The Emerging Role of the Microenvironment in Endometrial Cancer




Abstract
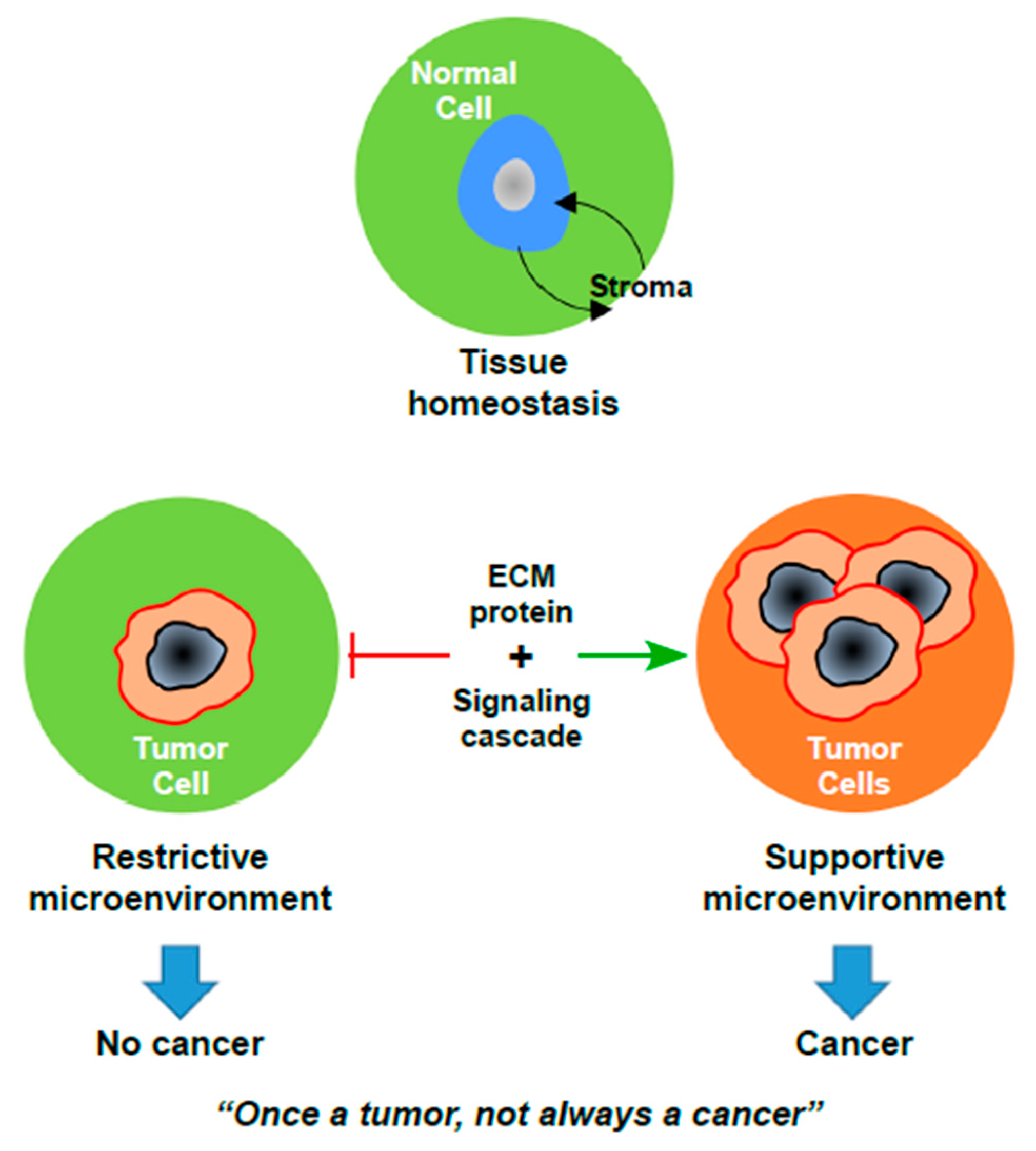
:1. Introduction
2. Endometrial Cancer Microenvironment
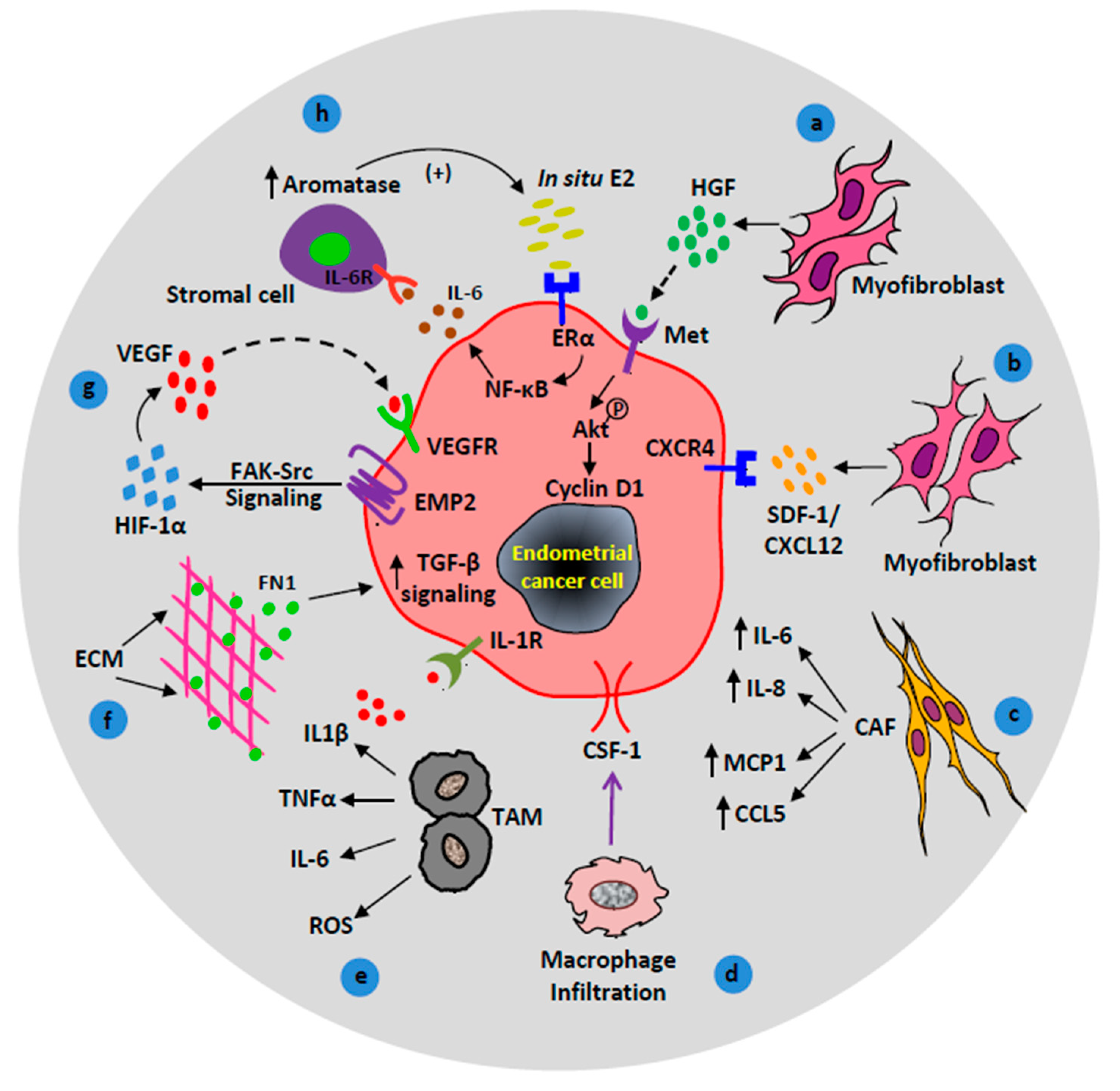
2.1. The Role of Stromal Myofibroblasts in EC Microenvironment
2.2. Macrophages in EC Microenvironment
2.3. Stromal Signaling in EC Microenvironment
2.3.1. ECM-Derived TGF-β Signaling
2.3.2. Stromal APC Signaling
2.3.3. Stromal LKB1 Signaling
2.3.4. Stromal HAND2 Signaling
2.3.5. Stromal VEGF Signaling
2.3.6. Stromal Estrogen Signaling
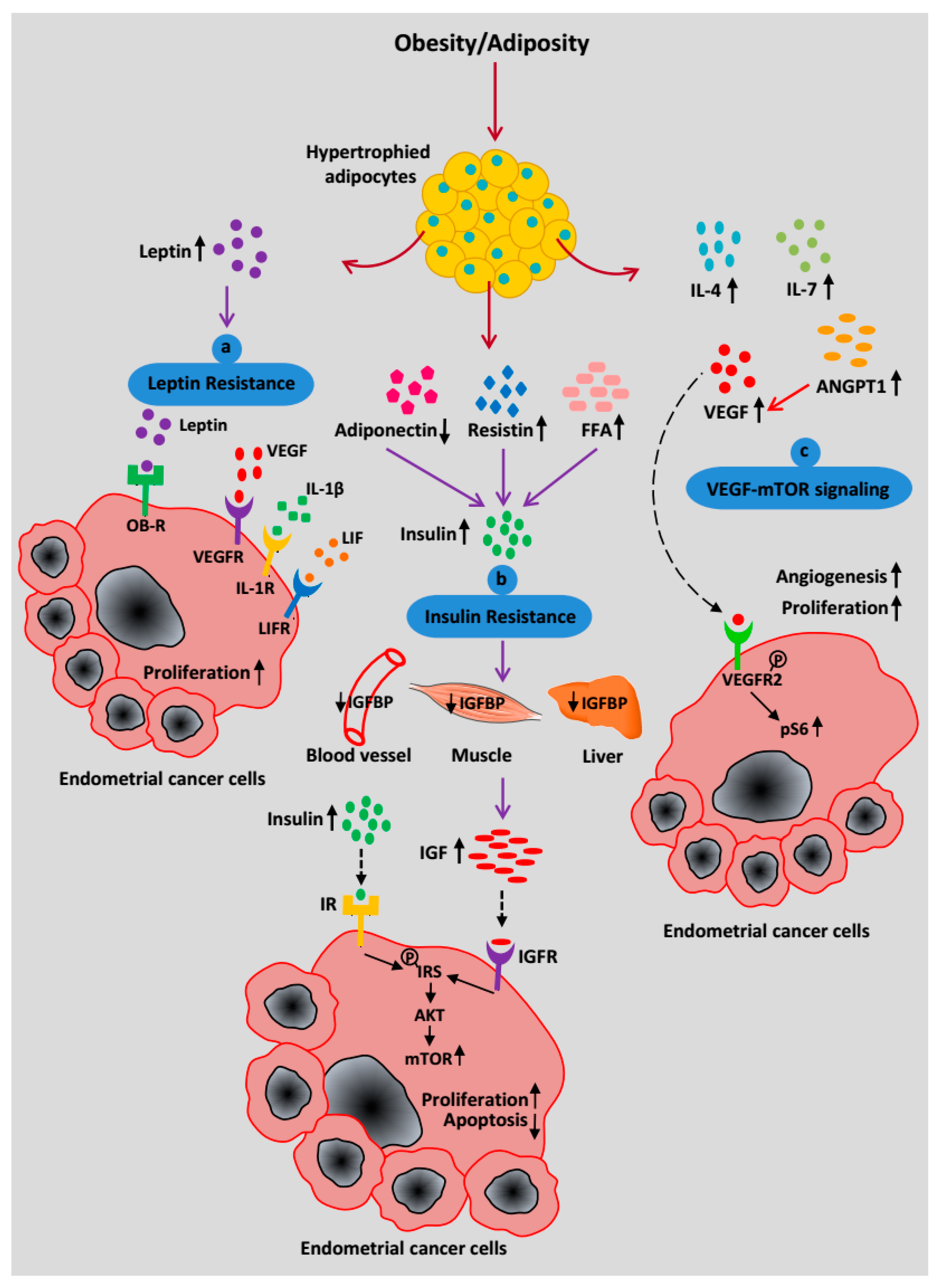
2.4. Paracrine Effects of Adipocytes in the EC Microenvironment
2.5. Mechanism Relating Obesity or Adiposity to EC Risk
2.5.1. Leptin Resistance
2.5.2. Insulin Resistance
2.5.3. EC Cell-Adipocyte Interactions
2.5.4. Adipose-Derived VEGF-mTOR Signaling
2.5.5. Adipocyte-Derived Estrogen Signaling
3. Targeting the EC Microenvironment for Chemoprevention
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metast. Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef]
- Ungefroren, H.; Sebens, S.; Seidl, D.; Lehnert, H.; Hass, R. Interaction of tumor cells with the microenvironment. Cell Commun. Signal. 2011, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmick, N.A.; Moses, H.L. Tumor-stroma interactions. Curr. Opin. Genet. Dev. 2005, 15, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, F.L.; Sikes, R.A. Insidious changes in stromal matrix fuel cancer progression. Mol. Cancer Res. 2014, 12, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Declerck, Y.A. Targeting the tumor microenvironment: From understanding pathways to effective clinical trials. Cancer Res. 2013, 73, 4965–4977. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Felix, A.S.; Weissfeld, J.; Edwards, R.; Linkov, F. Future directions in the field of endometrial cancer research: The need to investigate the tumor microenvironment. Eur. J. Gynaecol. Oncol. 2010, 31, 139–144. [Google Scholar] [PubMed]
