Modification of Epigenetic Histone Acetylation in Hepatocellular Carcinoma


Abstract
:1. Introduction
2. Aryl Hydrocarbon Receptor
2.1. Function
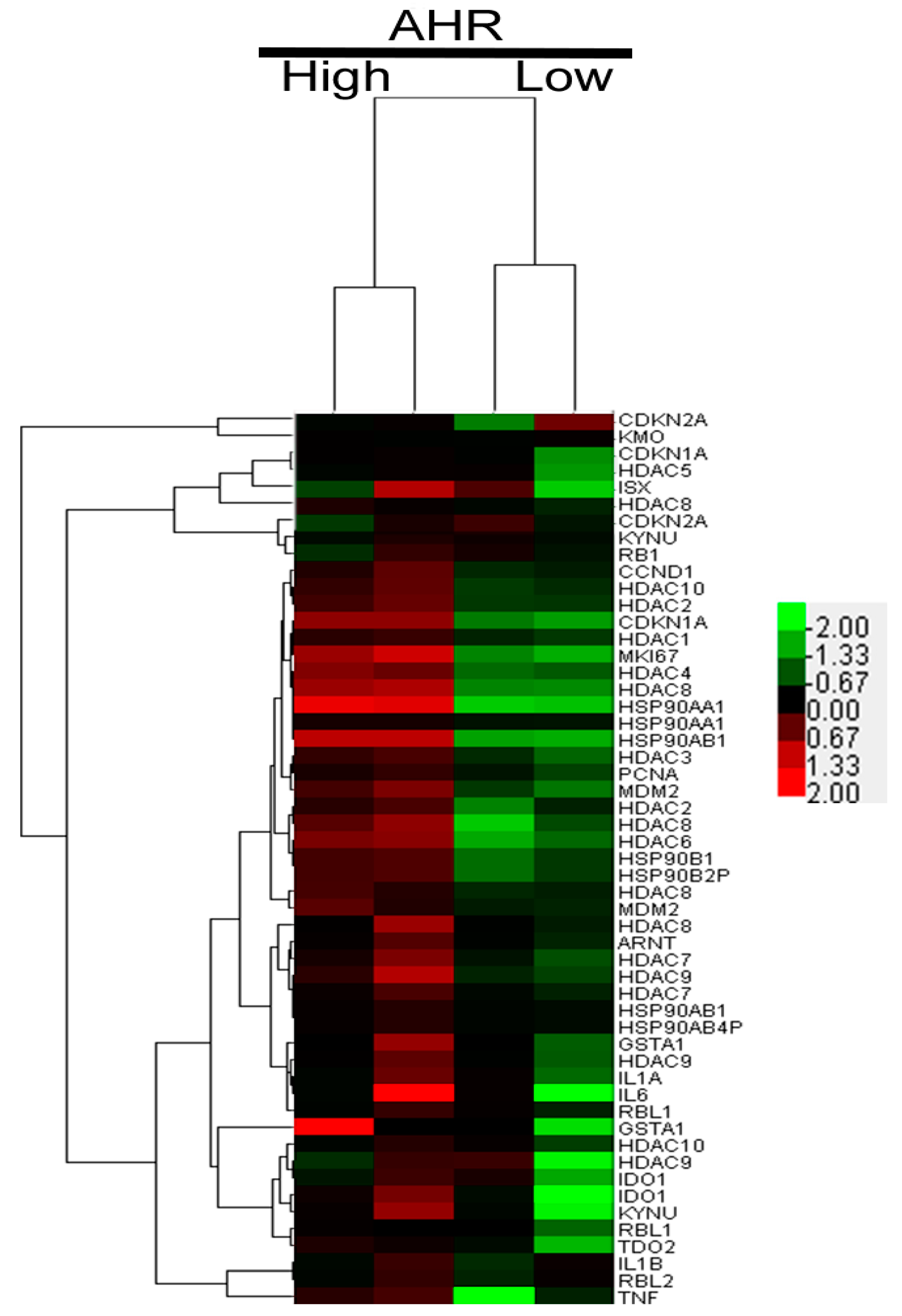
2.2. AHR Regulation in HCC
3. AHR Regulates Epigenetic Histone Acetylation in HCC
3.1. HDAC Classification
3.2. HDAC in HCC
4. HDAC Inhibitors as Clinical Trail of HCC
4.1. HDACi
4.2. Clinical Therapy of HDACi in HCC
4.3. Combination Therapy Involving HDACi
5. Discussion
Acknowledgments
Conflicts of Interest
References
- Dimitroulis, D.; Damaskos, C.; Valsami, S.; Davakis, S.; Garmpis, N.; Spartalis, E.; Athanasiou, A.; Moris, D.; Sakellariou, S.; Kykalos, S.; et al. From diagnosis to treatment of hepatocellular carcinoma: An epidemic problem for both developed and developing world. World J. Gastroenterol. 2017, 23, 5282–5294. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.Y.; Bass, N.M.; Nikolai, B.; Davern, T.J.; Kerlan, R.; Wu, V.; Ascher, N.L.; Roberts, J.P. Liver transplantation for hepatocellular carcinoma: Analysis of survival according to the intention-to-treat principle and dropout from the waiting list. Liver Transpl. 2002, 8, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Livraghi, T.; Meloni, F. Treatment of hepatocellular carcinoma by percutaneous interventional methods. Hepatogastroenterology 2002, 49, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Schwartz, L.; Ricci, S.; Amadori, D.; Santoro, A.; Figer, A.; De Greve, J.; Douillard, J.Y.; Lathia, C.; Schwartz, B.; et al. Phase ii study of sorafenib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2006, 24, 4293–4300. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.X.; Ribes, J.; Cleries, R.; Diaz, M. Epidemiology of hepatocellular carcinoma. Clin. Liver Dis. 2005, 9, 191–211. [Google Scholar] [CrossRef] [PubMed]
- Wogan, G.N. Impacts of chemicals on liver cancer risk. Semin. Cancer Biol. 2000, 10, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Balogh, J.; Victor, D., 3rd; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P., Jr. Hepatocellular carcinoma: A review. J. Hepatocell Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Li, W.W.; Yang, B.W.; Tao, Z.H.; Sun, H.C.; Wang, L.; Xia, J.L.; Qin, L.X.; Tang, Z.Y.; Fan, J.; et al. Aryl hydrocarbon receptor nuclear translocator is associated with tumor growth and progression of hepatocellular carcinoma. Int. J. Cancer 2012, 130, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Colonna, M. Aryl hydrocarbon receptor: Linking environment to immunity. Semin. Immunol. 2015, 27, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.; Chevallier, A.; Teixeira-Clerc, F.; Ambolet-Camoit, A.; Bui, L.C.; Bats, A.S.; Fournet, J.C.; Fernandez-Salguero, P.; Aggerbeck, M.; Lotersztajn, S.; et al. Aryl hydrocarbon receptor-dependent induction of liver fibrosis by dioxin. Toxicol. Sci. 2014, 137, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, G.D.; Nukaya, M.; Moran, S.M.; Glover, E.; Weinberg, S.; Balbo, S.; Hecht, S.S.; Pitot, H.C.; Drinkwater, N.R.; Bradfield, C.A. Liver tumor promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin is dependent on the aryl hydrocarbon receptor and TNF/IL-1 receptors. Toxicol. Sci. 2014, 140, 135–143. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Schroeder, J.C.; Francey, L.J.; Kusnadi, A.; Perdew, G.H. Mechanistic insights into the events that lead to synergistic induction of interleukin 6 transcription upon activation of the aryl hydrocarbon receptor and inflammatory signaling. J. Biol. Chem. 2010, 285, 24388–24397. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Dhar, D.; Nakagawa, H.; Font-Burgada, J.; Ogata, H.; Jiang, Y.; Shalapour, S.; Seki, E.; Yost, S.E.; Jepsen, K.; et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 2013, 155, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.H.; Wang, L.T.; Chai, C.Y.; Wu, C.C.; Hsi, E.; Chiou, S.S.; Wang, S.N. Aryl hydrocarbon receptor promotes hepatocellular carcinoma tumorigenesis by targeting intestine-specific homeobox expression. Mol. Carcinog. 2017, 56, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.T.; Chiou, S.S.; Chai, C.Y.; Hsi, E.; Yokoyama, K.K.; Wang, S.N.; Huang, S.K.; Hsu, S.H. Intestine-specific homeobox gene isx integrates IL6 signaling, tryptophan catabolism, and immune suppression. Cancer Res. 2017, 77, 4065–4077. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2016, 13, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Wick, W.; Van den Eynde, B.J. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res. 2012, 72, 5435–5440. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.T.; Chiou, S.S.; Chai, C.Y.; Hsi, E.; Wang, S.N.; Huang, S.K.; Hsu, S.H. Aryl hydrocarbon receptor regulates histone deacetylase 8 expression to repress tumor suppressive activity in hepatocellular carcinoma. Oncotarget 2017, 8, 7489–7501. [Google Scholar] [CrossRef] [PubMed]
- Fyodorov, D.V.; Zhou, B.R.; Skoultchi, A.I.; Bai, Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Seto, E. Acetylation/deacetylation modulates the stability of DNA replication licensing factor Cdt1. J. Biol. Chem. 2009, 284, 11446–11453. [Google Scholar] [CrossRef] [PubMed]
- Villagra, A.; Cheng, F.; Wang, H.W.; Suarez, I.; Glozak, M.; Maurin, M.; Nguyen, D.; Wright, K.L.; Atadja, P.W.; Bhalla, K.; et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jin, X.; Yan, Y.; Shao, Y.; Pan, Y.; Roberts, L.R.; Zhang, J.; Huang, H.; Jiang, J. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci. Rep. 2017, 7, 43864. [Google Scholar] [CrossRef] [PubMed]
- Quint, K.; Agaimy, A.; Di Fazio, P.; Montalbano, R.; Steindorf, C.; Jung, R.; Hellerbrand, C.; Hartmann, A.; Sitter, H.; Neureiter, D.; et al. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Arch. 2011, 459, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Ler, S.Y.; Leung, C.H.; Khin, L.W.; Lu, G.D.; Salto-Tellez, M.; Hartman, M.; Iau, P.T.; Yap, C.T.; Hooi, S.C. HDAC1 and HDAC2 independently predict mortality in hepatocellular carcinoma by a competing risk regression model in a southeast asian population. Oncol. Rep. 2015, 34, 2238–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Seo, D.; Choi, K.J.; Andersen, J.B.; Won, M.A.; Kitade, M.; Gomez-Quiroz, L.E.; Judge, A.D.; Marquardt, J.U.; Raggi, C.; et al. Antitumor effects in hepatocarcinoma of isoform-selective inhibition of HDAC2. Cancer Res. 2014, 74, 4752–4761. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, L.; Shan, J.; Shen, J.; Xu, Y.; Zhang, Q.; Yang, Z.; Wu, L.; Xia, F.; Bie, P.; et al. Histone deacetylase 3 participates in self-renewal of liver cancer stem cells through histone modification. Cancer Lett. 2013, 339, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wong, V.W.; Wong, G.L.; Yang, W.; Sun, H.; Shen, J.; Tong, J.H.; Go, M.Y.; Cheung, Y.S.; Lai, P.B.; et al. Histone deacetylase HDAC8 promotes insulin resistance and β-catenin activation in NAFLD-associated hepatocellular carcinoma. Cancer Res. 2015, 75, 4803–4816. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Lou, B.; Chen, W.; Zhang, J.; Lin, S.; Lv, F.F.; Chen, Y. Down-regulation of HDAC5 inhibits growth of human hepatocellular carcinoma by induction of apoptosis and cell cycle arrest. Tumour Biol. 2014, 35, 11523–11532. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, H.; Yin, M.; Ye, X.; Chen, G.; Zhou, X.; Yin, L.; Zhang, C.; Ding, B. MiR-376a and histone deacetylation 9 form a regulatory circuitry in hepatocellular carcinoma. Cell Physiol. Biochem. 2015, 35, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lawless, M.W. Resminostat: Opening the door to epigenetic treatments for liver cancer. Hepatology 2016, 63, 668–669. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, T.; Wang, Z.; Chen, X.; Liu, R. Histone deacetylase inhibitor quisinostat activates caspase signaling and upregulates p53 acetylation to inhibit the proliferation of HEPG2 cells. Mol. Med. Rep. 2017, 16, 6094–6101. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef] [PubMed]
- Gahr, S.; Peter, G.; Wissniowski, T.T.; Hahn, E.G.; Herold, C.; Ocker, M. The histone-deacetylase inhibitor MS-275 and the CDK-inhibitor CYC-202 promote anti-tumor effects in hepatoma cell lines. Oncol. Rep. 2008, 20, 1249–1256. [Google Scholar] [PubMed]
- Armeanu, S.; Pathil, A.; Venturelli, S.; Mascagni, P.; Weiss, T.S.; Gottlicher, M.; Gregor, M.; Lauer, U.M.; Bitzer, M. Apoptosis on hepatoma cells but not on primary hepatocytes by histone deacetylase inhibitors valproate and ITF2357. J. Hepatol. 2005, 42, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Pathil, A.; Armeanu, S.; Venturelli, S.; Mascagni, P.; Weiss, T.S.; Gregor, M.; Lauer, U.M.; Bitzer, M. HDAC inhibitor treatment of hepatoma cells induces both trail-independent apoptosis and restoration of sensitivity to trail. Hepatology 2006, 43, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Nam, J.Y.; Chang, Y.; Cho, H.; Kang, S.H.; Cho, Y.Y.; Cho, E.; Lee, J.H.; Yu, S.J.; Kim, Y.J.; et al. Synergistic effect of cytokine-induced killer cell with valproate inhibits growth of hepatocellular carcinoma cell in a mouse model. Cancer Biol. Ther. 2017, 18, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Pan, Y.; Dorfman, R.G.; Chen, Z.; Liu, F.; Zhou, Q.; Huang, S.; Zhang, J.; Yang, D.; Liu, J. AR-42 induces apoptosis in human hepatocellular carcinoma cells via HDAC5 inhibition. Oncotarget 2016, 7, 22285–22294. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.P.; Sandhu, D.S.; Moser, C.D.; Cazanave, S.C.; Oseini, A.M.; Shire, A.M.; Shridhar, V.; Sanderson, S.O.; Roberts, L.R. Additive effect of apicidin and doxorubicin in sulfatase 1 expressing hepatocellular carcinoma in vitro and in vivo. J. Hepatol. 2009, 50, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Sajadian, S.O.; Ehnert, S.; Vakilian, H.; Koutsouraki, E.; Damm, G.; Seehofer, D.; Thasler, W.; Dooley, S.; Baharvand, H.; Sipos, B.; et al. Induction of active demethylation and 5hmC formation by 5-azacytidine is TET2 dependent and suggests new treatment strategies against hepatocellular carcinoma. Clin. Epigenetics 2015, 7, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zopf, S.; Ocker, M.; Neureiter, D.; Alinger, B.; Gahr, S.; Neurath, M.F.; Di Fazio, P. Inhibition of DNA methyltransferase activity and expression by treatment with the pan-deacetylase inhibitor panobinostat in hepatocellular carcinoma cell lines. BMC Cancer 2012, 12, 386. [Google Scholar] [CrossRef] [PubMed]
- Cervoni, N.; Szyf, M. Demethylase activity is directed by histone acetylation. J. Biol. Chem. 2001, 276, 40778–40787. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.N.; Torrisani, J.; Unterberger, A.; Provencal, N.; Shikimi, K.; Karimi, M.; Ekstrom, T.J.; Szyf, M. Histone deacetylase inhibitor trichostatin a induces global and gene-specific DNA demethylation in human cancer cell lines. Biochem. Pharmacol. 2007, 73, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Detich, N.; Bovenzi, V.; Szyf, M. Valproate induces replication-independent active DNA demethylation. J. Biol. Chem. 2003, 278, 27586–27592. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. Fda approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Carlisi, D.; Lauricella, M.; D’Anneo, A.; Emanuele, S.; Angileri, L.; Di Fazio, P.; Santulli, A.; Vento, R.; Tesoriere, G. The histone deacetylase inhibitor suberoylanilide hydroxamic acid sensitises human hepatocellular carcinoma cells to TRAIL-induced apoptosis by TRAIL-DISC activation. Eur. J. Cancer 2009, 45, 2425–2438. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Annan, D.A.; Morita, T.; Li, W.; Muroyama, R.; Matsubara, Y.; Ito, S.; Nakagawa, R.; Tanoue, Y.; Jinushi, M.; et al. Novel chemoimmunotherapeutic strategy for hepatocellular carcinoma based on a genome-wide association study. Sci. Rep. 2016, 6, 38407. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.J.; Huang, H.; He, B.; Hu, D.H.; Li, P.H.; Yu, Y.J.; Zhou, X.H.; Lv, Z.; Zhou, L.; Hu, T.Y.; et al. Romidepsin induces G2/M phase arrest via Erk/cdc25C/cdc2/cyclinB pathway and apoptosis induction through JNK/c-Jun/caspase3 pathway in hepatocellular carcinoma cells. Biochem. Pharmacol. 2017, 127, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar] [PubMed]
- Frye, R.; Myers, M.; Axelrod, K.C.; Ness, E.A.; Piekarz, R.L.; Bates, S.E.; Booher, S. Romidepsin: A new drug for the treatment of cutaneous T-cell lymphoma. Clin. J. Oncol. Nurs. 2012, 16, 195–204. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.; Jimeno, A. Belinostat for the treatment of peripheral T-cell lymphomas. Drugs Today (Barc. 1998) 2014, 50, 337–345. [Google Scholar]
