Targeting the MYC Oncogene in Burkitt Lymphoma through HSP90 Inhibition

 ,
,
Abstract
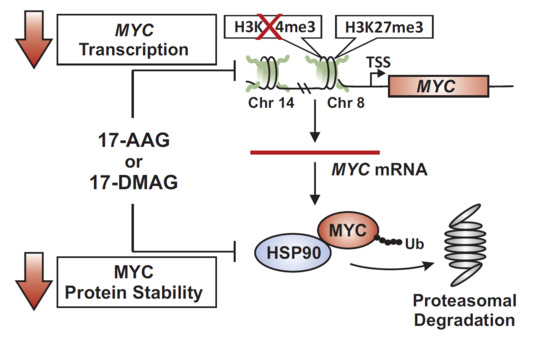
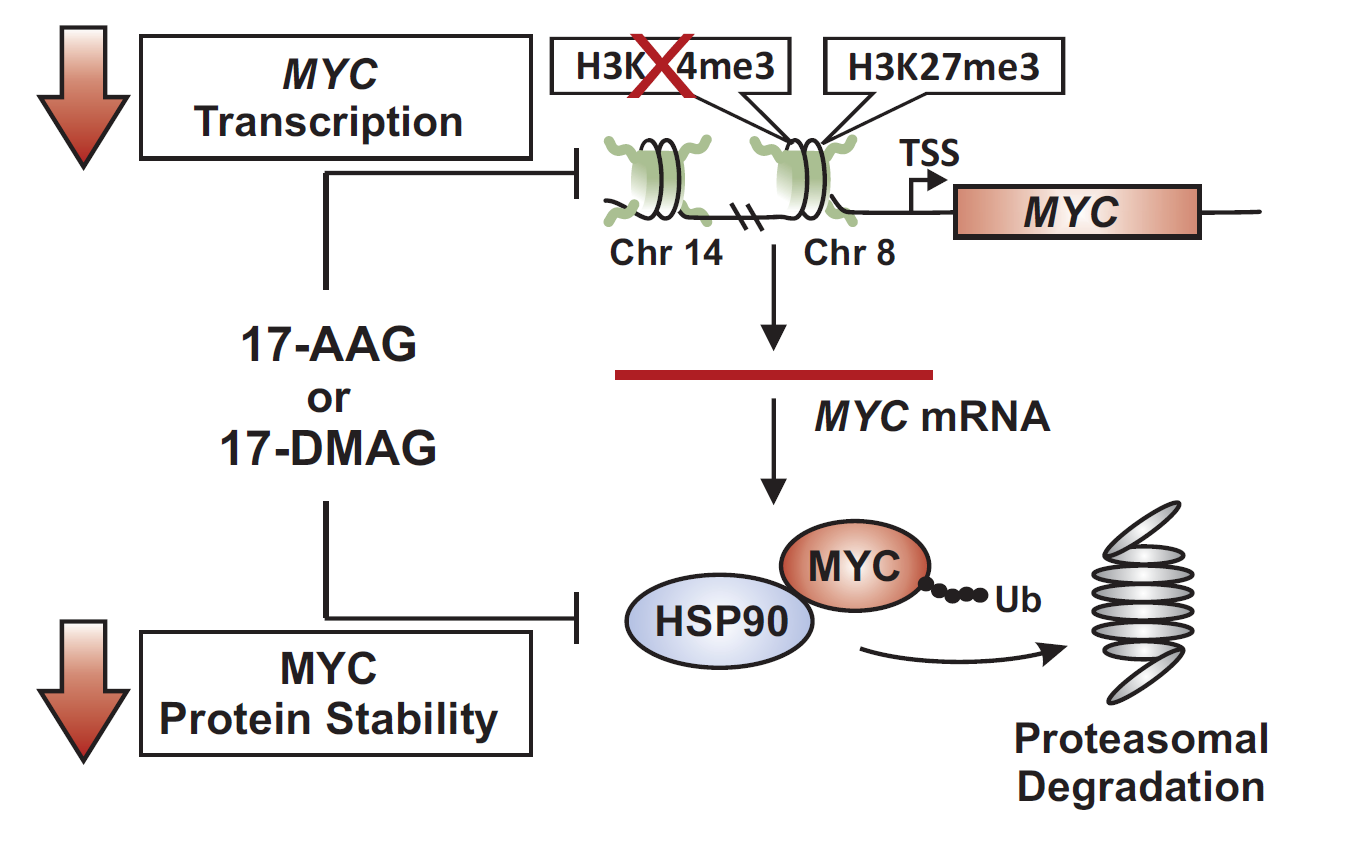
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
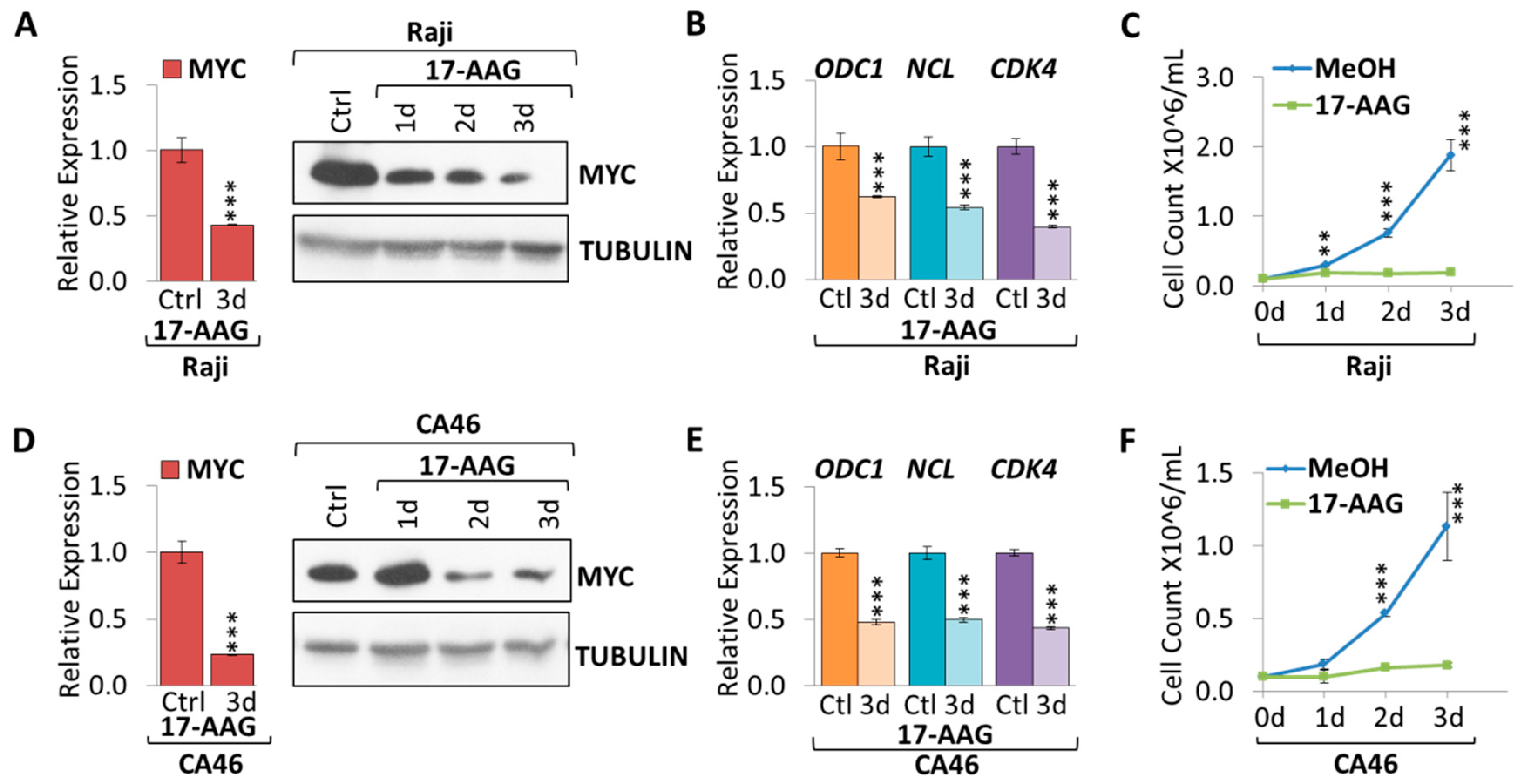
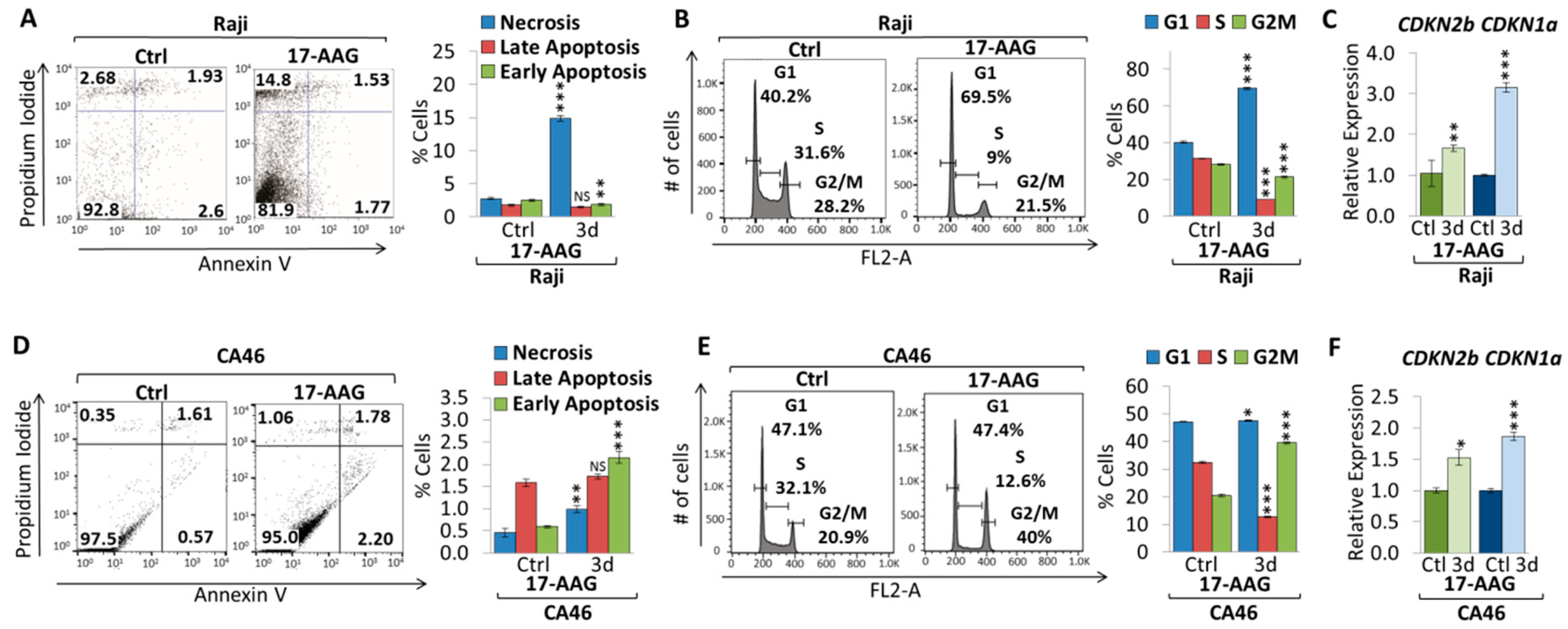
2.1. 17-AAG Treatment Downregulates MYC Expression in Burkitt Lymphoma
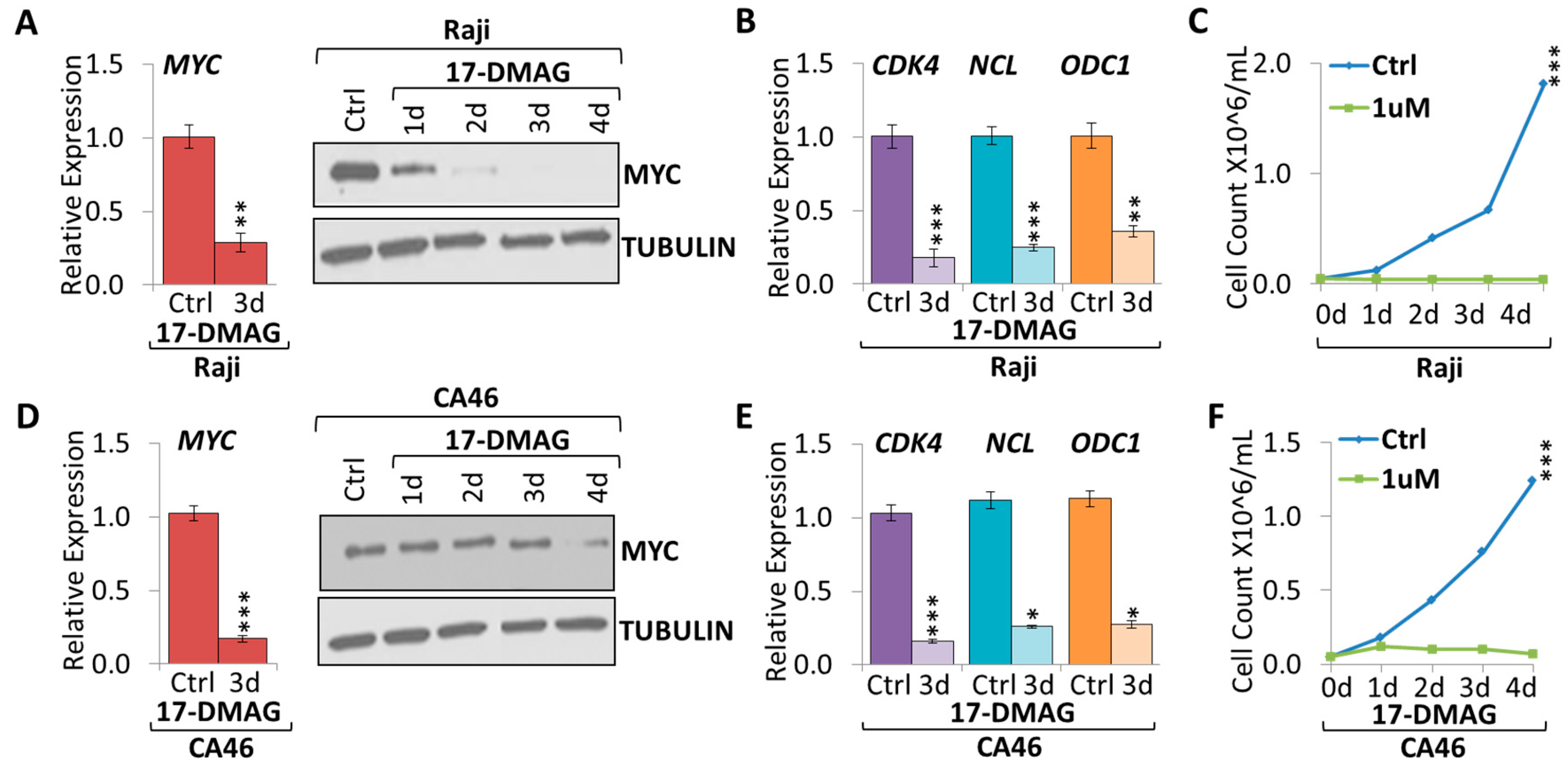
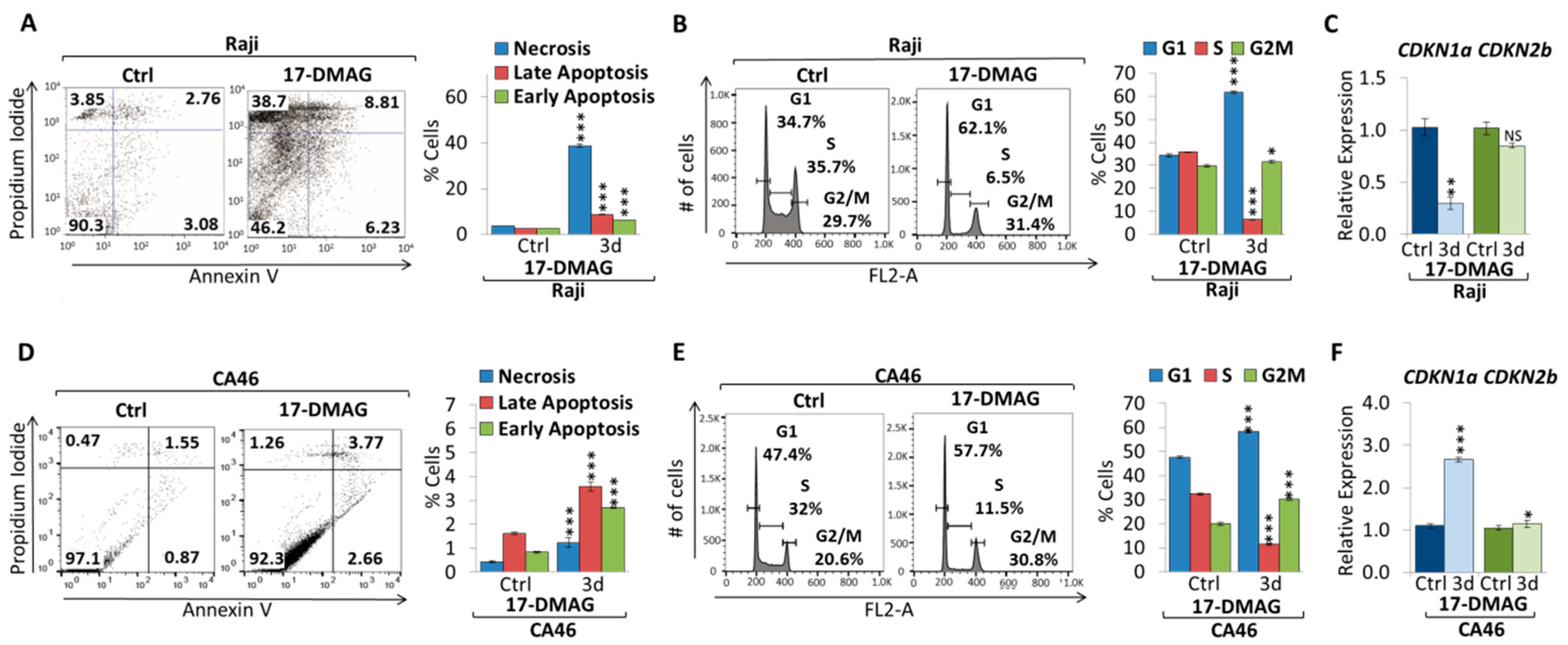
2.2. 17-DMAG Treatment Downregulates MYC Expression in Burkitt Lymphoma
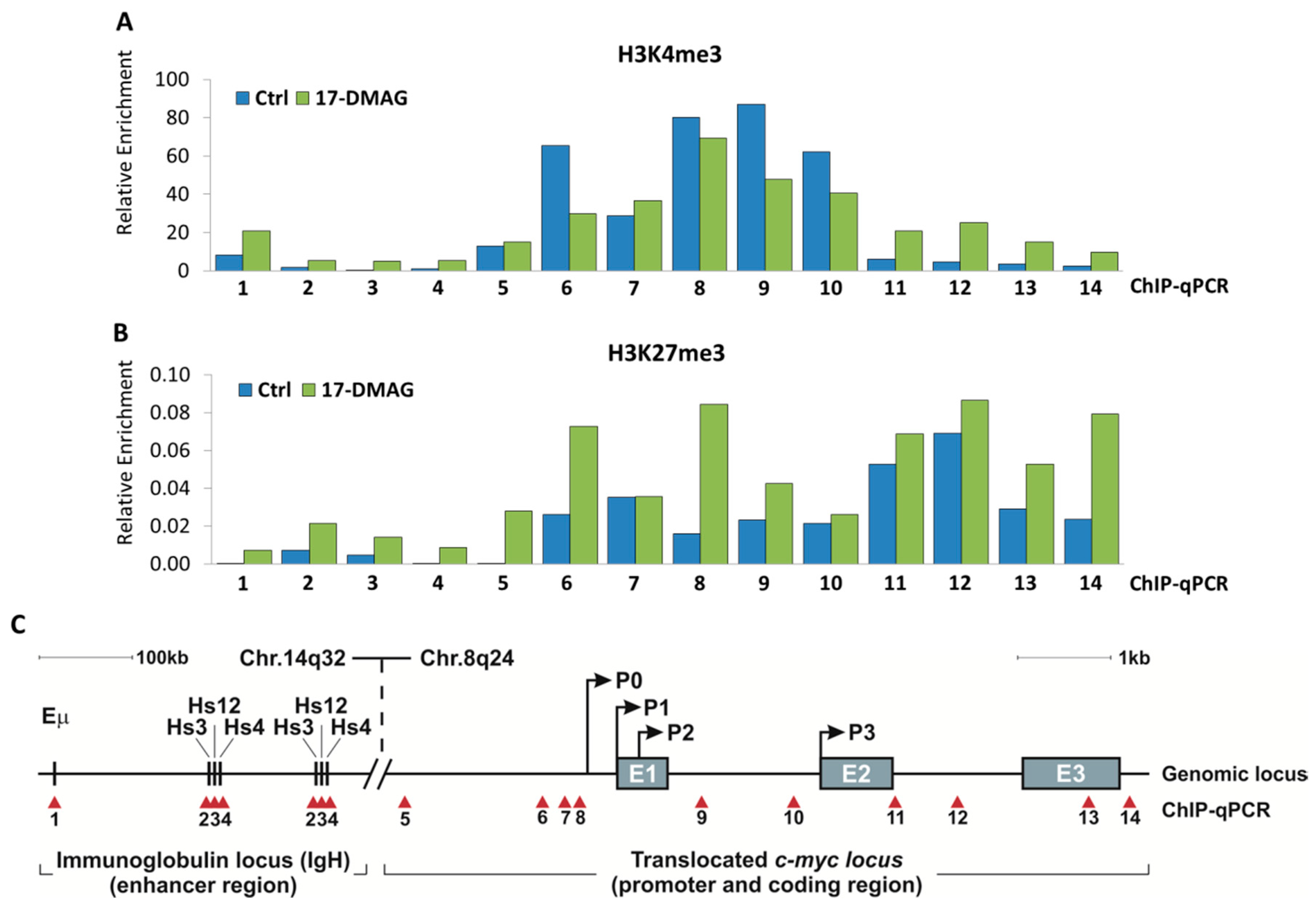
2.3. 17-DMAG Treatment Decreases Active, While Increasing Inactive Chromatin Marks at the Translocated MYC Locus in Burkitt Lymphoma
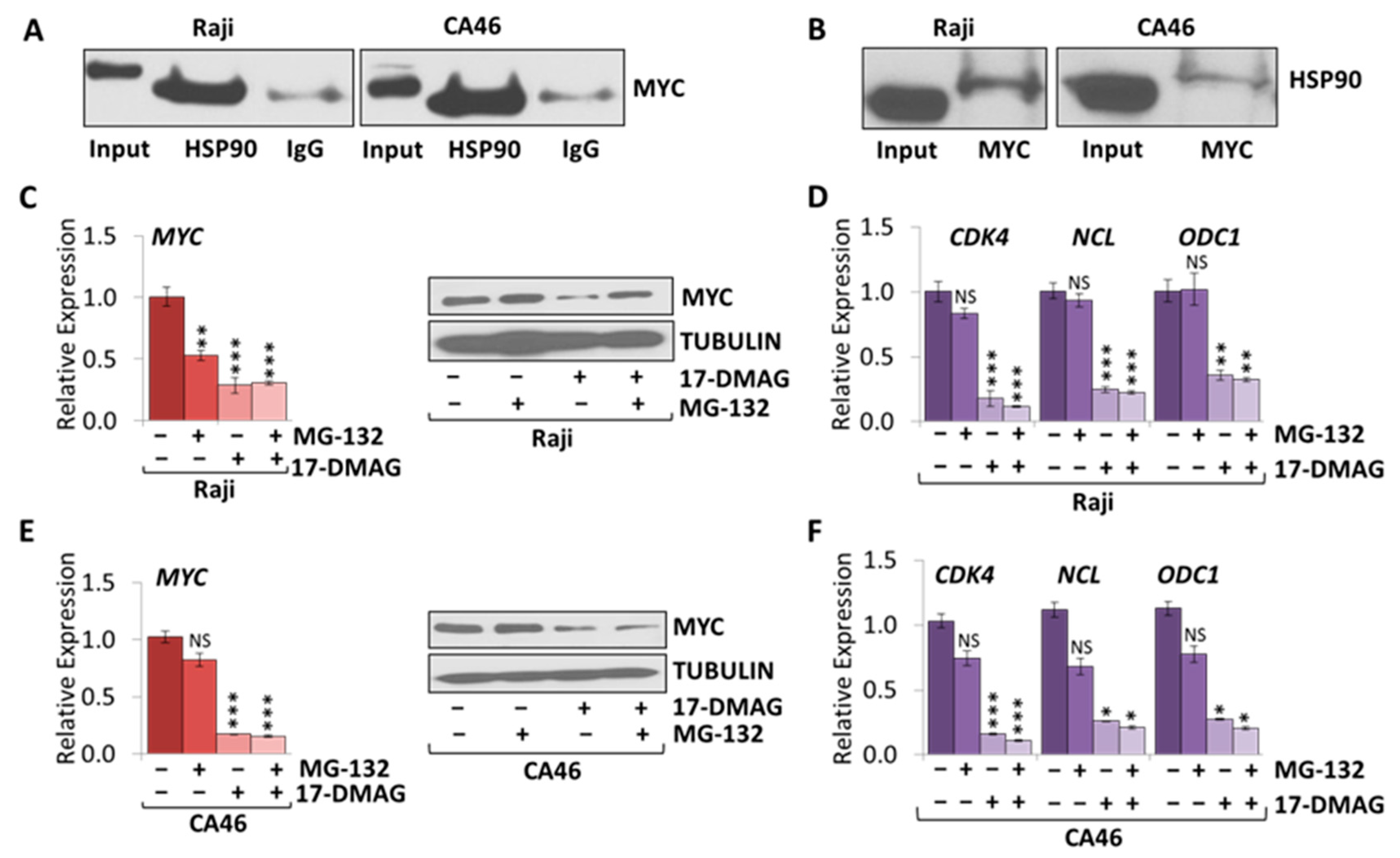
2.4. MYC Is a HSP90 Client Protein in Burkitt Lymphoma
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Treatment Conditions
4.3. RNA Extraction and Analysis of Gene Expression
4.4. Cell Extracts and Western Blot Analysis
4.5. Cell Cycle Analysis Using Propidium Iodide
4.6. Flow Cytometric Analysis of Cell Death
4.7. Chromatin Immunoprecipitation (ChIP) Analysis
4.8. Co-Immunoprecipitation
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The myc/max/mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef] [PubMed]
