approximately 15 times). In principle, the host lattice absorbs the excitation energy which is transferred to the Gd
3+ ions which in turn transfers it to the Dy
3+ ions. The CIE chromaticity diagram exhibited a cold-white emission (Dy
3+-Gd
3+ doped samples) and a warm-white emission (Dy
3+ doped samples), which shows the potential of these films for generating white light coatings for solid state lighting (SSL) applications.
The PL and structural properties of co-doped ZrO
2: Eu
3+ + Tb
3+ films, were also reported in 2015 [
69]. The PL spectra showed the typical emission bands associated with the Tb
3+ and Eu
3+ ions, as well as a broad emission, peaked at 440 nm associated to radiative transitions within the ZrO
2 host lattice. These films displayed multicolored emissions depending of the ratio Eu
3+/Tb
3+ and the excitation wavelength. The observed colors were: Blue (from the host lattice), green (from the ZrO
2:Tb
3+ films), red-orange (from the ZrO
2:Eu
3+ films), yellow (from the ZrO
2:Eu
3+ + Tb
3+ films, excited with 288 nm) and bluish-white and yellowish white (from the ZrO
2: Eu
3+ + Tb
3+ films, excited with 368 or 380 nm). The CIE coordinates of the double-doped ZrO
2:Tb
3+ (10 at.%) + Eu
3+ (5 at.%) films lie in the white light region of the chromaticity diagram and show good potential for lighting devices and photonic applications.
4.2. Al2O3
A pioneering work on luminescent Al
2O
3:Tb
3+ films appeared in 1992 [
70]. The films were deposited by the PSP technique on either plain or conductive oxide coated glass substrates at deposition temperatures in the range of 270–450 °C. PL emission from these films showed well-defined peaks at 490 and 550 nm, which were associated to the electronic transitions corresponding to Tb
3+ ions. The relative emission intensity was strongly dependent on the type of substrate, the deposition temperature and the amount of Tb
3+ ions incorporated in the films. Two years later, an investigation on Al
2O
3:CeCl
3 films was published in 1994 [
71]. PL spectra (excited with 300 nm light) showed a broad emission formed by two overlapping peaks at 365 and 395 nm. It was suggested that these bands originate from the 5d to 4f electronic energy levels of Ce in the CeC1
3 molecule. The PL emission intensity of these peaks was strongly dependent on the doping concentration and the substrate temperature. The films with greater intensity were those deposited at the lowest temperature, where there is a greater amount of CeCl
3 incorporated in the films. As the temperature increases, the concentration of CeCl
3 molecules decreases and so does the PL emission intensity—therefore, the presence of this molecule is essential for an optimal emission of blue light. Also, a quenching of the PL is observed for CeCl
3 concentrations higher than 1 at.%. Another research on Al
2O
3:Eu
3+ films was published in 2000 [
72]. These films were deposited by the USP technique at substrate temperatures from 300 to 540 °C and the Eu
3+ doping concentration was varied. All films were amorphous in structure and the PL spectra were measured as a function of substrate temperature and doping concentration. The excitation spectrum showed an intense peak centered at 395 nm. All the PL emission spectra (excited by 395 nm) showed bands located at 587, 600, 612, and 648 nm—typical of the electronic transitions in Eu
3+ ions. It was observed a concentration quenching of the PL emission intensity at values of above 1.5 at.% in the films. Thus, it was shown that Al
2O
3 is a suitable host lattice to support RE ions (such as Eu
3+) to generate strong PL emissions.
In 2003, a new research in Al
2O
3:Tb
3+ films was published [
73]. In this case, the transparency of the films was up to 88% on the 400 to 700 nm range. These was possible because the use of organic source reactive for both aluminum and terbium (acetylacetonates) that were dissolved in dimethylformamide and sprayed, deposited at temperatures up to 600 °C. These films were mostly amorphous in the range of deposition temperatures studied with an average roughness of 14 Å or less; which was perfect for the design and development of microdevices integrating this type of films. PL and CL spectra, studied as a function of the deposition parameters such as doping concentrations and substrate temperatures, were typical of the transitions among the electronic energy levels of the Tb
3+ ions. Thus, from this work, it is clear that the use of acetylacetonates as precursors, generates the formation of high transmittance films with low roughness, as described in the dielectric section thin films, in contrast to those films synthesized from chlorides, nitrates or acetates (dissolved in water) which are, in general, very rough and opaque.
An energy transfer mechanism between Ce
3+ and Mn
2+ ions in alumina films was reported in 2005 [
74]. Blue and red light emitting Al
2O
3:Ce
3+:Mn
2+ films, under ultraviolet light excitation, were investigated in this case. The blue emission is due to transitions from the excited state 5d to the split ground state
2F of the Ce
3+ ions. The usually weak Mn
2+ ions red emission, attributed to intra 3d transitions, was enhanced by an efficient energy transfer from the Ce
3+ ions. The energy transfer mechanism was an electric dipole–quadrupole interaction with a quantum efficiency estimated to be near to 100%, which makes these films interesting phosphors for the design of microdevices based on luminescent layers in flat-panel displays. Other studies on this type of amorphous Al
2O
3:Ce
3+:Mn
2+ films were also published [
75,
76]. However, in this case, the precursors were AlCl
3, CeCl
3 and MnCl
2 dissolved in deionized water (Ce: 10 at.%; Mn: 1, 3, 5, 7 and 10 at.%), deposited at a substrate temperature of 300 °C. The chemical composition and the profile distribution of the dopant ions across the films were determined by Rutherford backscattering (RBS). A homogeneous depth profile of both Ce
3+ and Mn
2+ ions was found within the films, and the overall measured quantities were as expected from the solution concentrations. Chlorine, which plays a significant role in luminescent properties, was detected in important quantities, something that was expected due to the low deposition temperatures used in this case. The red emission from manganese-doped samples was strongly enhanced with the co-doping with Ce due to the efficient energy transfer mechanism from Ce
3+ to Mn
2+ ions. From XPS analysis, it was determined that a considerable amount of Mn ions remains linked to chlorine, while Ce is mostly in an oxidized state.
In 2010, alumina was used to host three ions (Tb
3+, Ce
3+, and Mn
2+) to generate white light when excited by ultraviolet light [
77]. These amorphous films were also deposited at 300 °C. Sensitization of Tb
3+ and Mn
2+ ions by Ce
3+ ions gave rise to blue, green and red luminescent emission when the film was excited with UV radiation. The overall efficiency of such energy transfer was about 85% upon excitation with 312 nm light. Energy transfer from Ce
3+ to Tb
3+ ions through an electric dipole–quadrupole interaction mechanism appeared to be more probable than the electric dipole–dipole one. A strong white light emission from the Al
2O
3:Ce
3+ (1.3 at.%):Tb
3+ (0.2 at.%):Mn
2+ (0.3 at.%) films under UV excitation was obtained. The high efficiency of energy transfer from Ce
3+ to Tb
3+ and Mn
2+ ions, resulted in a cold white light emission (
x = 0.30 and
y = 0.32). Thus, these films resulted interesting material for the design of efficient UV pumped phosphors for white light generation which could be integrated in light emitting microdevices.
Similarly, alumina co-doped with Dy
3+ and Ce
3+ ions was reported in 2011 [
78]. The PL properties of these films were studied through excitation, emission spectra measurements and decay time spectroscopy. These films emitted a combination of blue and yellow colors through an efficient energy transfer (77%) from Ce
3+ to Dy
3+ ions. It was inferred that such energy transfer was non-radiative, taking place between Ce
3+ and Dy
3+ clusters, through a short-range interaction mechanism. Ce
3+ doped single films emitted in the violet-purplish-blue region; whereas co-doped films the presented a cold-white light emission. The PL properties of tri-doped Al
2O
3:Ce
3+:Dy
3+:Mn
2+ films were published in 2012 [
79]. Nonradiative energy transfer from Ce
3+ to Dy
3+ and Mn
2+ was reported upon UV excitation at 278 nm. From lifetime data, it was deducted that the energy transfer was nonradiative in nature. Simultaneous emission of all co-dopant ions in the blue, yellow and red regions, resulted in white light emission with CIE 1931 chromaticity coordinates,
x = 0.34 and
y = 0.23, with a color temperature of 4900 K. Thin films as these might contribute to the development of materials that, pumped with AlGaN-based LEDs, could generate white light emission.
Also, in 2012, a study on the PL characteristics, under continuous and pulsed excitation of Eu-doped alumina films was reported [
80]. It was determined that localized states in the undoped Al
2O
3 host lattice, excited with 250 nm radiation, emit a violet color (broad band centered at 415 nm) associated to a radiative recombination process involving F centers. When Eu
3+ ions were incorporated into these films, a charge transfer mechanism to these ions from the localized states seems to occur predominantly. The Eu
3+ related emission, generated in this way, results intensified and luminescence decay time extended as compared to that obtained when the excitation is achieved through an inter-electronic energy level transition in the Eu
3+ ion, excited by 395 nm radiation.