- Ma, B.B.; Sung, F.; Tao, Q.; Poon, F.F.; Lui, V.W.; Yeo, W.; Chan, S.L.; Chan, A.T. The preclinical activity of the histone deacetylase inhibitor PXD101 (belinostat) in hepatocellular carcinoma cell lines. Invest. New Drugs 2010, 28, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Yeo, W.; Chung, H.C.; Chan, S.L.; Wang, L.Z.; Lim, R.; Picus, J.; Boyer, M.; Mo, F.K.; Koh, J.; Rha, S.Y.; et al. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: A multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the mayo phase II consortium and the cancer therapeutics research group. J. Clin. Oncol. 2012, 30, 3361–3367. [Google Scholar] [PubMed]
- Richardson, P.G.; Laubach, J.P.; Lonial, S.; Moreau, P.; Yoon, S.S.; Hungria, V.T.; Dimopoulos, M.A.; Beksac, M.; Alsina, M.; San-Miguel, J.F. Panobinostat: A novel pan-deacetylase inhibitor for the treatment of relapsed or relapsed and refractory multiple myeloma. Expert. Rev. Anticancer Ther. 2015, 15, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Di Fazio, P.; Schneider-Stock, R.; Neureiter, D.; Okamoto, K.; Wissniowski, T.; Gahr, S.; Quint, K.; Meissnitzer, M.; Alinger, B.; Montalbano, R.; et al. The pan-deacetylase inhibitor panobinostat inhibits growth of hepatocellular carcinoma models by alternative pathways of apoptosis. Cell Oncol. 2010, 32, 285–300. [Google Scholar] [PubMed]
- Montalbano, R.; Waldegger, P.; Quint, K.; Jabari, S.; Neureiter, D.; Illig, R.; Ocker, M.; Di Fazio, P. Endoplasmic reticulum stress plays a pivotal role in cell death mediated by the pan-deacetylase inhibitor panobinostat in human hepatocellular cancer cells. Transl. Oncol. 2013, 6, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Di Fazio, P.; Waldegger, P.; Jabari, S.; Lingelbach, S.; Montalbano, R.; Ocker, M.; Slater, E.P.; Bartsch, D.K.; Illig, R.; Neureiter, D.; et al. Autophagy-related cell death by pan-histone deacetylase inhibition in liver cancer. Oncotarget 2016, 7, 28998–29010. [Google Scholar] [CrossRef] [PubMed]
- Di Fazio, P.; Montalbano, R.; Quint, K.; Alinger, B.; Kemmerling, R.; Kiesslich, T.; Ocker, M.; Neureiter, D. The pan-deacetylase inhibitor panobinostat modulates the expression of epithelial-mesenchymal transition markers in hepatocellular carcinoma models. Oncol. Lett. 2013, 5, 127–134. [Google Scholar]
- Henrici, A.; Montalbano, R.; Neureiter, D.; Krause, M.; Stiewe, T.; Slater, E.P.; Quint, K.; Ocker, M.; Di Fazio, P. The pan-deacetylase inhibitor panobinostat suppresses the expression of oncogenic miRNAs in hepatocellular carcinoma cell lines. Mol. Carcinog. 2015, 54, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Di Fazio, P.; Montalbano, R.; Neureiter, D.; Alinger, B.; Schmidt, A.; Merkel, A.L.; Quint, K.; Ocker, M. Downregulation of HMGA2 by the pan-deacetylase inhibitor panobinostat is dependent on hsa-let-7b expression in liver cancer cell lines. Exp. Cell Res. 2012, 318, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, J.; Zheng, T.; Song, R.; Liang, Y.; Bhatta, N.; Yin, D.; Pan, S.; Liu, J.; Jiang, H.; et al. LBH589 inhibits proliferation and metastasis of hepatocellular carcinoma via inhibition of gankyrin/STAT3/Akt pathway. Mol. Cancer 2013, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Gahr, S.; Mayr, C.; Kiesslich, T.; Illig, R.; Neureiter, D.; Alinger, B.; Ganslmayer, M.; Wissniowski, T.; Fazio, P.D.; Montalbano, R.; et al. The pan-deacetylase inhibitor panobinostat affects angiogenesis in hepatocellular carcinoma models via modulation of CTGF expression. Int. J. Oncol. 