- Boxer, L.M.; Dang, C.V. Translocations involving c-myc and c-myc function. Oncogene 2001, 20, 5595–5610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, J.L.; Aster, J.C. Molecular biology of burkitt’s lymphoma. J. Clin. Oncol. 2000, 18, 3707–3721. [Google Scholar] [CrossRef] [PubMed]
- Pelengaris, S.; Khan, M. The many faces of c-myc. Arch. Biochem. Biophys. 2003, 416, 129–136. [Google Scholar] [CrossRef]
- Poole, C.J.; van Riggelen, J. Myc-master regulator of the cancer epigenome and transcriptome. Genes 2017, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- Felsher, D.W.; Bishop, J.M. Reversible tumorigenesis by myc in hematopoietic lineages. Mol. Cell 1999, 4, 199–207. [Google Scholar] [CrossRef]
- Felsher, D.W. Oncogene addiction versus oncogene amnesia: Perhaps more than just a bad habit? Cancer Res. 2008, 68, 3081–3086. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef] [PubMed]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taldone, T.; Gozman, A.; Maharaj, R.; Chiosis, G. Targeting HSP90: Small-molecule inhibitors and their clinical development. Curr. Opin. Pharmacol. 2008, 8, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Prodromou, C.; Workman, P. The HSP90 molecular chaperone: An open and shut case for treatment. Biochem. J. 2008, 410, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhang, L.L.; Wu, W.; Guo, H.; Li, Y.; Sukhanova, M.; Venkataraman, G.; Zhang, H.; Alikhan, M.; Lu, P.; et al. Activation of myc, a bona fide client of HSP90, contributes to intrinsic ibrutinib resistance in mantle cell lymphoma. Blood Adv. 2018, 2, 2039–2051. [Google Scholar] [CrossRef] [PubMed]