Subsequently, in 2013, a contribution on the white light emission from Al
2O
3:Ce
3+:Tb
3+:Mn
2+ and HfO
2:Ce
3+:Tb
3+:Mn
2+ films was published [
81]. These oxide films doped with CeCl
3/TbCl
3/MnCl
2 were deposited at 300 °C. XRD measurements exhibited a very broadband typical of non-crystalline materials. Non-radiative energy transfer from Ce
3+ to Tb
3+ and Mn
2+ ions is observed upon UV excitation at 280 nm; the energy transfer could take place in Ce
3+-Tb
3+ and Ce
3+-Mn
2+ clusters through an electric dipole-quadrupole interaction mechanism. This energy transfer gives place to a simultaneous emission of the donor and acceptor ions in the blue, green, yellow and red regions, resulting white light emission. The chromaticity coordinates for Al
2O
3:Ce
3+:Tb
3+:Mn
2+ films and color temperatures were: (0.30, 0.32) and 7300 K (cold-white color). The chromaticity coordinates for HfO
2:Ce
3+:Tb
3+:Mn
2+ films and color temperatures were (0.32, 0.37) and 6000 K (warm-white color).
Another study on PL emission (white emission) from single and double layered Al
2O
3:Ce
3+:Tb
3+:Eu
3+ films was presented in 2013 [
10]. These films were deposited using acetylacetonates (dissolved in dimethylformamide) as precursors. Eu
3+ and Tb
3+ doped films showed the typical emissions of these trivalent ions (red and green, respectively). Ce doped films showed two broad bands associated with the 5d to 4f transitions of the Ce
3+ ion, centered at ~400 and 510 nm. As expected from films deposited with organic precursors, these films had low surface roughness (lower than 3 nm) and thicknesses between 50 and 260 nm. The double layer stacks involved first an Eu
3+ doped film followed by a second Ce
3+-Tb
3+ co-doped layer. The films were transparent in the visible region, with an optical bandgap of approximately 5.63 eV. The PL of these stacks was an overlap of the emissions corresponding to all the dopants when excited with 300 nm light, resulting in an intense white light emission, which would be suitable for the design of electroluminescent microdevices.
The PL characteristics of Eu
3+ doped alumina films co-doped with Bi
3+ and Li
+ were published in 2015 [
82]. In this case, the incorporation of Bi
3+ and Li
+ ions as co-dopants in Al
2O
3:Eu
3+ films and its effect on the luminescence characteristics of this material were described. Both Bi
3+ and Li
+ do not introduce new luminescence features but affect the luminescence intensity of the Eu
3+ related emission spectra as well as the excitation spectra. The introduction of Bi
3+ generates localized states in the aluminum oxide host that result in a quenching of the luminescence intensity, while Li
+ and Bi
3+ co-doping increases the luminescence intensity of these films. It was found that the Eu
3+ ions emission intensity in these films, when Bi
3+ ions were added together with Li
+, produce an increase of 62% in the emission intensity. It was suggested that the role of Li
+ co-doping was to redirect the energy paths back to the Eu
3+ ions from the Bi
3+ ions. Analysis of time decay measurements of the Eu
3+ related emission in the amorphous alumina films indicated the presence of two type of sites in the short-range surroundings of the Eu
3+ ions that could be correlated with those around this ion in α or γ Al
2O
3 crystalline phases.
4.3. HfO2
Luminescent HfO
2:Mn
2+ films (deposited by Ultrasonic Spray Pyrolysis technique) were reported for the first time, in 2004 [
83]. The deposited films were amorphous at deposition temperatures up to 300 °C; for higher temperatures a polycrystalline material was obtained with a monoclinic HfO
2 phase. The cathodoluminescence (CL) spectra showed blue–green and red bands associated with the electronic transitions
4T
1(
4G) →
6A
1(
6S) of the Mn
2+ ions. A dependence of the CL emissions, as a function of the doping concentration, substrate temperature and electron accelerating voltage was reported. It was determined that both amorphous and polycrystalline hafnium oxide make efficient host for Mn
2+ ions, and that the relative content of chlorine in the processed films have an important role on the luminescent emission intensity of the studied materials.
USP deposited HfO
2:CeCl
3 films luminescent properties were published in 2007 [
84]. These films were deposited from hafnium dichloride oxide and CeCl
3 dissolved in deionized water (18 MΩ/cm). The PL characteristics of the HfO
2:CeCl
3 films were studied as a function of doping concentrations and substrate temperature. XRD measurements showed the monoclinic phase of HfO
2 for samples deposited at deposition temperatures higher than 400 °C. These films showed a violet–blue PL emission that could easily be seen with the naked eye in normal room light. Also, PL emission and excitation spectra evidence the presence of two different Ce
3+ centers in HfO
2. A complete concentration quenching of the luminescence of one of the two centers is observed at high concentration of CeCl
3 (15 at.% in the start solution), which suggests a fast energy transfer from the high-energy to the low energy centers. Finally, it was confirmed that HfO
2 is an adequate host matrix for rare earth ions as active centers to generate strong violet–blue PL emissions.
Also, in 2007, a work on PL properties of HfO
2:Tb
3+films was published [
85]. The PL properties of these films were studied as a function of deposition temperature and Tb
3+ ions concentration. The films were deposited the USP technique from aqueous solution of Hafnium and Terbium chlorides. Results showed that crystalline structure of HfO
2:Tb
+3 films depends on the deposition temperature. PL excitation spectrum showed a wide band centered at 262 nm while the PL emission spectra showed bands centered at 488, 542, 584 and 621 nm, which correspond to the electronic transitions:
5D
4 →
7Fj (j = 3, 4,5, 6) typical of trivalent terbium ions. The dominant emission intensity corresponds to the green color (542 nm), which depended on the terbium concentration incorporated in the host lattice; the optimum doping concentration was 5 at.% Tb
+3 in the spraying solution.
The PL and CL characteristics of HfO
2:Sm
3+ films were published in 2008 [
86]. These films were deposited by the USP technique on Corning glass substrates at deposition temperatures ranging from 300 to 550 °C using chlorides as precursor materials. Scanning electron microcopy (SEM) micrographs revealed rough surfaces morphology with spherical particles. The PL and CL spectra exhibited four main bands centered at 570, 610, 652 and 716 nm, which are due to the well-known intra-4f transitions of the Sm
+3 ions. It was found that the overall emission intensity rose as the deposition temperature was increased. Moreover, a concentration quenching of the emission intensity was observed for doping concentration higher than 0.7 at.% as measured by EDS. These films showed good adherence to the substrate and a high deposition rate of up to 2 µm per minute. In addition, The CL emission intensity was found to increase as the electron accelerating voltage was raised.
Also, in 2008, HfO
2 films doped with CeCl
3 and/or MnCl
2 were deposited at 300 °C by the USP technique [
87]. The XRD results revealed that the films were predominantly amorphous. HfO
2: CeCl
3 showed a violet-blue emission. The weak green–red emissions of Mn
2+ ions was enhanced through an efficient energy transfer from Ce
3+ to Mn
2+ ions in the co-doped films. Spectroscopic data indicated that this energy transfer was nonradiative in nature and it could occur in Ce
3+ and Mn
2+ clusters through a short-range interaction mechanism. The efficiency of this energy transfer increases with the Mn
2+ ion concentration, so that an efficiency of about 78% is achieved for a 5 at.% of MnCl
2 concentration. The HfO
2:CeCl
3:MnCl
2 films are interesting phosphors for the design of luminescent layers emitting simultaneously in the three primary colors: Violet-blue, green and red.
The HfO
2 host lattice was also used to house, simultaneously, ions such as Ce
3+, Tb
3+ and Mn
2+ to generate cold white light [
88]. These films were either doubly doped with CeCl
3 and TbCl
3 or tri-doped with CeCl
3, TbCl
3, and MnCl
2 and deposited at 300 °C. In the doubly doped films, energy transfer from Ce
3+ to Tb
3+ ions could take place in Ce
3+-Tb
3+ clusters through an electric dipole-quadrupole interaction; the efficiency of this transfer was about 81% upon excitation with 270 nm light. In the triply doped films, both Tb
3+ and Mn
2+ ions, can be sensitized by Ce
3+ ions. The efficiency of energy transfer from Ce
3+ to Tb
3+ and Mn
2+ ions was enhanced by increasing the Mn
2+ concentration, up to about 76% for the films with the highest Mn
2+ ions content (1.6 at.%). The simultaneous emission of these ions under UV excitation resulted in white light luminescence.
The PL and TL properties of HfO
2 films were investigated [
89], these films were synthesized from hafnium chloride as raw material in deionized water as solvent and were deposited at temperatures from 300 to 600 °C. SEM images showed that the film’s surface resulted very rough with semi-spherical promontories. UV irradiation was used in order to perform the thermo-luminescent (TL) characterization of these films; the 240 nm wavelength irradiation induced the best response. The PL spectra showed emission bands, centered at 425, 512 and 650 nm, associated to impurities such as chlorine and/or structural defects. As the substrate temperature was raised, a higher intensity of the band centered at 425 nm was observed. The TL experimental results showed that HfO
2 films could be useful in UV radiation dosimetry applications, using the TL method mainly in the interval of 200–400 nm; indicating an advantage over other ultraviolet dosimeters currently used.
