IRF4 Mediates the Oncogenic Effects of STAT3 in Anaplastic Large Cell Lymphomas

 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
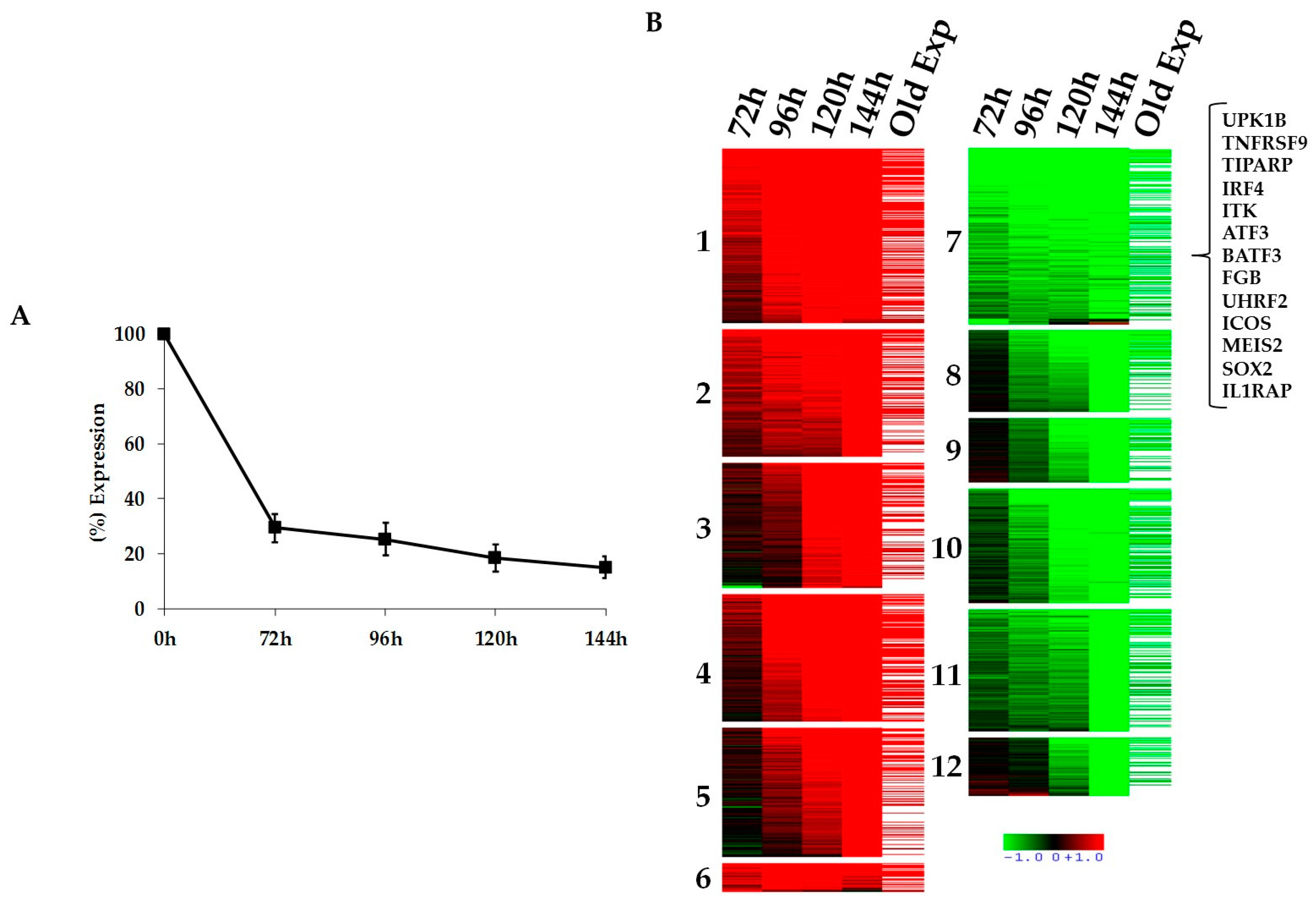
2.1. Kinetics of STAT3-Regulated Genes in ALCL Cell Lines
2.2. Functional Validation of STAT3 Target Genes
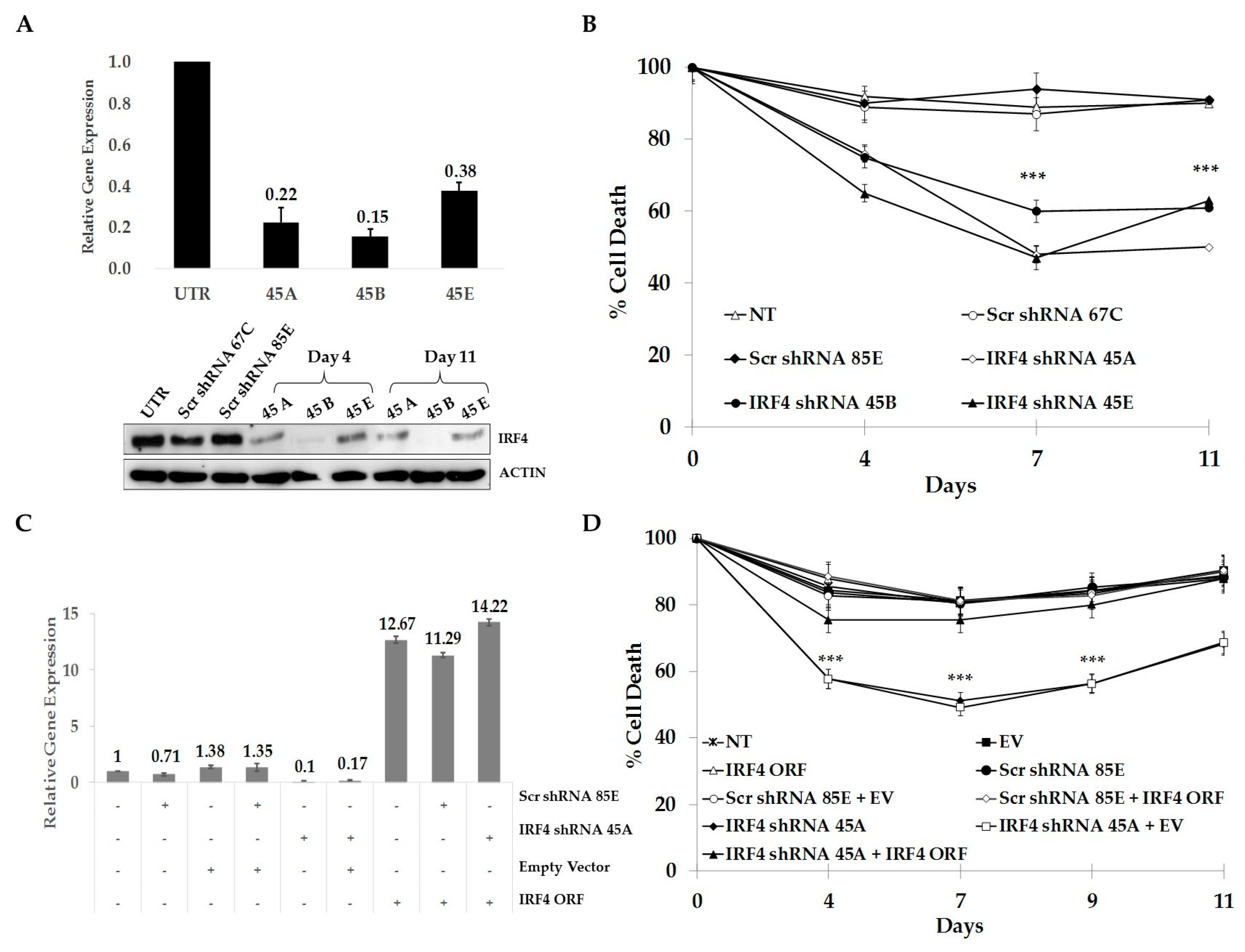
2.3. IRF4 is Required for Proliferation and Survival of ALCL Cells
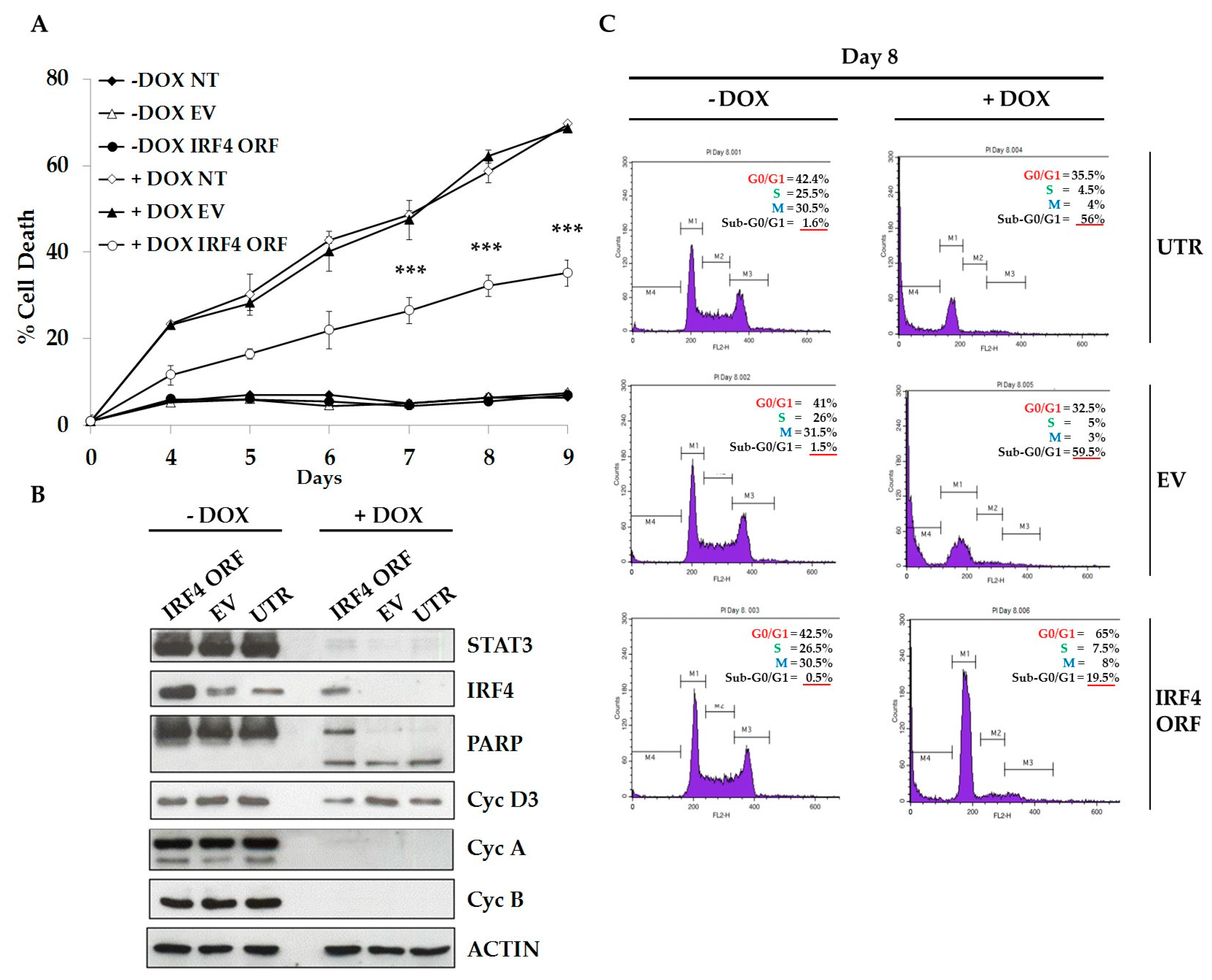
2.4. IRF4 Partially Mediates STAT3 Oncogenic Properties in ALCL Cells
2.5. ALCL Cell Lines Display Heterogeneous Sensitivity to Immunomodulatory Drugs
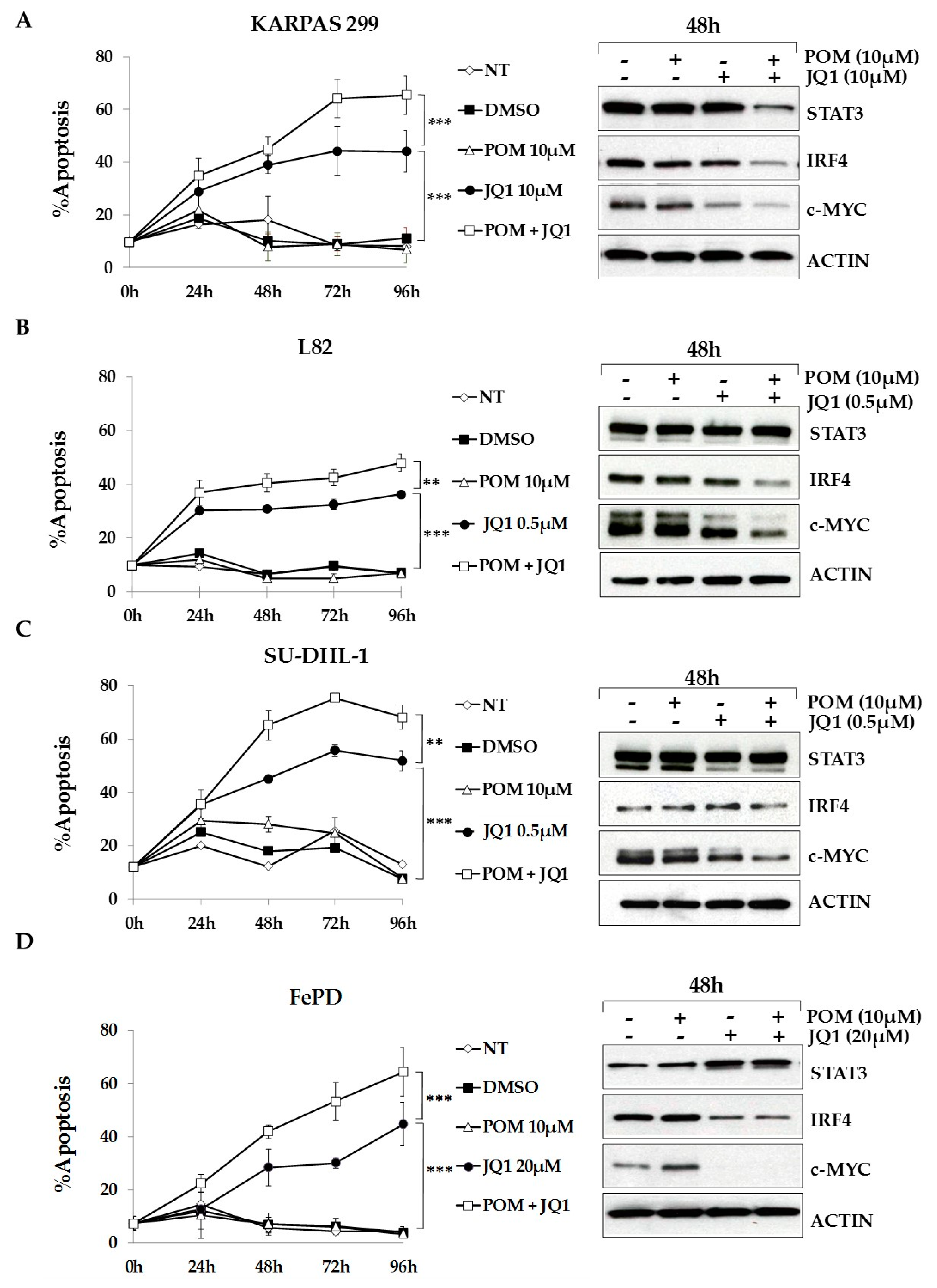
2.6. The Bromodomain and Extra-Terminal (BET)-Inhibitor JQ1 Sensitizes ALCL Cells to Pomalidomide Treatment
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. shRNA Sequences, cDNA, and Plasmid Constructs
4.3. Virus Production and In Vitro Transduction
4.4. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.5. Immunoblotting
4.6. Flow Cytometry Analysis of Apoptosis and Cell Cycle
4.7. ATPlite Assay
4.8. Lenalidomide, Pomalidomide and JQ1 Treatments
4.9. Gene Expression Profiling
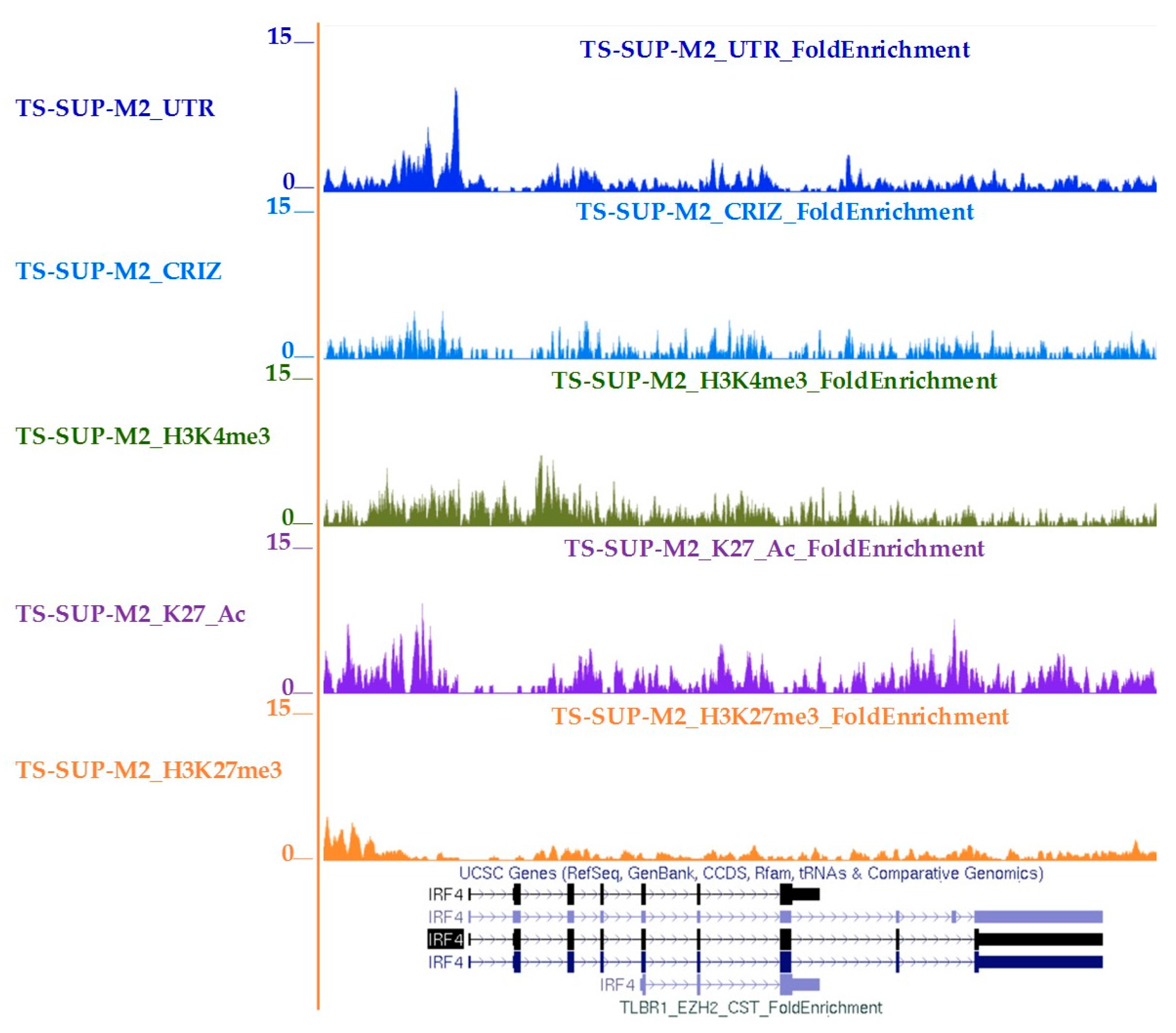
4.10. Chromatin Immunoprecipitation-Sequencing (ChIP-seq)
4.11. Statistics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stein, H.; Mason, D.Y.; Gerdes, J.; O’Connor, N.; Wainscoat, J.; Pallesen, G.; Gatter, K.; Falini, B.; Delsol, G.; Lemke, H.; et al. The expression of the hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: Evidence that reed-sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985, 66, 848–858. [Google Scholar] [PubMed]
- Turner, S.D.; Lamant, L.; Kenner, L.; Brugieres, L. Anaplastic large cell lymphoma in paediatric and young adult patients. Br. J. Haematol. 2016, 173, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the world health organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Iwahara, T.; Fujimoto, J.; Wen, D.; Cupples, R.; Bucay, N.; Arakawa, T.; Mori, S.; Ratzkin, B.; Yamamoto, T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Naeve, C.; Mathew, P.; James, P.L.; Kirstein, M.N.; Cui, X.; Witte, D.P. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997, 14, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Kruczynski, A.; Delsol, G.; Laurent, C.; Brousset, P.; Lamant, L. Anaplastic lymphoma kinase as a therapeutic target. Exp. Opin. Ther. Targets 2012, 16, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Lee, W.; Obenauf, A.C.; Ran, L.; Murali, R.; Zhang, Q.F.; Wong, E.W.; Hu, W.; Scott, S.N.; Shah, R.H.; et al. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature 2015, 526, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Martinengo, C.; Mastini, C.; Ambrogio, C.; D’Escamard, V.; Forni, G.; Inghirami, G. The anaplastic lymphoma kinase is an effective oncoantigen for lymphoma vaccination. Nat. Med. 2008, 14, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Gong, J.Z.; Guasparri, I.; Pesci, A.; Cai, J.; Liu, J.; Simmons, W.J.; Dhall, G.; Howes, J.; Piva, R.; et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood 2003, 101, 1919–1927. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Pellegrino, E.; Mattioli, M.; Agnelli, L.; Lombardi, L.; Boccalatte, F.; Costa, G.; Ruggeri, B.A.; Cheng, M.; Chiarle, R.; et al. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J. Clin. Investig. 2006, 116, 3171–3182. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Albom, M.S.; Lu, L.; Quail, M.R.; Becknell, N.C.; Weinberg, L.R.; Reddy, D.R.; Holskin, B.P.; Angeles, T.S.; Underiner, T.L.; et al. Anaplastic lymphoma kinase activity is essential for the proliferation and survival of anaplastic large-cell lymphoma cells. Blood 2006, 107, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Zamo, A.; Chiarle, R.; Piva, R.; Howes, J.; Fan, Y.; Chilosi, M.; Levy, D.E.; Inghirami, G. Anaplastic lymphoma kinase (ALK) activates STAT3 and protects hematopoietic cells from cell death. Oncogene 2002, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Simmons, W.J.; Cai, H.; Dhall, G.; Zamo, A.; Raz, R.; Karras, J.G.; Levy, D.E.; Inghirami, G. STAT3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med. 2005, 11, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raghunath, P.N.; Xue, L.; Majewski, M.; Carpentieri, D.F.; Odum, N.; Morris, S.; Skorski, T.; Wasik, M.A. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J. Immunol. 2002, 168, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Ruchatz, H.; Coluccia, A.M.; Stano, P.; Marchesi, E.; Gambacorti-Passerini, C. Constitutive activation of JAK2 contributes to proliferation and resistance to apoptosis in NPM/ALK-transformed cells. Exp. Hematol. 2003, 31, 309–315. [Google Scholar] [CrossRef]