- Regan, P.L.; Jacobs, J.; Wang, G.; Torres, J.; Edo, R.; Friedmann, J.; Tang, X.X. HSP90 inhibition increases p53 expression and destabilizes mycn and myc in neuroblastoma. Int. J. Oncol. 2011, 38, 105–112. [Google Scholar] [PubMed]
- Giulino-Roth, L.; van Besien, H.J.; Dalton, T.; Totonchy, J.E.; Rodina, A.; Taldone, T.; Bolaender, A.; Erdjument-Bromage, H.; Sadek, J.; Chadburn, A.; et al. Inhibition of HSP90 suppresses pi3k/akt/mtor signaling and has antitumor activity in burkitt lymphoma. Mol. Cancer Ther. 2017, 16, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of HSP90 confers tumour selectivity on HSP90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Ikebe, E.; Kawaguchi, A.; Tezuka, K.; Taguchi, S.; Hirose, S.; Matsumoto, T.; Mitsui, T.; Senba, K.; Nishizono, A.; Hori, M.; et al. Oral administration of an HSP90 inhibitor, 17-dmag, intervenes tumor-cell infiltration into multiple organs and improves survival period for atl model mice. Blood Cancer J. 2013, 3, e132. [Google Scholar] [CrossRef] [PubMed]
- Hertlein, E.; Wagner, A.J.; Jones, J.; Lin, T.S.; Maddocks, K.J.; Towns, W.H., 3rd; Goettl, V.M.; Zhang, X.; Jarjoura, D.; Raymond, C.A.; et al. 17-dmag targets the nuclear factor-kappab family of proteins to induce apoptosis in chronic lymphocytic leukemia: Clinical implications of HSP90 inhibition. Blood 2010, 116, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Nussbaumer, U.; Chen, Y.; Beisel, C.; Paro, R. Trithorax requires HSP90 for maintenance of active chromatin at sites of gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Carystinos, G.D.; Kandouz, M.; Alaoui-Jamali, M.A.; Batist, G. Unexpected induction of the human connexin 43 promoter by the ras signaling pathway is mediated by a novel putative promoter sequence. Mol. Pharmacol. 2003, 63, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Chang, W.W.; Chen, Y.Y.; Tsai, Y.H.; Chou, Y.H.; Tseng, H.C.; Chen, H.L.; Wu, C.C.; Chang-Chien, J.; Lee, H.T.; et al. HSP90alpha mediates bmi1 expression in breast cancer stem/progenitor cells through facilitating nuclear translocation of c-myc and ezh2. Int. J. Mol. Sci. 2017, 18, 1986. [Google Scholar] [CrossRef] [PubMed]
- Diamanti, P.; Cox, C.V.; Moppett, J.P.; Blair, A. Dual targeting of HSP90 in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2018, 180, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E.; Gojo, I.; Burton, M.; Quinn, M.; Tighe, S.M.; Kersey, K.; Zhong, Z.; Albitar, M.X.; Bhalla, K.; Hannah, A.L.; et al. Phase i study of the heat shock protein 90 inhibitor alvespimycin (kos-1022, 