- Lax, S.F.; Kendall, B.; Tashiro, H.; Slebos, R.J.; Hedrick, L. The frequency of p53, K-ras mutations, and microsatellite instability differs in uterine endometrioid and serous carcinoma: Evidence of distinct molecular genetic pathways. Cancer 2000, 88, 814–824. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.S.; Kim, H.J.; Yoon, J.H.; Yoo, S.C.; Jo, H.; Lee, S.Y.; Min, C.K.; Ryu, H.S. Endometrial cancer invasion depends on cancer-derived tumor necrosis factor-alpha and stromal derived hepatocyte growth factor. Int. J. Cancer 2009, 124, 2528–2538. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.J.; Coleman, D.T.; Cardelli, J.A. The HGF-met signaling axis: Emerging themes and targets of inhibition. Curr. Protein Pept. Sci. 2011, 12, 12–22. [Google Scholar] [PubMed]
- Li, M.; Xin, X.; Wu, T.; Hua, T.; Wang, H. HGF and c-Met in pathogenesis of endometrial carcinoma. Front. Biosci. 2015, 20, 635–643. [Google Scholar]
- Li, M.; Xin, X.; Wu, T.; Hua, T.; Wang, H.; Wang, H. Stromal cells of endometrial carcinoma promotes proliferation of epithelial cells through the HGF/c-Met/Akt signaling pathway. Tumour Biol. 2015, 36, 6239–6248. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metast. Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Gelmini, S.; Mangoni, M.; Castiglione, F.; Beltrami, C.; Pieralli, A.; Andersson, K.L.; Fambrini, M.; Taddei, G.L.; Serio, M.; Orlando, C. The CXCR4/CXCL12 axis in endometrial cancer. Clin. Exp. Metast. 2009, 26, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Kodama, J.; Hasengaowa; Seki, N.; Kusumoto, T.; Hiramatsu, Y. Expression of the CXCR4 and CCR7 chemokine receptors in human endometrial cancer. Eur. J. Gynaecol. Oncol. 2007, 28, 370–375. [Google Scholar] [PubMed]
- Paunescu, V.; Bojin, F.M.; Tatu, C.A.; Gavriliuc, O.I.; Rosca, A.; Gruia, A.T.; Tanasie, G.; Bunu, C.; Crisnic, D.; Gherghiceanu, M.; et al. Tumour-associated fibroblasts and mesenchymal stem cells: More similarities than differences. J. Cell. Mol. Med. 2011, 15, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, K.S.; Tham, S.T.; Mohamed, Z.; Woo, Y.L.; Mat Adenan, N.A.; Chung, I. Cancer-associated fibroblasts promote proliferation of endometrial cancer cells. PLoS ONE 2013, 8, e68923. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, K.S.; Omar, I.S.; Kwong, S.C.; Mohamed, Z.; Woo, Y.L.; Mat Adenan, N.A.; Chung, I. Cancer-associated fibroblasts promote endometrial cancer growth via activation of interleukin-6/STAT-3/c-Myc pathway. Am. J. Cancer Res. 2016, 6, 200–213. [Google Scholar] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Dun, E.C.; Hanley, K.; Wieser, F.; Bohman, S.; Yu, J.; Taylor, R.N. Infiltration of tumor-associated macrophages is increased in the epithelial and stromal compartments of endometrial carcinomas. Int. J. Gynecol. Pathol. 