- Amin, H.M.; Medeiros, L.J.; Ma, Y.; Feretzaki, M.; Das, P.; Leventaki, V.; Rassidakis, G.Z.; O’Connor, S.L.; McDonnell, T.J.; Lai, R. Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene 2003, 22, 5399–5407. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Medeiros, L.J.; Rassidakis, G.Z.; Yared, M.A.; Tsioli, P.; Leventaki, V.; Schmitt-Graeff, A.; Herling, M.; Amin, H.M.; Lai, R. Differential expression and clinical significance of tyrosine-phosphorylated STAT3 in ALK+ and ALK− anaplastic large cell lymphoma. Clin. Cancer Res. 2003, 9, 3692–3699. [Google Scholar] [PubMed]
- Chen, J.; Zhang, Y.; Petrus, M.N.; Xiao, W.; Nicolae, A.; Raffeld, M.; Pittaluga, S.; Bamford, R.N.; Nakagawa, M.; Ouyang, S.T.; et al. Cytokine receptor signaling is required for the survival of ALK—anaplastic large cell lymphoma, even in the presence of JAK1/STAT3 mutations. Proc. Natl. Acad. Sci. USA 2017, 114, 3975–3980. [Google Scholar] [CrossRef] [PubMed]
- Schlette, E.J.; Medeiros, L.J.; Goy, A.; Lai, R.; Rassidakis, G.Z. Survivin expression predicts poorer prognosis in anaplastic large-cell lymphoma. J. Clin. Oncol. 2004, 22, 1682–1688. [Google Scholar] [CrossRef] [PubMed]
- Wasik, M.A.; Zhang, Q.; Marzec, M.; Kasprzycka, M.; Wang, H.Y.; Liu, X. Anaplastic lymphoma kinase (ALK)-induced malignancies: Novel mechanisms of cell transformation and potential therapeutic approaches. Semin. Oncol. 2009, 36, S27–S35. [Google Scholar] [CrossRef] [PubMed]
- Marzec, M.; Liu, X.; Wong, W.; Yang, Y.; Pasha, T.; Kantekure, K.; Zhang, P.; Woetmann, A.; Cheng, M.; Odum, N.; et al. Oncogenic kinase NPM/ALK induces expression of HIF1α mRNA. Oncogene 2011, 30, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Ambrogio, C.; Voena, C.; Manazza, A.D.; Martinengo, C.; Costa, C.; Kirchhausen, T.; Hirsch, E.; Inghirami, G.; Chiarle, R. The anaplastic lymphoma kinase controls cell shape and growth of anaplastic large cell lymphoma through CDC42 activation. Cancer Res. 2008, 68, 8899–8907. [Google Scholar] [CrossRef] [PubMed]
- Spaccarotella, E.; Pellegrino, E.; Ferracin, M.; Ferreri, C.; Cuccuru, G.; Liu, C.; Iqbal, J.; Cantarella, D.; Taulli, R.; Provero, P.; et al. STAT3-mediated activation of microrna cluster 17~92 promotes proliferation and survival of ALK-positive anaplastic large cell lymphoma. Haematologica 2014, 99, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Hoareau-Aveilla, C.; Merkel, O.; Meggetto, F. Microrna and ALK-positive anaplastic large cell lymphoma. Front. Biosci. 2015, 7, 217–225. [Google Scholar]