17-dmag) administered intravenously twice weekly to patients with acute myeloid leukemia. Leukemia 2010, 24, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Posternak, V.; Cole, M.D. Strategically targeting myc in cancer. F1000Res. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, A.; Brickner, J.H. Epigenetic transcriptional memory. Curr. Genet. 2017, 63, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; van Riggelen, J.; Yetil, A.; Fan, A.C.; Bachireddy, P.; Felsher, D.W. Cellular senescence is an important mechanism of tumor regression upon c-myc inactivation. Proc. Natl. Acad. Sci. USA 2007, 104, 13028–13033. [Google Scholar] [CrossRef] [PubMed]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. Myc inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Arvanitis, C.; Chu, K.; Dewey, W.; Leonhardt, E.; Trinh, M.; Sundberg, C.D.; Bishop, J.M.; Felsher, D.W. Sustained loss of a neoplastic phenotype by brief inactivation of myc. Science 2002, 297, 102–104. [Google Scholar] [CrossRef] [PubMed]
- Zeller, K.I.; Jegga, A.G.; Aronow, B.J.; O’Donnell, K.A.; Dang, C.V. An integrated database of genes responsive to the myc oncogenic transcription factor: Identification of direct genomic targets. Genome Biol. 2003, 4, R69. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Lee, P.; Fiskus, W.; Yang, Y.; Joshi, R.; Wang, Y.; Buckley, K.; Balusu, R.; Chen, J.; Koul, S.; et al. Co-treatment with heat shock protein 90 inhibitor 17-dimethylaminoethylamino-17-demethoxygeldanamycin (dmag) and vorinostat: A highly active combination against human mantle cell lymphoma (mcl) cells. Cancer Biol. Ther. 2009, 8, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Rockliffe, N.; Johnson, G.G.; Sherrington, P.D.; Pettitt, A.R. HSP90 inhibition has opposing effects on wild-type and mutant p53 and induces p21 expression and cytotoxicity irrespective of p53/atm status in chronic lymphocytic leukaemia cells. Oncogene 2008, 27, 2445–2455. [Google Scholar] [CrossRef] [PubMed]
- Duthu, A.; Debuire, B.; Romano, J.; Ehrhart, J.C.; Fiscella, M.; May, E.; Appella, E.; May, P. P53 mutations in raji cells: Characterization and localization relative to other burkitt’s lymphomas. Oncogene 1992, 7, 2161–2167. [Google Scholar] [PubMed]
- Farrell, P.J.; Allan, G.J.; Shanahan, F.; Vousden, K.H.; Crook, T. P53 is frequently mutated in burkitt’s lymphoma cell lines. EMBO J. 1991, 10, 2879–2887. [Google Scholar] [CrossRef] [PubMed]
- Wiman, K.G.; Magnusson, K.P.; Ramqvist, T.; Klein, G. Mutant p53 detected in a majority of burkitt lymphoma cell lines by monoclonal antibody pab240. Oncogene 1991, 6, 1633–1639. [Google Scholar] [PubMed]