2013, 32, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, I.; Jose Carnicer, M.; Catasus, L.; Canet, B.; D’Angelo, E.; Zannoni, G.F.; Prat, J. Myometrial invasion and lymph node metastasis in endometrioid carcinomas: Tumor-associated macrophages, microvessel density, and HIF1A have a crucial role. Am. J. Surg. Pathol. 2010, 34, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.G.; Francisco, A.M.; Cimic, A.; Wofford, A.; Fitzgerald, N.C.; Yu, J.; Taylor, R.N. Type 2 Endometrial Cancer is Associated With a High Density of Tumor-Associated Macrophages in the Stromal Compartment. Reprod. Sci. 2015, 22, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Kubler, K.; Ayub, T.H.; Weber, S.K.; Zivanovic, O.; Abramian, A.; Keyver-Paik, M.D.; Mallmann, M.R.; Kaiser, C.; Serce, N.B.; Kuhn, W.; et al. Prognostic significance of tumor-associated macrophages in endometrial adenocarcinoma. Gynecol. Oncol. 2014, 135, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.; Gao, F.; Jegga, A.G.; Das, S.K. Estrogen mediated epithelial proliferation in the uterus is directed by stromal Fgf10 and Bmp8a. Mol. Cell. Endocrinol. 2015, 400, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Shiga, N.; Utsunomiya, H.; Miki, Y.; Nagase, S.; Kobayashi, R.; Matsumoto, M.; Niikura, H.; Ito, K.; Yaegashi, N. Local biosynthesis of estrogen in human endometrial carcinoma through tumor-stromal cell interactions. Clin. Cancer Res. 2009, 15, 6028–6034. [Google Scholar] [CrossRef] [PubMed]
- Che, Q.; Liu, B.Y.; Liao, Y.; Zhang, H.J.; Yang, T.T.; He, Y.Y.; Xia, Y.H.; Lu, W.; He, X.Y.; Chen, Z.; et al. Activation of a positive feedback loop involving IL-6 and aromatase promotes intratumoral 17beta-estradiol biosynthesis in endometrial carcinoma microenvironment. Int. J. Cancer 2014, 135, 282–294. [Google Scholar] [CrossRef] [PubMed]