An investigation on the luminescent properties of HfO
2 films co-doped with Ce
3+ and several concentrations of Dy
3+ was presented in 2011 [
90]. The deposition temperature was 300 °C. PL emissions from Dy
3+ ions centered at 480 nm (blue) and 575 nm (yellow) associated with the
4F
9/2 →
6H
15/2 and
4F
9/2 →
6H
13/2 electronic transitions, respectively, were observed upon UV (280 nm) excitation via a non-radiative energy transfer from Ce
3+ to Dy
3+ ions. Such energy transfer via an electric dipole–quadrupole interaction appeared to be the most probable transfer mechanism. The efficiency of this transfer increases up to 86 ± 3% for the film with the highest Dy
3+ content (1.9 ± 0.1 at.% as measured by EDS). The possibility of achieving the coordinates of ideal white light with increasing the concentration of Dy
3+ ions was demonstrated.
The PL, CL, and TL characteristics of HfO
2:Dy
3+ films were also reported in 2014 [
91]. The films were deposited at temperatures ranging from 300 to 600 °C, using chlorides as precursor reagents. XRD diffraction studies showed the presence of HfO
2 monoclinic phase in the films deposited at substrate temperatures greater than 400 °C. The surface morphology of films showed a veins shaped microstructure at low deposition temperatures, while at higher temperatures the formation of spherical particles was observed. The PL (excitation = 248 nm) and CL spectra of the doped films showed the highest emission in the band centered at 575 nm (yellow) corresponding to the transitions
4F
9/2→
6H
13/2, which is a typical transition of Dy
3+ ions. Regarding the TL behavior, the glow curve of HfO
2:Dy
+3 films exhibited spectrum with one broad band centered at about 150 °C. The highest intensity TL response was observed on the films deposited at 500 °C. A concentration quenching was observed and the optimum DyCl
3 concentration was 1 at.% in the initial solution. It was also determined that substrate temperature for the sample with maximum PL emission intensity was 600 °C. The PL (yellowish-white emission) is intense since it can be observed by the naked eyes with normal ambient illumination.
HfO
2 films co-doped with Tb
3+ or Eu
3+ ions using acetylacetonates as precursors, were studied [
92]. The films presented transmittance values in the visible region ≅90% and surface roughness less than 3.9 nm. These films were polycrystalline with a monoclinic phase for films deposited at substrate temperatures higher than 500 °C. The luminescent emissions (PL and CL) were typical of Tb
3+ and Eu
3+ ions with a luminescence concentration quenching observed for both Tb
3+ and Eu
3+ ions at 5 and 10 at.%, respectively. The peak PL and CL emission intensities for single doped films were observed for HfO
2:Tb
3+ (5 at.%) and HfO
2:Eu
3+ (10 at.%) films deposited at 500 °C. The refractive index observed in these films was between 1.97 and 2.04 and an optical band gap of 5.4 eV. The PL decay time measurements was measured on some HfO
2:Tb
3+, Eu
3+ samples. QE around 35% and 25% were obtained using excitation wavelengths of 204 nm for Tb
3+ and 215 nm for Eu
3+, respectively. HfO
2 films co-doped with Tb
3+ and Eu
3+ ions were synthesized at substrate temperatures from 400 to 600 °C using chlorides as reactive source materials [
93]. These films became polycrystalline at 600 °C exhibiting the HfO
2 monoclinic phase. Tuning by the means of the excitation wavelength and the relative concentration of the co-dopants, PL spectra with several shades, from blue to yellow (including white light) were obtained due to the combined emissions of Tb
3+ (green), Eu
3+ (red) ions and the host lattice (HfO
2) violet-blue emission. The best white light emission (
x = 0.3343,
y = 0.3406) was obtained with 382 nm excitation light and 1.35 and 0.88 at.% of Tb and Eu in the films, respectively. The CL emission spectra for these films also showed emissions from green to red (including yellow, orange, and other intermediate emissions depending on the relative content of Tb and Eu in the film). Quantum efficiency values between 47% and 78% were obtained for these films, depending on the excitation wavelength and co-doping concentrations.
4.4. Y2O3
The first publication on Y
2O
3:Eu
3+particles (synthesized by the spray pyrolysis process) was registered in the year 2000 [
94]. These particles were prepared from high solution concentrations which had a more hollow and porous structure than those prepared from low-concentration solutions. The PL spectra showed a prominent peak at 612 nm (pure red color). The colloidal seed-assisted spray pyrolysis introduced in this paper was found to be applicable to the control of morphology of phosphor particles when the stock solution concentration was high. For the colloidal seed-assisted spray pyrolysis, the stable colloidal solution should be used for homogeneity of phase and morphology of the phosphor particles. The colloidal solution of Y and Gd hydroxy carbonate sol obtained by the liquid phase reaction method, using urea, was appropriate for the preparation of Y
2O
3:Eu
3+ particles of filled and non-porous structure at high concentration of the precursor solution. The fine particles size prepared from the colloidal solution compared to those of the aqueous solution also revealed that the particles prepared from colloidal solution are much less hollow.
CL of USP deposited Y
2O
3 thin films doped separately with Eu
3+, Tb
3+ and Tm
3+ were reported in 2001 [
95]. CL spectra for films doped with Eu
3+, Tb
3+ or Tm
3+ ions presented red, green, and blue light emissions, respectively. The blue emission of Y
2O
3:Tm
3+ films had dominant peak at 476 nm. The CL intensity of these films depended strongly on annealing conditions and thulium doping concentration, presenting a maximum luminance of 30.4 cd/m
2. For the Eu
3+-doped films, a luminance of 255 cd/m
2 was obtained with a dominant peak centered at 604 nm. The luminance for the Tb
3+-doped film was 72 cd/m
2 with a dominant peak at 547 nm.
The role of LiCl added as flux on the luminescence properties of USP synthesized Y
2O
3:Eu
3+ phosphors was investigated in 2002 [
96]. The maximum PL intensity was obtained for phosphors prepared at 1300 °C from solution with LiCl flux, their intensity was 50% higher than that of phosphors prepared from solution without flux. The PL intensities of phosphors prepared at 700 and 900 °C from flux solution were 200% and 134% of those phosphors processed from solutions without flux at the same synthesis temperatures. LiCl flux played the role of enhancing the luminescence of Eu
3+ ions into Y
2O
3 host lattice by reducing defects in the phosphor particles.
Furthermore, in 2002, a study on spherical particles of Y
2O
3:Eu
3+ was published [
97]. Y
2O
3:Eu
3+ luminescent particles of spherical shape, filled morphology, and high brightness were prepared by combination of colloidal seed assisted spray pyrolysis and flux-added spray pyrolysis. Y
2O
3:Eu
3+ particles processed from Y colloidal solution with 5 at.% LiCl/KCl flux showed completely spherical shape, filled morphology, high crystallinity, and significantly improved PL emission intensity, which was 30% higher than that of particles prepared by general spray pyrolysis.
Another study on Y
2O
3:Eu
3+ powders was published in 2005 [
98]. These powders were synthesized by spray pyrolysis process and annealed at several temperatures, in the range 900–1400 °C, to achieve crystallized luminescent materials. The microstructure and macrostructure of these powders were investigated by high resolution SEM images and XRD measurements. The luminescent properties were measured under VUV excitation (254 nm). The results of this work allowed to understand the influence of the phosphors’ microstructure on PL characteristics. The spray pyrolysis powder PL efficiencies excited at 254 nm were lower than that of the commercial phosphor but under a 600 mbar Ne–Xe plasma excitation (this measurement provides a characteristic close to the working conditions in plasma display panels); the powder the brightness was equal that of the commercial phosphor. The results allowed differentiating the microstructure and macrostructure influence on luminescence. Eventually, a suitable phosphor powder for plasma display panels less dense than the commercial one has been prepared by spray pyrolysis.
A control of the morphology of Y
2O
3:Eu
3+ phosphor particles in the spray pyrolysis process was attempted by using citric acid and polyethylene glycol (PEG) as additives in the spray precursors [
99]. Three different morphologies of phosphor particles were obtained: Smooth spheres, rods, and flakes (with the presence of PEG with different molecular weights or without the presence of PEG, respectively). It was shown that the spherical Y
2O
3:Eu
3+ particles, obtained through a two-step spray pyrolysis process, had higher PL intensity than those with other morphologies.
In a similar work to the previous ones, also published in 2005, it was demonstrated that the densified particles of Y
2O
3:Eu
3+ remarkably improved the intensity of PL emissions [
100]. High luminous Y
2O
3:Eu
3+ phosphor particles with spherical shape were synthesized by Spray Pyrolysis technique. A simple but effective preparation strategy for enhancing the PL intensity of these particles was implemented. The yttrium nitrate solution was modified using an organic additive, then non-hollow particles were reached, but they were very porous, and the PL intensity was not improved. To solve this disadvantage, a drying control chemical additive (DCCA) was used as a secondary additive. It was found that the surface area was greatly reduced, and the crystallite size was increased by the use of DCCA. As a consequence, densified Y
2O
3:Eu
3+ particles showed great improvement in their PL emission intensity.