- Florean, C.; Schnekenburger, M.; Lee, J.Y.; Kim, K.R.; Mazumder, A.; Song, S.; Kim, J.M.; Grandjenette, C.; Kim, J.G.; Yoon, A.Y.; et al. Discovery and characterization of isofistularin-3, a marine brominated alkaloid, as a new DNA demethylating agent inducing cell cycle arrest and sensitization to trail in cancer cells. Oncotarget 2016, 7, 24027–24049. [Google Scholar] [CrossRef] [PubMed]
- Richart, L.; Carrillo-de Santa Pau, E.; Rio-Machin, A.; de Andres, M.P.; Cigudosa, J.C.; Lobo, V.J.; Real, F.X. Bptf is required for c-myc transcriptional activity and in vivo tumorigenesis. Nat. Commun. 2016, 7, 10153. [Google Scholar] [CrossRef] [PubMed]
- Van Riggelen, J.; Muller, J.; Otto, T.; Beuger, V.; Yetil, A.; Choi, P.S.; Kosan, C.; Moroy, T.; Felsher, D.W.; Eilers, M. The interaction between myc and miz1 is required to antagonize tgfbeta-dependent autocrine signaling during lymphoma formation and maintenance. Genes Dev. 2010, 24, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Cumming, K.T.; Paulsen, G.; Wernbom, M.; Ugelstad, I.; Raastad, T. Acute response and subcellular movement of hsp27, alphab-crystallin and hsp70 in human skeletal muscle after blood-flow-restricted low-load resistance exercise. Acta Physiol. 2014, 211, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.M.; Lucas, J.M.; Gomez-Sarosi, L.A.; Coleman, I.; Zhao, S.; Coleman, R.; Nelson, P.S. DNA damage induces gdnf secretion in the tumor microenvironment with paracrine effects promoting prostate cancer treatment resistance. Oncotarget 2015, 6, 2134–2147. [Google Scholar] [CrossRef] [PubMed]