- Furtek, S.L.; Backos, D.S.; Matheson, C.J.; Reigan, P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem. Biol. 2016, 11, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Agnelli, L.; Pellegrino, E.; Todoerti, K.; Grosso, V.; Tamagno, I.; Fornari, A.; Martinoglio, B.; Medico, E.; Zamo, A.; et al. Gene expression profiling uncovers molecular classifiers for the recognition of anaplastic large-cell lymphoma within peripheral T-cell neoplasms. J. Clin. Oncol. 2010, 28, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Kasprzycka, M.; Marzec, M.; Liu, X.; Zhang, Q.; Wasik, M.A. Nucleophosmin/anaplastic lymphoma kinase (NPM/ALK) oncoprotein induces the T regulatory cell phenotype by activating STAT3. Proc. Natl. Acad. Sci. USA 2006, 103, 9964–9969. [Google Scholar] [CrossRef] [PubMed]
- Vallania, F.; Schiavone, D.; Dewilde, S.; Pupo, E.; Garbay, S.; Calogero, R.; Pontoglio, M.; Provero, P.; Poli, V. Genome-wide discovery of functional transcription factor binding sites by comparative genomics: The case of STAT3. Proc. Natl. Acad. Sci. USA 2009, 106, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, Y.; Anderson, E.M.; Birmingham, A.; Reynolds, A.; Karpilow, J.; Robinson, K.; Leake, D.; Marshall, W.S.; Khvorova, A. Off-target effects by sirna can induce toxic phenotype. RNA 2006, 12, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Girona, A.; Heintel, D.; Zhang, L.H.; Mendy, D.; Gaidarova, S.; Brady, H.; Bartlett, J.B.; Schafer, P.H.; Schreder, M.; Bolomsky, A.; et al. Lenalidomide downregulates the cell survival factor, interferon regulatory factor-4, providing a potential mechanistic link for predicting response. Br. J. Haematol. 2011, 154, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L.; Emre, N.C.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Weilemann, A.; Grau, M.; Erdmann, T.; Merkel, O.; Sobhiafshar, U.; Anagnostopoulos, I.; Hummel, M.; Siegert, A.; Hayford, C.; Madle, H.; et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood 2015, 125, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Boi, M.; Todaro, M.; Vurchio, V.; Yang, S.N.; Moon, J.; Kwee, I.; Rinaldi, A.; Pan, H.; Crescenzo, R.; Cheng, M.; et al. Therapeutic efficacy of the bromodomain inhibitor OTX015/MK-8628 in ALK-positive anaplastic large cell lymphoma: An alternative modality to overcome resistant phenotypes. Oncotarget 2016, 7, 79637–79653. [Google Scholar] [CrossRef] [PubMed]