The luminescent characteristics of Y
2O
3:Eu
3+ (5 and 10 at.%) submicron particles, synthesized from the pure nitrate solutions at 900 °C, was also reported in 2010 [
101]. The synthesis conditions (gradual increase of temperature within triple zone reactor and extended residence time) assured formation of spherical, dense, non-agglomerated particles with a crystallite size about 20 nm with a cubic Y
2O
3 crystalline phase. PL emission spectra were studied under excitation with 393 nm and together with the decay lifetimes for Eu
3+ ion
5D
0 and
5D
1 levels revealed the effect of nanocrystalline nature on the luminescent properties of the powders. The PL emission spectra showed typical Eu
3+ 5D
0 →
7F
i (i = 0, 1, 2, 3, 4) electronic transitions with dominant red emission at 611 nm, while the lifetime measurements revealed the quenching effect with the rise of dopant concentration and its more consistent distribution into host lattice due to the thermal treatment. The nanostructured Y
2O
3:Eu
3+ phosphors possess favorable morphological properties for applications as red phosphor in optoelectronic microdevices, for example for luminescent displays.
Y
2O
3 powders doped with Yb
3+ and co-doped either with Tm
3+ or Ho
3+ were synthesized and reported in 2012 [
102]. These powders were processed at 900 °C using 0.1 M nitrates precursor solution and a cubic structure with space group Ia-3 was confirmed for all samples. Spherical particles with average size about 400 nm were generated with certain degree of porosity which alters their morphology during additional thermal treatment. The up-conversion emission spectra after excitation with 978 nm, as well as emission lifetimes and up-converted emission intensity dependence on excitation power were investigated. Dominant green (
5F
4,
5S
2 →
5I
8) and blue (
1G
4 →
3H
6) emissions were found for Ho
3+ and Tm
3+ samples, respectively. The enhanced emission intensities and lifetime in thermally treated samples were correlated with morphological and structural changes observed.
The enhancement of the PL emission intensity from Y
2O
3:Er
3+ thin films with Li
+ as co-dopant was published in 2013 [
103]. These films were deposited using 0.03 M of yttrium acetylacetonate, dissolved in
N,
N-dimethylformamide. The doping of the films with Er was achieved by adding erbium (III) acetate in the solution at 1.5% in relation to the Y content. The co-doping with Li was achieved adding lithium acetylacetonate to the spraying solution; the Li contents studied were 0, 0.5, 1, 2, 3, 3.5, and 4 at.% in relation to the Y content. The films were deposited at 500 °C on (1 0 0) silicon wafers. These films were polycrystalline with a pure Y
2O
3 cubic phase. The typical Er
3+ related emission spectra showed an intensity increase by a factor of ~4–5 times with the addition of 2% of Li
+. This behavior is attributed to the distortion of the local crystalline field induced by the incorporation of Li
+ ions. The addition of Li
+ reduces the intensity of the diffraction peaks after 1%, and shifts the main diffraction peak toward large angles for Li
+ doping less than 3%. The distortion of the crystalline field leads to an increment of the efficiency of intra-4f transitions by permitting the otherwise parity forbidding transitions and reducing alternative nonradiative processes. These results showed that the low-cost ultrasonic spray pyrolysis technique was a simple way to obtain rare earth doped metallic oxide films co-doped with Li
+ ions as a strategy to improve their PL emission intensity.
The enhancement of the PL emission from Y
2O
3:Er
3+ films, with the incorporation of Li
+ ions, was reported in 2014 [
104] for both visible and IR characteristic emissions of Er
3+ ions. The presence of Li
+ ions in the USP deposited films was inferred from Fourier transform infrared (FTIR) spectroscopy and also measured by Ion Beam Analysis (EBS), in which the high energies α particle yield from the
7Li(p,
α)
4He nuclear reaction was used to determine the content of Li
+ inside the films. The average content of Li
+ inside the films, as determined by EBS, increases from 0 up to 18.5 at.% for un-doped to 4 at.% Li
+ co-doped samples. The Li-C-O absorption band in the IR region was directly proportional to the Li
+ content inside the films and a calibration curve was generated with the EBS analysis. In a related work [
105], the effect of Li
+ co-doping on PL time decay characteristics of Y
2O
3:Er
3+ was reported for films deposited at 500 °C. The Er
3+ content, in this case, was fixed at 1.5 at.% while the Li
+ content in the spraying solution was varied from 1 to 4 at.% in relation to Y
3+ content. The addition of Li
+ content up to 2 at.%, besides resulting in an increase of the luminescence emission intensity, modified the luminescence time decay behavior as well. A simple model in which charge transfer from localized centers to the Er
3+ ions was proposed to describe the temporal evolution of the PL emission. The introduction of Li
+ ions in Y
2O
3:Er
3+ had an impact on the charge transfer (CT) process and on the total number of Er
3+ ions contributing to the PL emission. The PL time decay characteristics of Y
2O
3:Er
3+ films under 207 nm or 414 nm excitation light were analyzed with a simple model in which, in addition to the radiative recombination sites associated with Er
3+ ions, a CT process from localized states was considered.
Luminescent and structural characteristics of Y
2O
3:Tb
3+ thin films deposited from β-diketonates as precursors on c-Si substrates, at temperatures in the 400–550 °C range, were reported in 2014 [
106]. The PL and CL spectra intensity depended strongly on substrate temperature, the thickness of the films and the Tb
3+ doping concentration. Y
2O
3:Tb
3+ thin films exhibited one main band centered at 547 nm due to the
5D
4 →
7F
5 electronic transition of the Tb
3+ ion. A concentration quenching of the luminescence intensity was observed. At high temperatures the cubic crystalline phase of Y
2O
3 was observed as well as a reduction of organic residues. Also, at elevated temperatures, a low average surface roughness was obtained in the films with a high transmittance in the visible region.
PL and CL from Y
2O
3 doped with Tb
3+ and Eu
3+ ions films results were published in 2015 [
107]. The deposition conditions were similar to those of the work described above. The optical and structural characterization of these films was carried out as a function of substrate temperature and Tb
3+ and Eu
3+ concentrations. Films deposited above 450 °C exhibited the typical PL bands associated with either Tb
3+ or Eu
3+ intra electronic energy levels transitions. The most intense PL and CL emissions were found for dopant concentration of 10 at.% for Tb
3+ and at 8 at.% for Eu
3+ ions in spraying solution. Higher substrate temperatures improved the crystallinity of Y
2O
3 films, and showed a low average surface roughness (62 Å for Y
2O
3:Tb
3+, and 25 Å for Y
2O
3:Eu
3+ thin films). The films reported in this work were dense, and showed high refraction index (1.81), as well as a high optical transmittance in the UV-Vis range (about 90%) of the electromagnetic spectrum. These results suggest the possibility of applying those films in electroluminescent microdevices.
Recently, in 2017, an investigation on luminescent (PL and TL) Y
2O
3:Sm
3+, Li nanostructured thin films was presented [
108]. XRD measurements confirmed the cubic structure of Y
2O
3 thin films. Li ions were successfully incorporated into the Y
2O
3 host lattice and it served as a sensitizer for better crystallization. The crystallites sizes are found to be ~50 nm. Surface morphology appeared as carved sculptures of particles with agglomeration. Optical absorption spectrum exhibited a prominent absorption peak at 270 nm and the corresponding energy gap was found to be ~5.53 eV. A broad PL emission was observed in the range 560–690 nm with peaks at 595, 608 and 622 nm corresponding to characteristic electronic transitions in the Sm
3+ ions. These films were irradiated with γ-rays in a dose range 187–563 Gy; TL glow curve is deconvoluted into three peaks with temperature maxima at 400, 460 and 580 K. The activation energy and frequency factor of these TL glows were found to be in the order of ~0.58 eV and ~10
6 s
-1, respectively. Trap depths for the three luminescent centers were calculated and dose response was found to be linear in the range of 422–469 Gy.
4.5. ZnO
Zinc oxide (ZnO) is one of the most studied materials due to the various areas in which it is used. This material in the form of films and powders has also been frequently synthesized by the spray pyrolysis technique. One of the first studies on luminescent films deposited by the PSP technique of this material was on ZnO:TbCl
3 films published in 1987 [
8]. Both intrinsic and ZnO:TbCl
3 films were deposited at atmospheric pressure, using air as the carrier gas. The substrate temperature during deposition was varied from 270 to 400 °C. The solution flow rate was changed in the range of 4–16 cm
3/min and the carrier gas flow rate was kept constant throughout the deposition process at 10 I/min; the deposition time was 10 minutes in all cases and the TbCl
3 concentration was 10 at.%. These films were polycrystalline with a hexagonal wurtzite structure. The PL spectra from un-doped films showed a peak centered at 510 nm [
109], while ZnO:TbCl
3 films showed a peak at 550 nm associated to electronic transitions in the Tb
3+ ions. Later in a follow up study about these films [
110] it was reported that the light emission of the ZnO:TbCl
3 decreased with time of exposure of the sample to the excitation radiation. The phenomenon was interpreted in terms of a simple model in which a competitive process of hole trapping and photo-detrapping occurred at a radiative recombination center generated by the presence of TbC1
3.
The luminescence of undoped ZnO films, deposited from zinc nitrate solution, was also published in 1998 [
111]. The films had a polycrystalline hexagonal wurtzite type structure with no preferred orientation. Green and orange PL (excited by 320 nm light) with emission intensity strongly dependent on the deposition and annealing temperatures was reported. The best green (broad band peaked at 510 nm) luminescent films had a porous structure while orange (band peaked at 640 nm) films consisted of close-packed grains with diameters of up to more than 1 micrometer. Green and orange PL bands resulted from oxygen-poor and oxygen-rich states, respectively, in ZnO. In the case of the green films, the vacancies did not appear to penetrate deeply into the crystallites.