- Poole, C.J.; Zheng, W.; Lodh, A.; Yevtodiyenko, A.; Liefwalker, D.; Li, H.; Felsher, D.W.; van Riggelen, J. Dnmt3b overexpression contributes to aberrant DNA methylation and myc-driven tumor maintenance in t-all and burkitt’s lymphoma. Oncotarget 2017, 8, 76898–76920. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poole, C.J.; Zheng, W.; Lee, H.; Young, D.; Lodh, A.; Chadli, A.; Van Riggelen, J. Targeting the MYC Oncogene in Burkitt Lymphoma through HSP90 Inhibition. Cancers 2018, 10, 448. https://doi.org/10.3390/cancers10110448
Poole CJ, Zheng W, Lee H, Young D, Lodh A, Chadli A, Van Riggelen J. Targeting the MYC Oncogene in Burkitt Lymphoma through HSP90 Inhibition. Cancers. 2018; 10(11):448. https://doi.org/10.3390/cancers10110448
Chicago/Turabian StylePoole, Candace J., Wenli Zheng, Haesung Lee, Danielle Young, Atul Lodh, Ahmed Chadli, and Jan Van Riggelen. 2018. "Targeting the MYC Oncogene in Burkitt Lymphoma through HSP90 Inhibition" Cancers 10, no. 11: 448. https://doi.org/10.3390/cancers10110448
APA StylePoole, C. J., Zheng, W., Lee, H., Young, D., Lodh, A., Chadli, A., & Van Riggelen, J. (2018). Targeting the MYC Oncogene in Burkitt Lymphoma through HSP90 Inhibition. Cancers, 10(11), 448. https://doi.org/10.3390/cancers10110448