- He, Y.Y.; Cai, B.; Yang, Y.X.; Liu, X.L.; Wan, X.P. Estrogenic G protein-coupled receptor 30 signaling is involved in regulation of endometrial carcinoma by promoting proliferation, invasion potential, and interleukin-6 secretion via the MEK/ERK mitogen-activated protein kinase pathway. Cancer Sci. 2009, 100, 1051–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristancho, A.G.; Lazar, M.A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Hlavna, M.; Kohut, L.; Lipkova, J.; Bienertova-Vasku, J.; Dostalova, Z.; Chovanec, J.; Vasku, A. Relationship of resistin levels with endometrial cancer risk. Neoplasma 2011, 58, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carino, C.; Olawaiye, A.B.; Cherfils, S.; Serikawa, T.; Lynch, M.P.; Rueda, B.R.; Gonzalez, R.R. Leptin regulation of proangiogenic molecules in benign and cancerous endometrial cells. Int. J. Cancer 2008, 123, 2782–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wincewicz, A.; Koda, M.; Sulkowska, M.; Kanczuga-Koda, L.; Sulkowski, S. Comparison of STAT3 with HIF-1alpha, Ob and ObR expressions in human endometrioid adenocarcinomas. Tissue Cell 2008, 40, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.C. IGF binding proteins in cancer: Mechanistic and clinical insights. Nat. Rev. Cancer 2014, 14, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhou, L.; Shangguan, A.J.; Bulun, S.E. Aromatase expression and regulation in breast and endometrial cancer. J. Mol. Endocrinol. 2016, 57, R19–R33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa, I.; Catasus, L.; Emanuela, D.; Mozos, A.; Pedrola, N.; Bertolo, C.; Ferrer, I.; Zannoni, G.F.; West, R.B.; van de Rijn, M.; et al. Stromal signatures in endometrioid endometrial carcinomas. Mod. Pathol. 2014, 27, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Friedenreich, C.M.; Langley, A.R.; Speidel, T.P.; Lau, D.C.; Courneya, K.S.; Csizmadi, I.; Magliocco, A.M.; Yasui, Y.; Cook, L.S. Case-control study of inflammatory markers and the risk of endometrial cancer. Eur. J. Cancer Prev. 2013, 22, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.S.; Quah, M.Y.; Nielsen, S.; Atkins, J.; Au, G.G.; Cairns, M.J.; Nahar, P.; Lombard, J.M.; Tanwar, P.S. Inhibition of extracellular matrix mediated TGF-beta signalling suppresses endometrial cancer metastasis. Oncotarget 2017, 8, 71400–71417. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, P.S.; Zhang, L.; Roberts, D.J.; Teixeira, J.M. Stromal deletion of the APC tumor suppressor in mice triggers development of endometrial cancer. Cancer Res. 2011, 71, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, P.S.; Kaneko-Tarui, T.; Zhang, L.; Tanaka, Y.; Crum, C.P.; Teixeira, J.M. Stromal liver kinase B1 [STK11] signaling loss induces oviductal adenomas and endometrial cancer by activating mammalian Target of Rapamycin Complex 1. PLoS Genet. 