- Hassler, M.R.; Pulverer, W.; Lakshminarasimhan, R.; Redl, E.; Hacker, J.; Garland, G.D.; Merkel, O.; Schiefer, A.I.; Simonitsch-Klupp, I.; Kenner, L.; et al. Insights into the pathogenesis of anaplastic large-cell lymphoma through genome-wide DNA methylation profiling. Cell Rep. 2016, 17, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Schiefer, A.I.; Vesely, P.; Hassler, M.R.; Egger, G.; Kenner, L. The role of AP-1 and epigenetics in ALCL. Front. Biosci. 2015, 7, 226–235. [Google Scholar]
- Jundt, F.; Anagnostopoulos, I.; Forster, R.; Mathas, S.; Stein, H.; Dorken, B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood 2002, 99, 3398–3403. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Hinz, M.; Anagnostopoulos, I.; Krappmann, D.; Lietz, A.; Jundt, F.; Bommert, K.; Mechta-Grigoriou, F.; Stein, H.; Dorken, B.; et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-κB. EMBO J. 2002, 21, 4104–4113. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, L.; Mereu, E.; Pellegrino, E.; Limongi, T.; Kwee, I.; Bergaggio, E.; Ponzoni, M.; Zamo, A.; Iqbal, J.; Piccaluga, P.P.; et al. Identification of a 3-gene model as a powerful diagnostic tool for the recognition of ALK-negative anaplastic large-cell lymphoma. Blood 2012, 120, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Lollies, A.; Hartmann, S.; Schneider, M.; Bracht, T.; Weiss, A.L.; Arnolds, J.; Klein-Hitpass, L.; Sitek, B.; Hansmann, M.L.; Kuppers, R.; et al. An oncogenic axis of STAT-mediated BATF3 upregulation causing MYC activity in classical Hodgkin lymphoma and anaplastic large cell lymphoma. Leukemia 2018, 32, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Grumont, R.J.; Gerondakis, S. Rel induces interferon regulatory factor 4 (IRF-4) expression in lymphocytes: Modulation of interferon-regulated gene expression by Rel/nuclear factor κB. J. Exp. Med. 2000, 191, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Mittrucker, H.W.; Matsuyama, T.; Grossman, A.; Kundig, T.M.; Potter, J.; Shahinian, A.; Wakeham, A.; Patterson, B.; Ohashi, P.S.; Mak, T.W. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science 1997, 275, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, V.; Kannanganat, S.; Maienschein-Cline, M.; Cook, S.L.; Chen, J.; Bahroos, N.; Sievert, E.; Corse, E.; Chong, A.; Sciammas, R. The IRF4 gene regulatory module functions as a read-write integrator to dynamically coordinate T helper cell fate. Immunity 2017, 47, 481–497.e7. [Google Scholar] [CrossRef] [PubMed]
