PHD3 Acts as Tumor Suppressor in Mouse Osteosarcoma and Influences Tumor Vascularization via PDGF-C Signaling


 and
and
Abstract
:1. Introduction
2. Results
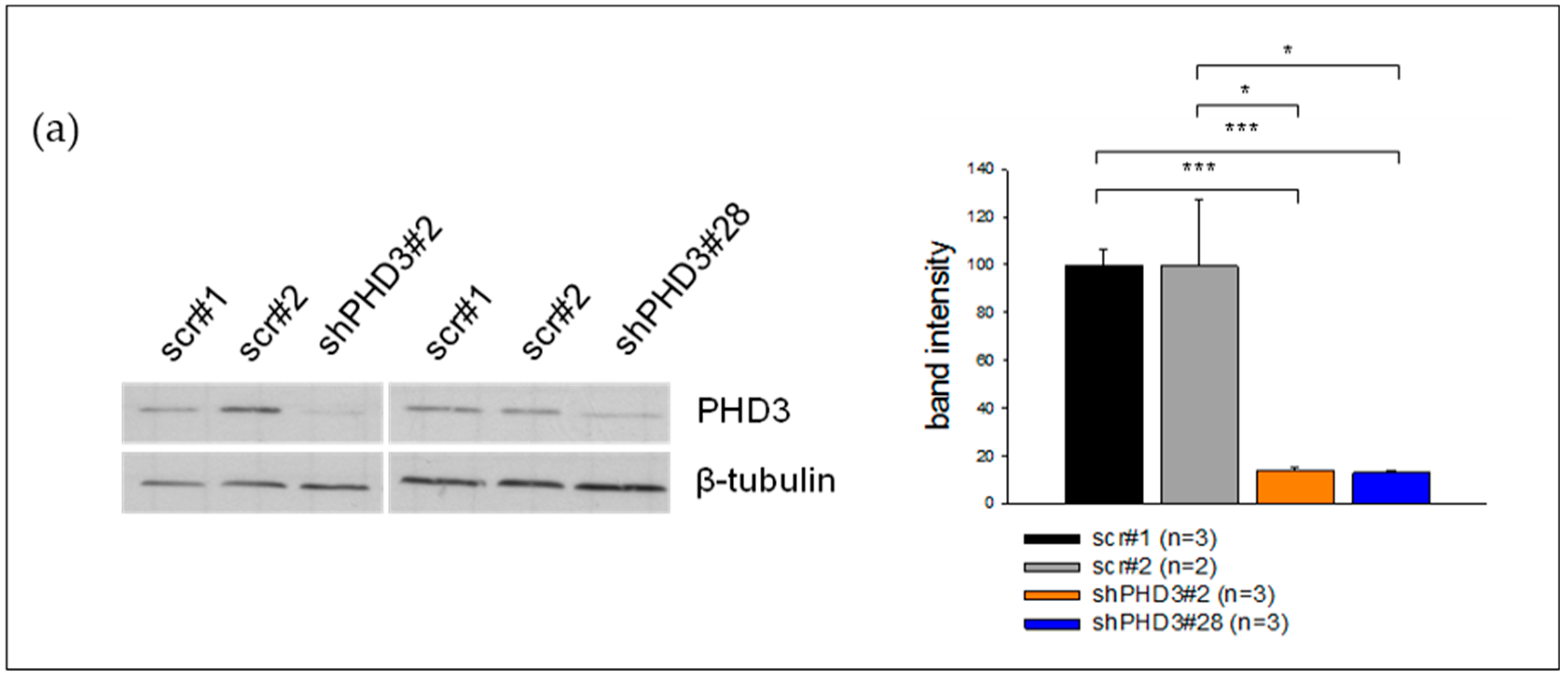
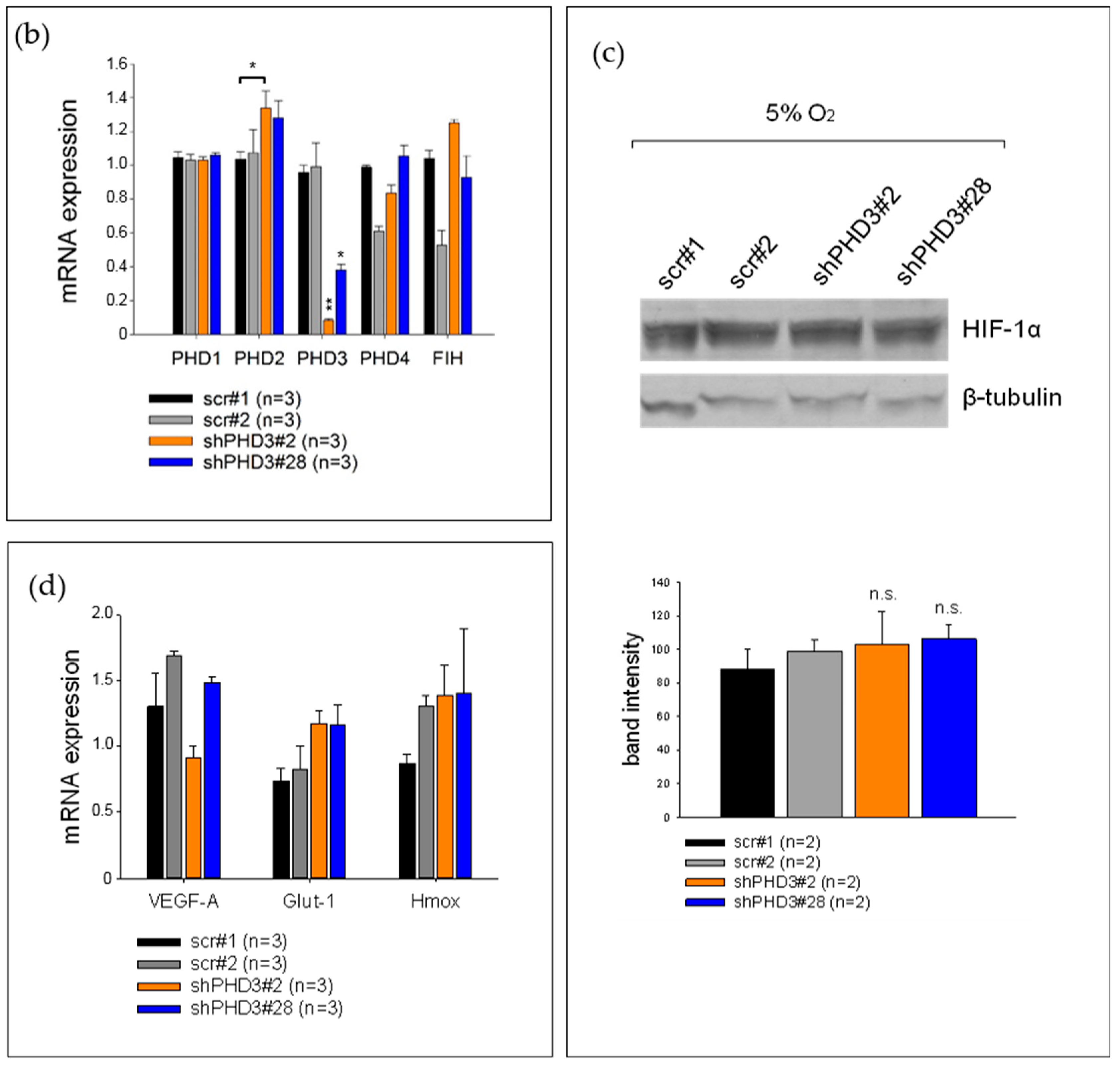
2.1. PHD3 Silencing Does not Affect HIF-1α Protein Levels or HIF-Target Gene Expression
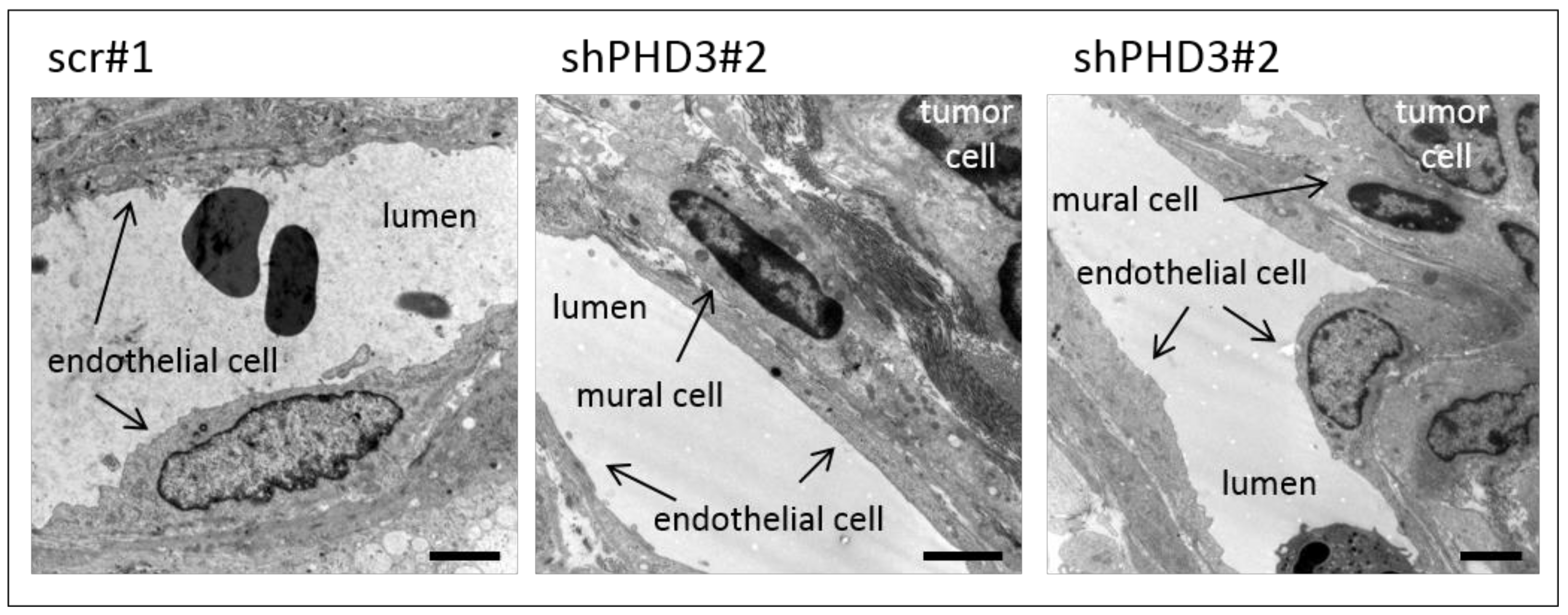
2.2. Reduction of PHD3 Leads to Accelerated Tumor Growth and Enlargement of Tumor Vessels
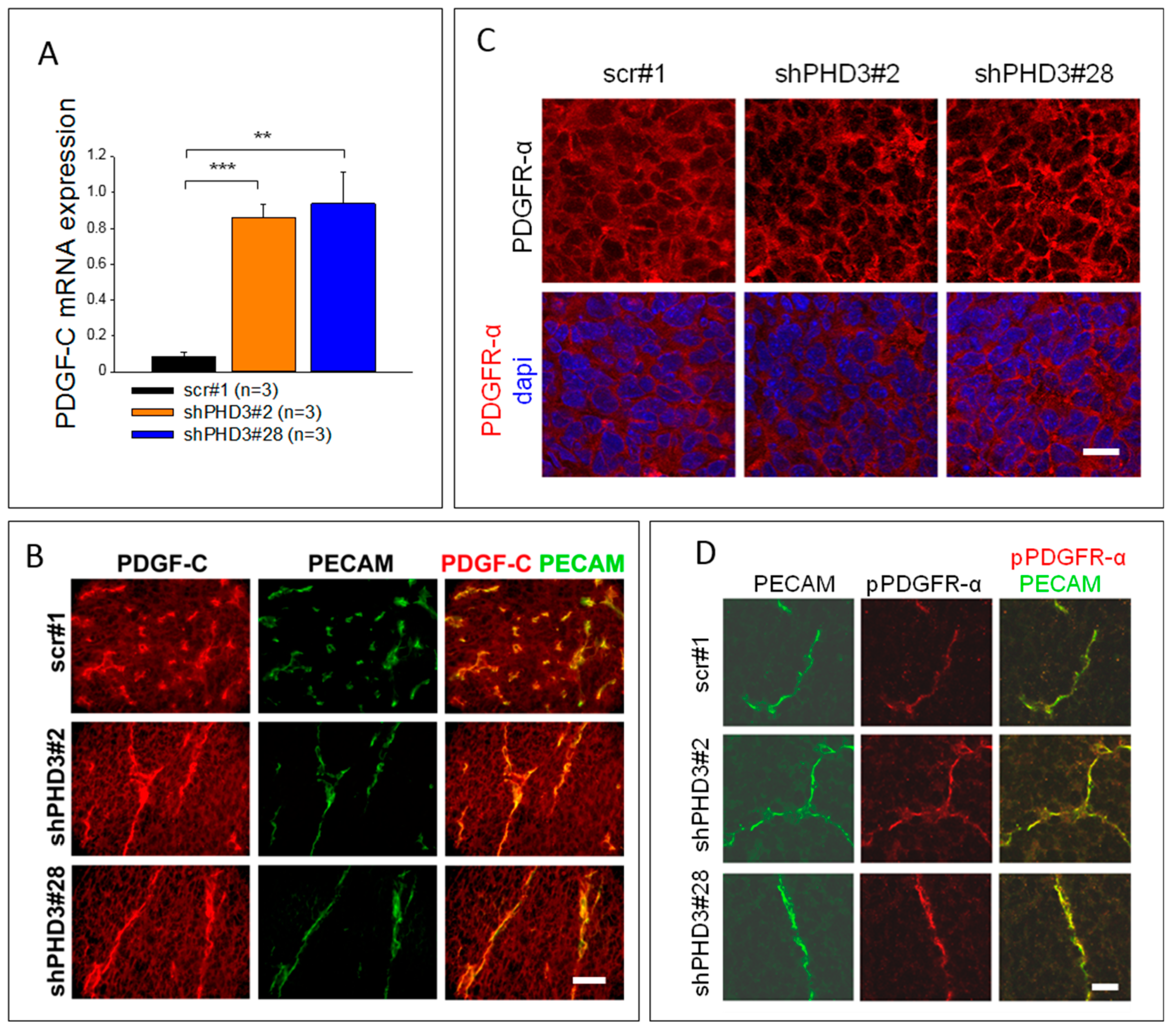
2.3. PDGF-C Signaling Is Strongly Increased in LM8-shPHD3 Cells and Tumors
2.4. Knock-Down of PDGF-C in shPHD3 Tumors Reduces Tumor Growth
2.5. The shPHD3 Vessel Phenotype Is Rescued in shPHD3+shPDGF-C Tumors
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Virus Production
4.2. Silencing of PHD3 and PDGF-C
4.3. Tumor Model and Mice
4.4. RNA Extraction and qRT-PCR
4.5. Western Blot