2015, 47, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Lachenmayer, A.; Toffanin, S.; Cabellos, L.; Alsinet, C.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Tsai, H.W.; Ward, S.C.; Thung, S.; et al. Combination therapy for hepatocellular carcinoma: Additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J. Hepatol. 2012, 56, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, S.; Armeanu, S.; Pathil, A.; Hsieh, C.J.; Weiss, T.S.; Vonthein, R.; Wehrmann, M.; Gregor, M.; Lauer, U.M.; Bitzer, M. Epigenetic combination therapy as a tumor-selective treatment approach for hepatocellular carcinoma. Cancer 2007, 109, 2132–2141. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, H.; Shimizu, M.; Shirakami, Y.; Sakai, H.; Yasuda, Y.; Tsurumi, H.; Moriwaki, H. Acyclic retinoid synergises with valproic acid to inhibit growth in human hepatocellular carcinoma cells. Cancer Lett. 2009, 285, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Li, D.Q.; He, A.R.; Motwani, M.; Vasiliou, V.; Eswaran, J.; Mishra, L.; Kumar, R. Synergistic inhibition of hepatocellular carcinoma growth by cotargeting chromatin modifying enzymes and poly (ADP-ribose) polymerases. Hepatology 2012, 55, 1840–1851. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.Y.; Xiong, M.; Ji, G.B.; Zhang, E.L.; Zhang, Z.Y.; Dong, K.S.; Chen, X.P.; Huang, Z.Y. Synergistic suppressive effect of PARP-1 inhibitor PJ34 and HDAC inhibitor SAHA on proliferation of liver cancer cells. Med. Sci. 2015, 35, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhan, Q.; Wan, Y.J. Enrichment of Nur77 mediated by retinoic acid receptor β leads to apoptosis of human hepatocellular carcinoma cells induced by fenretinide and histone deacetylase inhibitors. Hepatology 2011, 53, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.P.; Hong, Y.H.; Yang, P.M. In silico and in vitro identification of inhibitory activities of sorafenib on histone deacetylases in hepatocellular carcinoma cells. Oncotarget 2017, 8, 86168–86180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Park, M.A.; Mitchell, C.; Hamed, H.; Rahmani, M.; Martin, A.P.; Curiel, D.T.; Yacoub, A.; Graf, M.; Lee, R.; et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin. Cancer Res. 2008, 14, 5385–5399. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.T.; Liu, Y.C.; Chiang, I.T.; Liu, R.S.; Wang, H.E.; Lin, W.J.; Hwang, J.J. Sorafenib increases efficacy of vorinostat against human hepatocellular carcinoma through transduction inhibition of vorinostat-induced ERK/NF-κB signaling. Int. J. Oncol. 2014, 45, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Knieling, F.; Waldner, M.J.; Goertz, R.S.; Strobel, D. Quantification of dynamic contrast-enhanced ultrasound in HCC: Prediction of response to a new combination therapy of sorafenib and panobinostat in advanced hepatocellular carcinoma. BMJ Case Rep. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Gahr, S.; Wissniowski, T.; Zopf, S.; Strobel, D.; Pustowka, A.; Ocker, M. Combination of the deacetylase inhibitor panobinostat and the multi-kinase inhibitor sorafenib for the treatment of metastatic hepatocellular carcinoma—Review of the underlying molecular mechanisms and first case report. J. Cancer 2012, 3, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, S.; Lauricella, M.; Carlisi, D.; Vassallo, B.; D’Anneo, A.; Di Fazio, P.; Vento, R.; Tesoriere, G. Saha induces apoptosis in hepatoma cells and synergistically interacts with the proteasome inhibitor bortezomib. Apoptosis 2007, 12, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Spratlin, J.L.; Pitts, T.M.; Kulikowski, G.N.; Morelli, M.P.; Tentler, J.J.; Serkova, N.J.; Eckhardt, S.G. Synergistic activity of histone deacetylase and proteasome inhibition against pancreatic and hepatocellular cancer cell lines. Anticancer Res. 2011, 31, 1093–1103. [Google Scholar] [PubMed]
- Lu, Y.S.; Chou, C.H.; Tzen, K.Y.; Gao, M.; Cheng, A.L.; Kulp, S.K.; Cheng, J.C. Radiosensitizing effect of a phenylbutyrate-derived histone deacetylase inhibitor in hepatocellular carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, e181–e189. [Google Scholar] [CrossRef] [PubMed]
- Il Yu, J.; Choi, C.; Shin, S.W.; Son, A.; Lee, G.H.; Kim, S.Y.; Park, H.C. Valproic acid sensitizes hepatocellular carcinoma cells to proton therapy by suppressing NRF2 activation. Sci. Rep. 2017, 7, 14986. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| HDACis | Specificity | Experimental Design | Clinical Trial of HCC | References |
|---|---|---|---|---|
| Resminostat (4SC-201) | Classes I and II | Patient with Hepatocelluler carcinoma | Phase II trial | [34] |
| Quisinostat (JNJ-26481585) | Class I and II HDACs | HCC cell lines | Preclinical | [35] |
| MPT0E028 | HDAC1, 2, 6 | Patient with Hepatocelluler carcinoma | Under Phase I trial | clinicaltrials.gov |
| CUDC-101 | Classes I and II HDAC, EGFR, HER2 | HCC cell lines | Preclinical | [36] |
| Entinostat (MS-275) | HDAC1, 2, 3 | HCC cell lines | Preclinical | [37] |
| Valproic acid (VPA) | Class I and II | HCC cell lines Mouse model | Preclinical | [38,39,40] |
| AR-42 | Class I and IIb | HCC cell lines | Preclinical | [41] |
| Apicidin | HDAC1, 2, 3 | HCC cell lines Mouse model | Preclinical | [42] |
| PCI-34051 | HDAC8 | HCC cell lines Mouse model | Preclinical | [19] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, K.-Y.; Wang, L.-T.; Hsu, S.-H. Modification of Epigenetic Histone Acetylation in Hepatocellular Carcinoma. Cancers 2018, 10, 8. https://doi.org/10.3390/cancers10010008
Liu K-Y, Wang L-T, Hsu S-H. Modification of Epigenetic Histone Acetylation in Hepatocellular Carcinoma. Cancers. 2018; 10(1):8. https://doi.org/10.3390/cancers10010008
Chicago/Turabian StyleLiu, Kwei-Yan, Li-Ting Wang, and Shih-Hsien Hsu. 2018. "Modification of Epigenetic Histone Acetylation in Hepatocellular Carcinoma" Cancers 10, no. 1: 8. https://doi.org/10.3390/cancers10010008
APA StyleLiu, K. -Y., Wang, L. -T., & Hsu, S. -H. (2018). Modification of Epigenetic Histone Acetylation in Hepatocellular Carcinoma. Cancers, 10(1), 8. https://doi.org/10.3390/cancers10010008