- Iida, S.; Rao, P.H.; Butler, M.; Corradini, P.; Boccadoro, M.; Klein, B.; Chaganti, R.S.; Dalla-Favera, R. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat. Genet. 1997, 17, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.L.; Law, M.; Remstein, E.D.; Macon, W.R.; Erickson, L.A.; Grogg, K.L.; Kurtin, P.J.; Dogan, A. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia 2009, 23, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, A.; Veckman, V.; Nikula, T.; Lahesmaa, R.; Kinnunen, L.; Matikainen, S.; Julkunen, I. Differential expression of ifn regulatory factor 4 gene in human monocyte-derived dendritic cells and macrophages. J. Immunol. 2005, 175, 6570–6579. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.X.; Braggio, E.; Shi, C.X.; Kortuem, K.M.; Bruins, L.A.; Schmidt, J.E.; Chang, X.B.; Langlais, P.; Luo, M.; Jedlowski, P.; et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDS in multiple myeloma. Blood 2014, 124, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shaffer, A.L., 3rd; Emre, N.C.; Ceribelli, M.; Zhang, M.; Wright, G.; Xiao, W.; Powell, J.; Platig, J.; Kohlhammer, H.; et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell 2012, 21, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Witzig, T.E.; Wiernik, P.H.; Moore, T.; Reeder, C.; Cole, C.; Justice, G.; Kaplan, H.; Voralia, M.; Pietronigro, D.; Takeshita, K.; et al. Lenalidomide oral monotherapy produces durable responses in relapsed or refractory indolent non-Hodgkin’s lymphoma. J. Clin. Oncol. 2009, 27, 5404–5409. [Google Scholar] [CrossRef] [PubMed]
- Zeidner, J.F.; Foster, M.C. Immunomodulatory drugs: IMiDS in acute myeloid leukemia (AML). Curr. Drug Targets 2015, 18, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Zhu, Y.; Shi, C.; Stewart, A.K. Mechanism of immunomodulatory drugs’ action in the treatment of multiple myeloma. Acta Biochim. Biophys. Sin. (Shanghai) 2014, 46, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.X.; Braggio, E.; Shi, C.X.; Bruins, L.A.; Schmidt, J.E.; Van Wier, S.; Chang, X.B.; Bjorklund, C.C.; Fonseca, R.; Bergsagel, P.L.; et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011, 118, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Heintel, D.; Rocci, A.; Ludwig, H.; Bolomsky, A.; Caltagirone, S.; Schreder, M.; Pfeifer, S.; Gisslinger, H.; Zojer, N.; Jager, U.; et al. High expression of cereblon (CRBN) is associated with improved clinical response in patients with multiple myeloma treated with lenalidomide and dexamethasone. Br. J. Haematol. 