The effect of Li ions incorporation on the luminescence of ZnO films was reported in 1990 [
112]. The spraying solution was 0.1 M zinc acetate in isopropyl alcohol and deionized water mixed in equal proportions. Lithium chloride was added to the spraying solution at a concentration of 10 at.%. All deposited films exhibited a hexagonal polycrystalline structure. The optical transmission depended on the deposition temperatures (Ts = 340–330 °C) which showed an absorption edge shifting to longer wavelengths with higher Ts. The PL spectra of samples deposited at low Ts showed two emissions located at 420 nm and 500 nm, associated with blue emission from the Pyrex glass substrate and the blue green emission typical of un-doped zinc oxide, respectively. Films deposited at high Ts showed an emission peak centered at 555 nm apparently associated with the localized states generated by incorporation of Li ions in the ZnO films.
The photoluminescence from PSP deposited indium doped ZnO films was reported in 1992 [
113]. This study was carried out as a function of the substrate temperature and solution flow rate. Deposition solution was 0.1 M zinc acetate in three parts of isopropyl alcohol mixed with one part of deionized water. Indium doping was achieved by adding InCl
3 to the spraying solution in a concentration of 2 at.%. The substrate temperature was varied from 260 to 320 °C. These films were polycrystalline with a hexagonal crystalline structure; high solution flow rates resulted in larger disorder on the orientation of the polycrystallites. The PL spectra from films deposited at low substrate temperature or with high solution flow rate showed a broad peak centered at 530 nm which was associated with (In
Zn Vz)-luminescent centers.
The green photoluminescence efficiency and free-carrier density in ZnO phosphor powders were investigated in 1997 [
114]. An aqueous zinc nitrate solution (10 at.% Zn) was utilized in the synthesis of all powders at processing temperatures from 700 to 900 °C. Electron paramagnetic resonance, optical absorption, and photoluminescence spectroscopy were combined to characterize ZnO powders. Green PL emission was generated and a good correlation between the 510 nm green emissions with the density of paramagnetic isolated oxygen vacancies was observed. Also, both quantities increase with free-carrier concentration n
e, as long as n
e < 1.4 × 10
18 cm
−3. At higher free-carrier concentrations, both quantities decrease. A model is proposed involving the isolated oxygen vacancy as the luminescence center. It was also shown that a free-carrier depletion layer, which forms at the surface of the powder particles, and the overall free-carrier concentration of the particles have a large impact on the green emission intensity of the ZnO powder.
PL from ZnO and ZnO:Li films, reported in 1997 [
115], showed the well-known blue-green emission typical of ZnO for the undoped films. The Li-doped films PL emission was a broad band composed of four overlapping peaks at 508, 590, 604 and 810 nm (the excitation wavelength was 365 nm); the PL excitation spectra indicated that the excitation mechanism is primarily due to electron-hole pair generation across the ZnO energy bandgap. The decay time measurements of the PL showed that the lifetime of the luminescence emission was 187 ns. The dependence of the luminescent intensity with temperature showed an activation energy of 0.057 eV for competitive non-radiative transitions. These results were indicative that the lithium was atomically incorporated giving rise to a donor level in the ZnO.
PL dependence on the deposition temperature, film thickness, and post heat treatment of ZnO films, deposited from 0.4 M solution of zinc acetate dihydrate in a mixture of deionized water and isopropyl alcohol, was reported in 2000 [
116]. Chlorine free ZnO films were obtained using zinc acetate as a precursor with the (002) oriented wurtzite structure in the substrate temperature range 250–350 °C. For films with the same thickness, the intensity of green emission decreased with an increment of the O/Zn ratio as determined by XPS. The green emission intensity was gradually enhanced with increasing film thickness. Increasing deposition temperatures resulted in a reduction of the O/Zn ratio and an increment of the intensity of the green PL emission. Also, as the annealing temperature was increased, the O/Zn ratio decreased, and the green emission was consequently enhanced.
The CL from ZnO and ZnO:F (5 at.%) films deposited from ZnCl
2 precursor solutions and fluorine doped by adding NH
4F to spraying solution was reported in 2002 [
117]. The optimal substrate temperature was 450 °C presenting a hexagonal close packed structure. The CL spectra of both ZnO and ZnO:F films exhibited near-ultra-violet band peaked at
λ = 382 nm, but they differ on the visible emissions; the undoped ZnO films emitted an intense blue-green light at 520 nm and a red emission at 672 nm, the fluorine doped samples presented a new band emission centered at 454 nm and no blue-green emission. This emission was interpreted as coming from a lattice modification of the Zn
2+ environment in the crystal that could be due to a total anionic substitution process of O by F species.
Luminescent properties of ZnO and ZnO:Sn (6 at.%) films were studied through cathodoluminescence as well, in 2003 [
118]. The spraying solutions (0.05 M) were prepared from Zn and Sn chlorides dissolved in deionized water. The substrate temperature was fixed at 450 °C. Luminescence films had a polycrystalline hexagonal wurtzite type structure. The CL measurements of the undoped films showed three bands centered at 382, 520 and 672 nm. Incorporation of tin ions extinguishes the blue–green band (520 nm) while appears a blue light at 463 nm and increases the value of the band-gap transition. CL imaging of ZnO films showed that the luminescence was located at defined sites giving rise to a grain-like structure inherent to the surface morphology. The presence of Sn inside the films led to great luminescent spots, attributed to large grain sizes.
The photoluminescent properties of Eu
2+ and Eu
3+ ions in ZnO phosphors were reported in 2004 [
119]. These particles were synthesized from a zinc acetate solution and europium nitrate as the europium ions source. The crystal structure (zincite) of samples depended on the europium ions and the synthesis temperature. It was identified the coexistence of Eu
2+ and Eu
3+ ions in the as prepared ZnO samples. With addition of a 0.5 mol% concentration of europium ions, only the Eu
2+ ion was detected inside the samples, while both Eu
2+ and Eu
3+ ions existed in samples using 1 mol% or higher concentration of europium ions. Changing the excitation wavelength, it was observed that both the blue and red PL can be obtained. The reduction of the Eu
3+ to Eu
2+ ions occurred in the particles prepared by the addition of a low concentration of europium ions. This reduction changed the color of PL from red to blue. Blue PL can be enhanced by increasing the synthesis temperature. At a high concentration of europium ions, the Eu
3+ created the Eu
2O
3 component forming a ZnO–Eu
2O
3 composite.
The origin of the well-known blue-green emission of ZnO thin films was discussed on the basis of variation of the properties induced by different treatment of these films, such as ion beam irradiation (120 MeV Au ions and 80 MeV Ni ions were used for ion beam irradiation), and doping (Indium) [
120]. PL studies of untreated thin films showed only one emission at 517 nm at room temperature while the irradiated films showed a decrease in this emission intensity. Indium doping also reduced the intensity of this emission; but additional emissions (centered at 407, 590 and 670 nm) were observed in these thin films. It was proposed that the blue-green emission was due to the transitions from the bottom of the ZnO conduction band to the level associated with an oxygen antisite (O
Zn).
Photoluminescence from Er-doped ZnO films were reported in 2008 [
121]. These ZnO:Er films were deposited on (1 0 0) MgO wafers at 550 °C; the concentration of Er ions in the deposition solution (from Zn and Er acetates in methanol at 0.1 M) varied from 1.0 to 3.0 at.%. The films were polycrystalline with a dominant [002] preferential orientation. The near-ultraviolet (n-UV) PL from undoped ZnO films, n-UV peaks at 3.375 and 3.360 eV were observed at 18 K, which were proposed to be originated by free excitons and donor-bound excitons, respectively. The peaks from the free exciton transitions disappeared at room temperature. However, Er-doping enhanced the room temperature n-UV emission of ZnO:Er films. ZnO:Er (2.0 at.%) films showed n-UV peaks which were ~15 times stronger than those of undoped ZnO films.
Also, the luminescence of ZnO and ZnO:Ag nanocrystalline films deposited on Si (1 0 0) substrates from aqueous solution prepared by Zinc acetate dehydrate and Silver nitrate (6 at.%) was reported in 2008 [
122]. Intrinsic samples deposited at 500 °C with spray rate of 0.15 mL/min presented the best near-band edge near-ultraviolet emission at 378 nm observed within a set of samples deposited at different deposition temperature and spray rates. The PL intensity ratio of the n-UV emission to the deep-level emission had a largest value of 470 and the full-width at half-maximum of n-UV peak had a smallest value of 10 nm (87 meV). In addition, the n-UV emission intensity of ZnO:Ag films (with the Ag:Zn atomic ratio = 3% in the precursor solution) is markedly enhanced and the ratio to the deep-level emission, increased to at least 700. However, a silver phase was detected and the n-UV luminescence became weak for ZnO:Ag films after the annealing at 700 °C in air for 1 h.