2012, 8, e1002906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena, C.G.; Nakada, Y.; Saatcioglu, H.D.; Aloisio, G.M.; Cuevas, I.; Zhang, S.; Miller, D.S.; Lea, J.S.; Wong, K.K.; DeBerardinis, R.J.; et al. LKB1 loss promotes endometrial cancer progression via CCL2-dependent macrophage recruitment. J. Clin. Investig. 2015, 125, 4063–4076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, A.; Teschendorff, A.E.; Li, Q.; Hayward, J.D.; Kannan, A.; Mould, T.; West, J.; Zikan, M.; Cibula, D.; Fiegl, H.; et al. Role of DNA methylation and epigenetic silencing of HAND2 in endometrial cancer development. PLoS Med. 2013, 10, e1001551. [Google Scholar] [CrossRef] [PubMed]
- Gordon, L.K.; Kiyohara, M.; Fu, M.; Braun, J.; Dhawan, P.; Chan, A.; Goodglick, L.; Wadehra, M. EMP2 regulates angiogenesis in endometrial cancer cells through induction of VEGF. Oncogene 2013, 32, 5369–5376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamat, A.A.; Merritt, W.M.; Coffey, D.; Lin, Y.G.; Patel, P.R.; Broaddus, R.; Nugent, E.; Han, L.Y.; Landen, C.N., Jr.; Spannuth, W.A.; et al. Clinical and biological significance of vascular endothelial growth factor in endometrial cancer. Clin. Cancer Res. 2007, 13, 7487–7495. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.L.; Mutter, G.L. Molecular and pathologic aspects of endometrial carcinogenesis. J. Clin. Oncol. 2006, 24, 4783–4791. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, G.K.; Pirie, K.; Beral, V.; Green, J.; Spencer, E.; Bull, D.; Million Women Study, C. Cancer incidence and mortality in relation to body mass index in the Million Women Study: Cohort study. BMJ 2007, 335, 1134. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Onstad, M.A.; Schmandt, R.E.; Lu, K.H. Addressing the Role of Obesity in Endometrial Cancer Risk, Prevention, and Treatment. J. Clin. Oncol. 2016, 34, 4225–4230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Fader, A.N.; Arriba, L.N.; Frasure, H.E.; von Gruenigen, V.E. Endometrial cancer and obesity: Epidemiology, biomarkers, prevention and survivorship. Gynecol. Oncol. 