2013, 161, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Kosek, J.; Wang, M.; Heise, C.; Schafer, P.H.; Chopra, R. Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. Br. J. Haematol. 2013, 160, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Pal, R.; Monaghan, S.A.; Schafer, P.; Ouyang, H.; Mapara, M.; Galson, D.L.; Lentzsch, S. Imid immunomodulatory compounds block C/EBPβ translation through EIF4E down-regulation resulting in inhibition of mm. Blood 2011, 117, 5157–5165. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, C.C.; Ma, W.; Wang, Z.Q.; Davis, R.E.; Kuhn, D.J.; Kornblau, S.M.; Wang, M.; Shah, J.J.; Orlowski, R.Z. Evidence of a role for activation of Wnt/β-catenin signaling in the resistance of plasma cells to lenalidomide. J. Biol. Chem. 2011, 286, 11009–11020. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, R.; Matta, H.; Tolani, B.; Triche, T., Jr.; Chaudhary, P.M. Immunomodulatory drugs target IKZF1-IRF4-MYC axis in primary effusion lymphoma in a cereblon-dependent manner and display synergistic cytotoxicity with BRD4 inhibitors. Oncogene 2016, 35, 1797–1810. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.T.; Ramachandran, J.; Yee, A.J.; Eda, H.; Santo, L.; Panaroni, C.; Mertz, J.A.; Sims Iii, R.J.; Cooper, M.R.; Raje, N. Preclinical activity of CPI-0610, a novel small-molecule bromodomain and extra-terminal protein inhibitor in the therapy of multiple myeloma. Leukemia 2017, 31, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Moros, A.; Rodriguez, V.; Saborit-Villarroya, I.; Montraveta, A.; Balsas, P.; Sandy, P.; Martinez, A.; Wiestner, A.; Normant, E.; Campo, E.; et al. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia 2014, 28, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Diaz, T.; Rodriguez, V.; Lozano, E.; Mena, M.P.; Calderon, M.; Rosinol, L.; Martinez, A.; Tovar, N.; Perez-Galan, P.; Blade, J.; et al. The BET bromodomain inhibitor CPI203 improves lenalidomide and dexamethasone activity in in vitro and in vivo models of multiple myeloma by blockade of ikaros and MYC signaling. Haematologica 2017, 102, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Du, Z.; Xing, Y.; Zhou, T.; Chen, T.; Shi, M. Interferon regulatory factor 4 (IRF4) is overexpressed in human nonsmall cell lung cancer (NSCLC) and activates the notch signaling pathway. Mol. Med. Rep. 2017, 16, 6034–6040. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Geuna, M. A flow cytometry assay simultaneously detects independent apoptotic parameters. Cytometry 2001, 45, 151–157. [Google Scholar] [CrossRef]
- Tsuboi, K.; Iida, S.; Inagaki, H.; Kato, M.; Hayami, Y.; Hanamura, I.; Miura, K.; Harada, S.; Kikuchi, M.; Komatsu, H.; et al. MUM1/IRF4 expression as a frequent event in mature lymphoid malignancies. Leukemia 2000, 14, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Boddicker, R.L.; Kip, N.S.; Xing, X.; Zeng, Y.; Yang, Z.Z.; Lee, J.H.; Almada, L.L.; Elsawa, S.F.; Knudson, R.A.; Law, M.E.; et al. The oncogenic transcription factor IRF4 is regulated by a novel CD30/NF-κ positive feedback loop in peripheral T-cell lymphoma. Blood 2015, 125, 3118–3127. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bandini, C.; Pupuleku, A.; Spaccarotella, E.; Pellegrino, E.; Wang, R.; Vitale, N.; Duval, C.; Cantarella, D.; Rinaldi, A.; Provero, P.; et al. IRF4 Mediates the Oncogenic Effects of STAT3 in Anaplastic Large Cell Lymphomas. Cancers 2018, 10, 21. https://doi.org/10.3390/cancers10010021
Bandini C, Pupuleku A, Spaccarotella E, Pellegrino E, Wang R, Vitale N, Duval C, Cantarella D, Rinaldi A, Provero P, et al. IRF4 Mediates the Oncogenic Effects of STAT3 in Anaplastic Large Cell Lymphomas. Cancers. 2018; 10(1):21. https://doi.org/10.3390/cancers10010021
Chicago/Turabian StyleBandini, Cecilia, Aldi Pupuleku, Elisa Spaccarotella, Elisa Pellegrino, Rui Wang, Nicoletta Vitale, Carlotta Duval, Daniela Cantarella, Andrea Rinaldi, Paolo Provero, and et al. 2018. "IRF4 Mediates the Oncogenic Effects of STAT3 in Anaplastic Large Cell Lymphomas" Cancers 10, no. 1: 21. https://doi.org/10.3390/cancers10010021
APA StyleBandini, C., Pupuleku, A., Spaccarotella, E., Pellegrino, E., Wang, R., Vitale, N., Duval, C., Cantarella, D., Rinaldi, A., Provero, P., Di Cunto, F., Medico, E., Bertoni, F., Inghirami, G., & Piva, R. (2018). IRF4 Mediates the Oncogenic Effects of STAT3 in Anaplastic Large Cell Lymphomas. Cancers, 10(1), 21. https://doi.org/10.3390/cancers10010021