The electrical resistivity and the photoluminescence of zinc oxide films were correlated and reported in 2009 [
123]. ZnO thin films were deposited, in this case, using zinc acetate dehydrate dissolved in methanol, ethanol, and deionized water within the substrate temperature range 320–420 °C. PL measurements showed that the as-grown ZnO thin films exhibited ultraviolet and green emission bands when excited by an Hg arc lamp using 313 nm as the excitation source. A red-shift in the near band edge was observed with the increase in the deposition temperature and was attributed to the compressive intrinsic stress present into the films. It is confirmed that oxygen vacancy (VO) is the most important factor that causes the broad visible emission. Furthermore, the visible emission and electrical resistivity of ZnO thin films are found to be a function of porosity. Additionally, it has been found that the intensity of the green emission at ~2.5 eV increased when ZnO films were deposited at 320 °C. The reason might be that the intrinsic stress, surface-to-volume ratio and porosity were incremented at low substrate temperatures. The resistivity presented similar behavior as the intensity of the green emission. A new luminescence mechanism based on the recombination related to oxygen vacancies in Zn-rich or stoichiometric conditions, was proposed.
Another study about ZnO:Li films was reported [
124] for thin films deposited on borosilicate glass substrates; the deposition temperature was kept at 250 °C. The spraying solution was 0.2 M zinc acetate in a mixture of equal proportion of isopropyl alcohol and deionized water. Lithium doping was achieved by adding required amount of lithium acetate to the spraying solution. The spray time was 2 min with solution flow rate of 18 cm
3·min
−1 and gas flow rate of 15 L·min
−1. The polycrystalline nature of the films was confirmed from XRD and TEM studies. A two-dimensional fringe moiré pattern with spacing of 1.2 nm was observed for the Li doped thin films. Lithium doping increased the roughness of the surface, thus making the film more passivated. Lithium was founded to play a key role in the excitonic as well as visible PL of ZnO films.
The effect of introducing Yb ions into ZnO films was reported in 2011 [
125]. Yb-doped ZnO thin films were deposited on glass substrates at 350 °C during 77 min with a flow rate of the solution fixed at 2.6 mL/min; the molar ratio of Yb in the spray solution was varied in the range of 0–5 at.%. XRD measurements showed that the undoped and Yb-doped ZnO films exhibit the hexagonal wurtzite crystal structure with a preferential orientation along [002] direction. All films exhibited a high transmittance. The PL measurements showed a band at 980 nm that is characteristic of Yb
3+ transition between the electronic levels
2F
5/2 and
2F
7/2. This was an experimental evidence for an efficient energy transfer from ZnO matrix to Yb
3+ ions. These films showed low resistivity and high carrier mobility which makes of interest to photovoltaic devices; all ZnO:Yb thin films were n-type semiconductor. Also, ZnO:Yb
3+ films had potential as candidates for photons down conversion process.
An investigation of structural, optical and luminescent properties of sprayed N-doped zinc (NZO) oxide thin films was reported in 2012 [
126]. The precursor solution (0.1 M of zinc acetate and
N,
N-dimethylformamide) was sprayed onto the preheated corning glass, and fluorine doped tin oxide substrates held at optimized substrate temperature of 450 °C. Influence of N doping on structural, optical and luminescence properties were studied. These films were nanocrystalline having hexagonal crystal structure. Raman analysis depicted an existence of N-Zn-O structure in NZO thin film. XPS spectrum of N 1s showed the 400 eV peak terminally bonded, well screened molecular nitrogen (γ-N
2). Lowest direct band gap of 3.17 eV was observed for 10 at.% NZO thin film. The UV, blue and green deep-level emissions in PL of NZO films were due to Zn interstitials and O vacancies. The intensity of UV emission band increased with the concentration of activated nitrogen impurities. Shifting of PL peak from 393 to 388 nm seemed to be associated with free electron to neutral acceptor transition or some LO phonon replicas, followed by free electron-acceptor transitions.
The effect that Ga has on the properties of ZnO films deposited with an aqueous solution of 0.1 M zinc acetate and gallium nitrate on corning glass substrates was reported in 2012 [
127]. XRD study depicted that the films were polycrystalline with hexagonal crystal structure and strong orientations along the (0 0 2) and (1 0 1) planes. Presence of E
high2 mode in Raman spectra indicated that the gallium doping does not affect the hexagonal structure. The ZnO:Ga thin films were adherent, compact, densely packed with hexagonal flakes and spherical grains. Optical transmittance was high (about 80%). PL spectra showed violet, blue and green emission in these films. The specific heat and thermal conductivity study showed that the phonon conduction behavior was dominant in these films. XPS analysis confirmed that the majority Zn atoms remain in the same formal valence state of Zn
2+ within an oxygen-deficient ZnO host lattice. The presence of zinc and oxygen vacancies was confirmed from PL results. The potential use of these films for optoelectronic microdevices was considered possible.
Optical and structural characteristics of ZnO:Al microrod films, obtained using different solvents (methanol and propanol), were published [
128]. Zinc chloride at 0.1 M concentration in methanol and propanol was used as spraying solution. The doping was achieved by the addition of Alq3 (tris(quinolin-8-olato) aluminum(III)) dissolved in chloroform with a concentration of 7 at.% Al; a 50 nm/min deposition rate on glass substrates, at 500 °C and a spray rate of about 5 mL/min, was achieved. Both undoped ZnO and ZnO:Al films were composed of microrods with hexagonal crystal structure and a (0 0 2) preferential orientation. SEM images revealed a quasi-aligned hexagonal shaped microrods with diameters varying between 0.7 and 1.3 micrometers. Optical studies showed that microrods had a low transmittance (~30%) and the band gap increased from 3.24 to 3.26 eV upon Al doping. PL measurements showed the two emission bands usually present in ZnO PL spectra: One sharp and intense peak at ~383 nm and one broadband ranging from 420 to 580 nm.
The lithium effect on the blue and red emissions of ZnO:Er thin films was reported in 2013 [
129]. These films were successfully deposited on heated (at 450 °C) glass substrates. The spraying solution was 0.05 M zinc chloride; erbium doping was achieved by adding ErCl
3 in concentrations of 2, 3, 5, and 7 at.%. Lithium was obtained from Li
2SO
4 in concentrations of 3, 5 and 7 at.%. This study was an investigation of the Li effect on the enhancement of CL emission intensity on Er-mono doped ZnO films. The Li–Er co-doped ZnO films showed a higher CL intensity of blue and red emissions than the Er-mono doped ZnO films. This behavior was attributed to the modification of the local symmetry of the Er
3+ ions, which increases the probabilities for the radiative intra 4f transition of the Er
3+ ions to occur. These results suggested that optimized Er–Li-codoped ZnO films could be used in data storage devices.
The blue luminescence of ZnO:Zn nanocrystals prepared from zinc acetate dihydrate aqueous solutions (0.05 M), and air as carrier gas with 1, 3, and 5 L/min flow rate was also reported in 2013 [
130]. The temperature of the tubular reactor was set at 500, 600, and 700 °C. The crystal sizes were about 14–22 nm with a zincite structure; the observed morphology was partially spherical with other particles of irregular shape. The highest PL intensity, peaked at 450 nm (excitation wavelength of 250 nm), was obtained from samples prepared using 5 L/min carrier gas at 700 °C. These PL emission was associated to oxygen vacancy in the ZnO:Zn nanocrystals.
PL emission from ZnO:Ag films, formed by nanorods (NRs) as a function of the measurement temperature (10–300 K), was published in 2014 [
131]. These films were deposited on soda-lime glass substrates at the deposition temperature of 400 °C and different deposition times (3, 5, and 10 min). The spraying solution (0.4 M) was prepared from zinc acetate and silver nitrate dissolved in in a mix of deionized water, acetic acid and methanol, a constant [Ag]/[Zn] ratio of 2 at.% was used for ZnO: Ag films deposition. The de position time variation permitted modifying the ZnO phase from the amorphous to crystalline, to change the size of ZnO:Ag NRs and to vary the PL emission spectra. PL spectra, versus temperature, revealed that the band related to the acceptor AgZn (LO phonon replicas of an acceptor bound exciton (2.877 eV)), and its second-order diffraction peak (1.44 eV) disappeared in the temperature range of 10–170 K with the formation of free exciton (FX). The PL intensity of defect related PL bands decreases monotonously in the range 10–300 K with the activation energy of 13 meV. The PL band (3.22 eV), related to the LO phonon replica of free exciton (FX-2LO) and its second-order diffraction peak (1.61 eV) increased in the range 10–300 K. FX related peak dominated in PL spectra at room temperature testifying the high quality of ZnO:Ag films deposited by the ultrasonic spray pyrolysis process.
A study on the role of substrates on the structural, optical, and morphological properties of ZnO films (nanotubes) was also reported in 2014 [
132]. The role of substrate on the properties of ZnO films was investigated; these films were deposited onto glass, ITO coated glass and sapphire substrate and annealed at 400 °C for 3 hours. Aqueous solution (0.1 M) of zinc acetate was used to deposit these films at 350 °C. In the characterization XRD, SEM, Atomic force microscopy (AFM), and PL measurements were employed. XRD measurements showed that the ZnO films deposited on sapphire and ITO substrates exhibited a strong c-axis orientation of grains with hexagonal wurtzite structure. Extremely high UV emission intensity was observed in the film on ITO. The different luminescence behavior was discussed, which would be caused by least value of strain in the film—it is well known that the visible emission of ZnO thin films is due to the lattice defects that form deep energy levels in the bandgap. Films grown on different substrates revealed differences in the morphology. ZnO films on ITO and sapphire substrates revealed better morphology than that of the films deposited on glass. AFM images of the films prepared on ITO showed uniform distribution of grains with large surface roughness, suitable for application in dye sensitized solar cells. It was concluded that the nature of substrate had significant effect on the crystal structure, PL spectra, and morphological characteristics of the deposited ZnO films.