2009, 114, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, S.S.; Lombard, J.M.; Ius, Y.; O’Sullivan, R.; Wood, L.G.; Nahar, P.; Jaaback, K.; Tanwar, P.S. Adipose-Derived VEGF-mTOR Signaling Promotes Endometrial Hyperplasia and Cancer: Implications for Obese Women. Mol. Cancer Res. 2018, 16, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Liu, M.; Wang, G.; Torroella-Kouri, M.; Gonzalez-Perez, R.R. Oncogenic role and therapeutic target of leptin signaling in breast cancer and cancer stem cells. Biochim. Biophys. Acta 2012, 1825, 207–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley-Brown, D.; Oprea-Ilies, G.M.; Lee, R.; Pattillo, R.; Gonzalez-Perez, R.R. Molecular cues on obesity signals, tumor markers and endometrial cancer. Horm. Mol. Biol. Clin. Investig. 2015, 21, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Mu, N.; Zhu, Y.; Wang, Y.; Zhang, H.; Xue, F. Insulin resistance: A significant risk factor of endometrial cancer. Gynecol. Oncol. 2012, 125, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Schmandt, R.E.; Iglesias, D.A.; Co, N.N.; Lu, K.H. Understanding obesity and endometrial cancer risk: Opportunities for prevention. Am. J. Obstet. Gynecol. 2011, 205, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirat, B.; Bochet, L.; Escourrou, G.; Valet, P.; Muller, C. Unraveling the obesity and breast cancer links: A role for cancer-associated adipocytes? Endocr. Dev. 2010, 19, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Simo, R.; Saez-Lopez, C.; Lecube, A.; Hernandez, C.; Fort, J.M.; Selva, D.M. Adiponectin upregulates SHBG production: Molecular mechanisms and potential implications. Endocrinology 2014, 155, 2820–2830. [Google Scholar] [CrossRef] [PubMed]
- Saloniemi, T.; Jarvensivu, P.; Koskimies, P.; Jokela, H.; Lamminen, T.; Ghaem-Maghami, S.; Dina, R.; Damdimopoulou, P.; Makela, S.; Perheentupa, A.; et al. Novel hydroxysteroid (17beta) dehydrogenase 1 inhibitors reverse estrogen-induced endometrial hyperplasia in transgenic mice. Am. J. Pathol. 2010, 176, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, P.; Nielsen, S.; Lombard, J.M.; Rassam, L.; Nahar, P.; Rueda, B.R.; Wilkinson, J.E.; Miller, R.A.; Tanwar, P.S. Overactive mTOR signaling leads to endometrial hyperplasia in aged women and mice. Oncotarget 2017, 8, 7265–7275. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.L.; Price, J.C.; Fogoros, S.; Godwin, A.K.; Sgroi, D.C.; Merino, M.J.; Bell, D.W. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin. Cancer Res. 2011, 17, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N.; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Ramos, P.; Bentires-Alj, M. Mechanism-based cancer therapy: Resistance to therapy, therapy for resistance. Oncogene 2015, 34, 3617–3626. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.J.; Thiel, K.W.; Yang, S.; Leslie, K.K. Catch it before it kills: Progesterone, obesity, and the prevention of endometrial cancer. Discov. Med. 2012, 14, 215–222. [Google Scholar] [PubMed]
- Decruze, S.B.; Green, J.A. Hormone therapy in advanced and recurrent endometrial cancer: A systematic review. Int. J. Gynecol. Cancer 2007, 17, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Janzen, D.M.; Rosales, M.A.; Paik, D.Y.; Lee, D.S.; Smith, D.A.; Witte, O.N.; Iruela-Arispe, M.L.; Memarzadeh, S. Progesterone receptor signaling in the microenvironment of endometrial cancer influences its response to hormonal therapy. Cancer Res. 2013, 73, 4697–4710. [Google Scholar] [CrossRef] [PubMed]
- Kokka, F.; Brockbank, E.; Oram, D.; Gallagher, C.; Bryant, A. Hormonal therapy in advanced or recurrent endometrial cancer. Cochrane Database Syst. Rev. 2010. [Google Scholar] [CrossRef] [PubMed]
- Longoria, T.C.; Eskander, R.N. Erratum to: Immunotherapy in endometrial cancer—An evolving therapeutic paradigm. Gynecol. Oncol. Res. Pract. 2016, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Vanderstraeten, A.; Tuyaerts, S.; Amant, F. The immune system in the normal endometrium and implications for endometrial cancer development. J. Reprod. Immunol. 2015, 109, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Vanderstraeten, A.; Luyten, C.; Verbist, G.; Tuyaerts, S.; Amant, F. Mapping the immunosuppressive environment in uterine tumors: Implications for immunotherapy. Cancer Immunol. Immunother. 2014, 63, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Boilly, B.; Faulkner, S.; Jobling, P.; Hondermarck, H. Nerve Dependence: From Regeneration to Cancer. Cancer Cell 2017, 31, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Jobling, P.; Pundavela, J.; Oliveira, S.M.; Roselli, S.; Walker, M.M.; Hondermarck, H. Nerve-Cancer Cell Cross-talk: A Novel Promoter of Tumor Progression. Cancer Res. 2015, 75, 1777–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Cell Type | Roles in Endometrial Cancer | References | |