A comprehensive review on the structure, optical, and luminescence properties of ZnO:RE nanophosphors, including up-conversion (UC) and down-conversion (DC) and/or down shifting PL, was published in 2017 [
133]. Some of ZnO:RE nanophosphors reviewed were synthesized by spray pyrolysis technique. The interest on RE doped ZnO for UC and DC nanophosphors has been motivated by the potential application of these materials in light emitting microdevices and photovoltaic cells. The two characteristic emissions observed in ZnO at the ultraviolet and visible regions are related, respectively, to excitonic recombination and intrinsic defects. XPS data demonstrated a correlation between the visible emission and intrinsic defects in these phosphors. In the case of the DC or down shifting processes, there was simultaneous emission related to intra-f level transitions of the RE ions and defects associated transitions in the ZnO host lattice. These emissions were mainly dependent on the synthesis process, annealing temperature, and RE ion concentration; only f → f transitions of RE ions were observed in the case of the UC process. These down and up conversion RE doped ZnO phosphors were evaluated for a possible application in solid state lighting devices and photovoltaic cells.
Also, in 2017 a study on the morphological, structural and optical properties (PL and CL) of ZnO thin films formed by nanoleafs or micron/submicron cauliflowers was reported [
134]. Precursor solution was composed of zinc acetate dihydrate in deionized water (resistivity: 18 MΩ·cm); solution concentration was 0.002–0.064 mol·dm
3 and reactor temperature was varied from 300 to 450 °C, in 50 °C steps. These films formed by nano and microstructures with hexagonal crystal phase were successfully synthesized on aluminum or silicon substrates. The morphology showed the presence of three types of particles: Nano-leafs, single microparticles, and particles formed by the aglomeration of microparticles. The largest zone was formed by nanoleafs with a width of 25 nm and a length 200 nm long regardless of the roughness of the substrate. Moreover, the energy bandgap (3.26 eV) was invariant to changes in synthesis parameters. The optical measurements showed no considerable differences between the luminescence properties of films formed by nanoleafs and cauliflower particles. Deconvolution of PL emission spectra made it possible to elucidate the existence of oxygen vacancies, interstitial oxygen, zinc vacancies and interstitial zinc, structural defects in nanoleafs, and micro-cauliflowers. Defects such as these play an important role into PL and CL emissions of ZnO because electronic transitions associated to these defects originated almost the 100% of these emissions.
In 2018, a paper on the enhancement of visible luminescence and photocatalytic activity of ZnO:Cu thin films was published [
135]. ZnO thin films doped with copper (0–4 at.%) were deposited on glass substrates maintaining a substrate temperature of 400 ± 10 °C. 0.4 M solution of zinc acetate and cupric acetate dissolved in a mixture of methanol, deionized water and acetic acid was used as the precursor for the deposition of these thin films. Hexagonal crystallinity (wurtzite) of the films improved at lower doping concentrations due to the easy fitting of Cu dopants in the Zn host lattice sites and preferred highly textured growth along the (0 0 2) plane. Higher doping concentration deteriorated the crystallinity and the optical transmission. EDX measurements confirmed the incorporation of Cu in the doped films. Optical energy gap red-shifted with the addition of Cu contents due to the exchange interactions and difference in iconicity of Zn and Cu. Cu doped films exhibited strong PL visible emission due to the modulation of the band structure and subsequently new levels acting as emission centers were formed in the forbidden bandgap of ZnO films. The addition of Cu ions increases the concentration of zinc interstitials, as well as zinc and oxygen vacancies which cause more intense emission in the visible region. ZnO:Cu thin films exhibited very good photocatalytic activity due to the efficient trapping of photo-generated electrons thereby suppressing the electron-hole recombination and higher doping level slightly decreased the degradation efficiency because excess dopants may act as recombination centers.
The effect of fluorine and boron co-doped ZnO thin films on the structural and luminescence properties was published in 2018 [
136]. Fluorine and boron co-doped zinc oxide (ZnO:B:F) thin films were deposited on the corning glass substrates at 400 ± 5 °C: The spraying solution was prepared by mixing zinc acetate, boric acid, and ammonium fluoride, dissolved in methanol and deionized water with a ratio of 3:1. After characterization it was found that ZnO:B:F films had high average optical transmittance; XRD patterns indicated that the obtained ZnO:B:F films had a hexagonal wurtzite type structure with (0 0 2) preferential orientation. The crystallite sizes were in the 18–40 nm range. Green emission and UV emission band are observed in PL spectra of ZnO and ZnO:B:F. Undoped ZnO films exhibited only one peak around 390 nm associated with near band ultra violet emission. It is well known that the UV emission peak usually originates from the near band-edge emission from the recombination of free exciton. Also, it was considered that the intensity ratio of UV to visible emission is commonly considered as a sign of perfect crystal quality and low defect concentration. A green emission peak was observed for ZnO:B:F films; the intensity of this peak centered at 520 nm increased while the B–F concentrations increased. The observed green emission is also due to the impurity levels related the oxygen vacancy (Vo) in ZnO:B:F films. The electrical resistivity, carrier concentration and Hall mobility also were measured. The highest Hall mobility of 13.22 cm
2 v
−1·s, and the lowest electrical resistivity of 3.13 × 10
−4 Ω·cm, were obtained at the optimal boron-fluorine co-doping concentration of 5 at.%. All of the results were appreciated in point of view of optoelectronic industry and photovoltaic solar cell applications and it was concluded that B-F co-doping has a positively effect on electrical properties.
4.6. ZnS
A research on the luminescence of ZnS, ZnS:TbC1
3 and ZnS:SmC1
3 films, deposited by the Pneumatic SP technique, was first reported in 1988 [
137]. The ZnS films were deposited using a spraying solution obtained by mixing in equal proportions solutions of 0.1 M of Zn acetate and 0.1 M of dimetylthiourea (C
3H
8N
2S), both dissolved in three parts of isopropyl alcohol and one part of deionized water. The doped films were prepared by adding TbCI
3; or SmCI
3, to the spraying solution at a 10 at.% concentration; the substrate temperature, during the deposition, was either 300, 330, 360, or 375 °C. The solution flow rate was 14 cm
3/minute and the nozzle substrate distance was 30 cm in all cases. The doped films exhibited strong PL emission with a blue dominant peak at about 460 nm. This peak is characteristic of chlorine-doped ZnS phosphors. The films had poor crystallinity with a cubic crystalline structure. The optical transmission (T about 80%) characteristics of these films showed an absorption edge shifted to shorter wavelengths compared with those of the undoped films. Photoluminescent characteristics of In-, Al-, and Cu-doped ZnS films were reported in 1989 [
138]. These films were deposited from a spraying solution formed by 0.1 M (CH
3COO)
2 + 0.1 M C
3H
8N
2S dissolved in a mixture of three parts of isopropyl alcohol plus one part of deionized water. Doping was reached adding InC1
3, A1C1
3, or CuCl to the starting solution. The substrate temperatures were either 270, 300 or 330 °C. The substrates were pyrex glass slides, pyrex glass coated with In
2O
3 and silicon oxide. All films showed polycrystalline features which could be associated to a wurtzite structure of ZnS. Also, the presence of chlorine was detected into the films in quantities that depended on the deposition parameters. The PL spectra measured at room temperature displayed different emission peaks for each one of the impurities. The PL spectra from the Al-doped ZnS films showed a peak centered at 470 nm. The In-doped ZnS films showed a peak about 545 nm and the PL spectrum from the Cu-doped films exhibited a peak at 570 nm. The shape and intensity of the PL spectra do not depend strongly on the type of substrate.
The luminescent properties of ZnS:Mn films deposited by the pyrolysis spray technique on glass substrates at atmospheric pressure using air as a carrier gas were reported, for the first time, in 1992 [
139]. The spraying solution in this case consisted of 0.1 M of Zn acetate and 0.1 M of dimethylthiourea in a mixture of three parts of isopropyl alcohol and one part of deionized water. The Mn doping was achieved by mixing MnCl
2 (0–20 at.%) in the spraying solutions; the deposition temperature was varied between 340 and 500 °C in steps of 20 °C. All films resulted polycrystalline with a wurtzite (hexagonal) structure. The PL spectra show, besides the characteristic light emission associated with Mn (yellow at 590 nm) in a ZnS host lattice, a peak associated with the self-activated emission (blue at 490 nm) observable at low substrate temperatures and/or long deposition times. The presence of chlorine impurities in the films was suggested to be associated with this emission. The Mn related luminescence showed a quenching effect with the Mn concentration (at concentrations higher than 3 at.% Mn in the spraying solution). The light emission at this center had an activation energy of 0.71 ± 0.05 eV with the deposition temperature. This energy was proposed to be related with the energy required for the Mn atoms to find a proper site during the growth process to form a Mn
2+ center. These films were incorporated in a Metal-Insulator-active layer- Insulator-Metal (M-I-S-I-M) structure and their electroluminescent features were reported in 1995 [
14]. These alternating current electroluminescent thin film structures were prepared using, for the first time, high-quality SiO
2 insulating thin films and spray pyrolyzed ZnS:Mn
2+ as the active layer. The structures prepared with 60 nm thick insulating films showed threshold voltages of 30 V (rms) and saturation voltages of about 56 V (rms). The electroluminescent emission spectra presented a peak centered at 590 nm (yellow emission) associated with the Mn
2+ center. The brightness-voltage characteristics were typical for a structure of the M-I-S-I-M type. The external efficiency calculated from the charge-voltage characteristics had a value of 1.8 Lumen/Watt.
Spray pyrolysis synthesis of ZnS nanoparticles (sub-10 nm) from a single-source precursor was published in 2009 [
140]. Here, it was reported the synthesis of cubic ZnS nanoparticles from a low-cost single-source precursor in a continuous spray pyrolysis reactor. In this study, a single-source precursor: Diethyldithiocarbamate, [(C
2H
5)
2NCS
2]
2Zn, dissolved in toluene was used to synthesize ZnS nanoparticles. The furnace setpoint temperature was typically 600–800 °C. In this method, the evaporation and decomposition of precursor and nucleation of particles occur sequentially. XRD indicated a Cubic ZnS (zinc blende) for the synthesized particles. High Resolution Transmission Electron Microscopy (HRTEM) images showed ZnS particles with diameters ranging from 2 to 7 nm were. As-synthesized ZnS nanoparticles (excited at 350 nm) exhibited blue photoluminescence near to 440 nm had quantum yields up to 15% after HF treatment. This demonstrated a potentially general approach for continuous low-cost synthesis of semiconductor quantum dots, and applications in solar cells, lasers and displays. Also, ZnS nanoparticles can be applied as phosphors, probes for bio-imaging, emitters in light emitting diodes and photocatalysts.




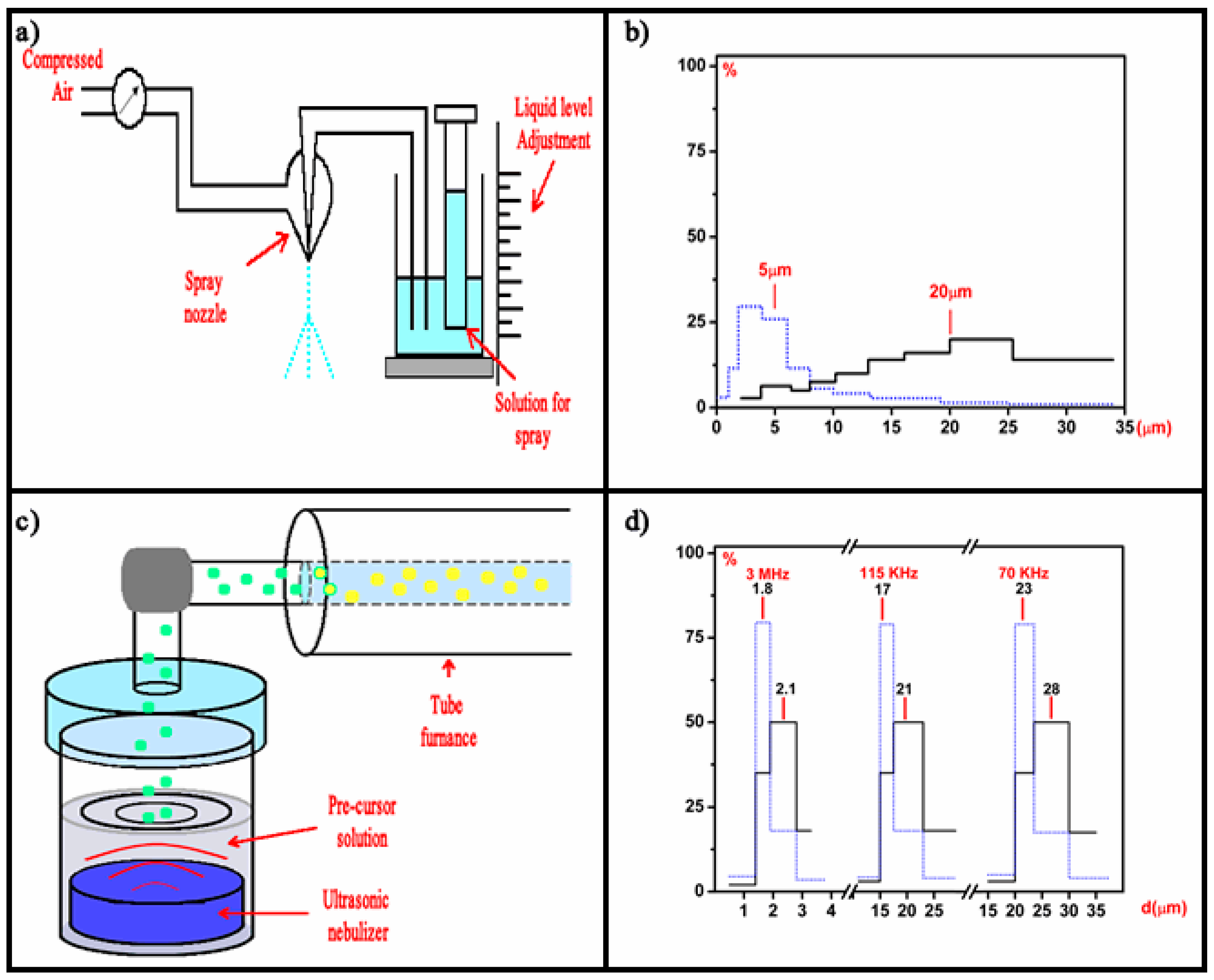{kind=link}
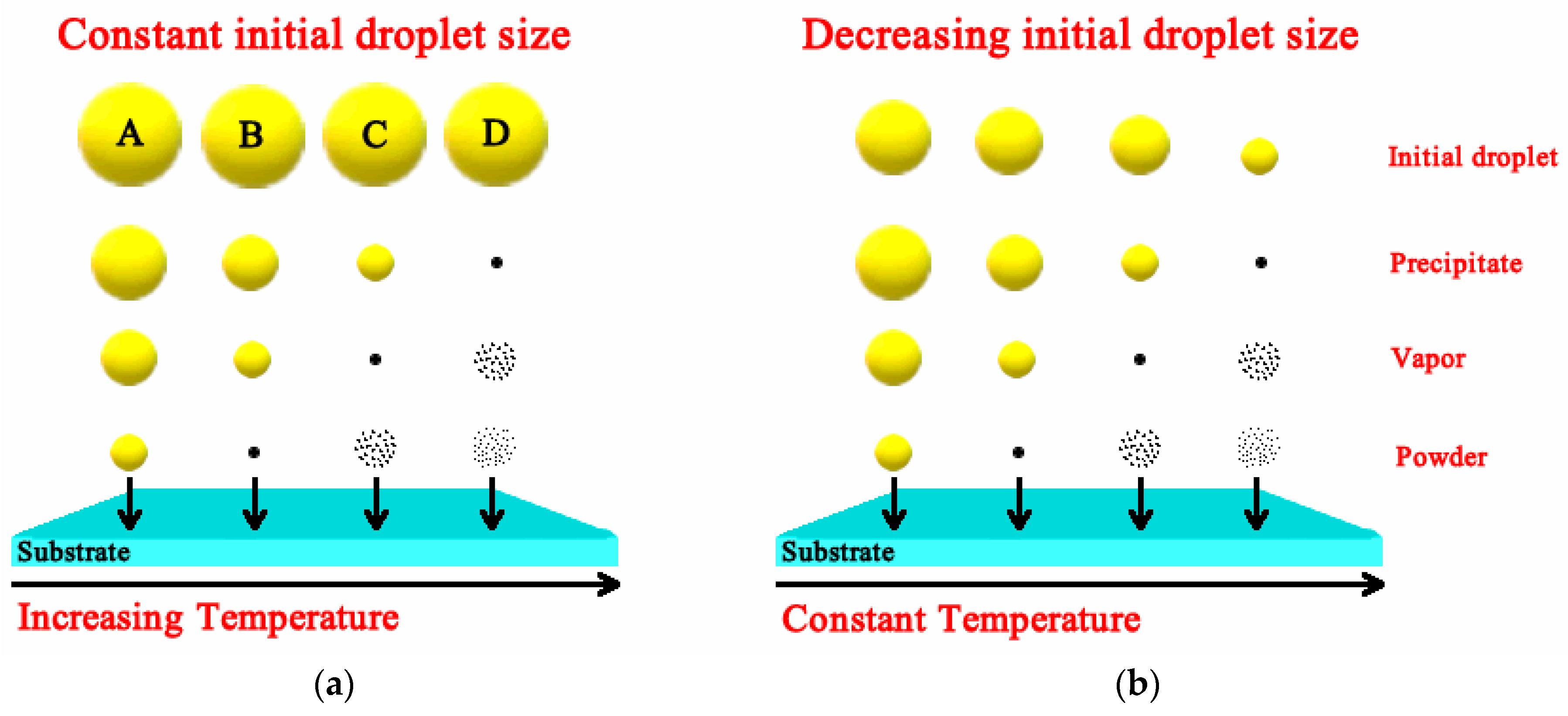{kind=link}