|---|---|---|---|
| Anti-Tumorigenic | Pro-Tumorigenic | ||
| Fibroblasts | Release growth factors and maintain tissue integrity | Limited | [17,18] |
| Myofibroblasts | Facilitate deposition of collagen fibers in ECM and involve in wound healing | Chronic secretion of HGF and CXCL12 promote EC cell proliferation and angiogenesis | [19,20,21,22,23,24,25] |
| Cancer-associated fibroblasts (CAF) | Limited | ECM remodeling Provide oncogenic signals and secrete cytokines for infiltration of tumor cells and macrophages | [26,27,28] |
| Macrophages (M1) | Provide pro-inflammatory response and secrete TH1 cytokines | Limited | [29] |
| Tumor-associated macrophages (M2) | Limited | Provide anti-inflammatory response and secrete TH2 cytokines Support angiogenesis and invasion | [30,31,32,33] |
| Uterine stroma | Provides structural support to endometrium | Expression of aromatase synthesizes in situ E2 to induce endometrial hyperplasia | [34,35,36,37] |
| Adipocytes | Function as an endocrine organ, accumulate lipids and store as energy | Limited | [38] |
| Cancer-associated adipocytes (CAA) | Limited | Chronic adipokine and cytokine secretion leads to leptin and insulin resistance Aromatase synthesis results in excess estrogen production | [39,40,41,42,43] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahoo, S.S.; Zhang, X.D.; Hondermarck, H.; Tanwar, P.S. The Emerging Role of the Microenvironment in Endometrial Cancer. Cancers 2018, 10, 408. https://doi.org/10.3390/cancers10110408
Sahoo SS, Zhang XD, Hondermarck H, Tanwar PS. The Emerging Role of the Microenvironment in Endometrial Cancer. Cancers. 2018; 10(11):408. https://doi.org/10.3390/cancers10110408
Chicago/Turabian StyleSahoo, Subhransu S., Xu Dong Zhang, Hubert Hondermarck, and Pradeep S. Tanwar. 2018. "The Emerging Role of the Microenvironment in Endometrial Cancer" Cancers 10, no. 11: 408. https://doi.org/10.3390/cancers10110408
APA StyleSahoo, S. S., Zhang, X. D., Hondermarck, H., & Tanwar, P. S. (2018). The Emerging Role of the Microenvironment in Endometrial Cancer. Cancers, 10(11), 408. https://doi.org/10.3390/cancers10110408