4.6. Immunostaining and Microscopy
4.7. Determination of Hypoxic Area, Vessel Perfusion, Leakage and Tumor Necrosis
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism: Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Span, P.N.; Bussink, J. Biology of hypoxia. Semin. Nucl. Med. 2015, 45, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.-C.; Qiu, Z.-J.; Liu, J.; Sun, J.; Jiang, T.; Huang, K.-J.; Yao, M.; Huang, C. Expression of hypoxia-inducible factor-1 alpha and associated proteins in pancreatic ductal adenocarcinoma and their impact on prognosis. Int. J. Oncol. 2007, 30, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Minardi, D.; Lucarini, G.; Santoni, M.; Mazzucchelli, R.; Burattini, L.; Conti, A.; Principi, E.; Bianconi, M.; Scartozzi, M.; Milanese, G.; et al. Survival in patients with clear cell renal cell carcinoma is predicted by HIF-1α expression. Anticancer Res. 2015, 35, 433–438. [Google Scholar]
- Ouyang, Y.; Li, H.; Bu, J.; Li, X.; Chen, Z.; Xiao, T. Hypoxia-inducible factor-1 expression predicts osteosarcoma patients’ survival: A meta-analysis. Int. J. Biol. Markers 2016, 31, e229–e234. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Shen, S.; Zhao, X.; Chen, G. Targeted genes and interacting proteins of HIF-1. Int. J. Biochem. Mol. Biol. 2012, 3, 165–178. [Google Scholar]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Ivan, M.; Haberberger, T.; Gervasi, D.C.; Michelson, K.S.; Gunzler, V.; Kondo, K.; Yang, H.; Sorokina, I.; Conaway, R.C.; Conaway, J.W.; et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc. Natl. Acad. Sci. USA 2002, 99, 13459–13464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oehme, F.; Ellinghaus, P.; Kolkhof, P.; Smith, T.J.; Ramakrishnan, S.; Hutter, J.; Schramm, M.; Flamme, I. Overexpression of PH-4, a novel putative proline 4-hydroxylase, modulates activity of hypoxia-inducible transcription factors. Biochem. Biophys. Res. Commun. 2002, 296, 343–349. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin Jr., W.G. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, C.; Chen, X.; Takada, M.; Fan, C.; Zheng, X.; Wen, H.; Liu, Y.; Wang, C.; Pestell, R.G.; et al. EglN2 associates with the NRF1-PGC1α complex and controls mitochondrial function in breast cancer. EMBO J. 2015, 34, 2953–2970. [Google Scholar] [CrossRef]
- Chan, D.A.; Kawahara, T.L.; Sutphin, P.D.; Chang, H.Y.; Chi, J.T.; Giaccia, A.J. Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell 2009, 15, 527–538. [Google Scholar] [CrossRef]
- Kozlova, N.; Wottawa, M.; Katschinski, D.M.; Kristiansen, G.; Kietzmann, T. Hypoxia-inducible factor prolyl hydroxylase 2 ( PHD2 ) is a direct regulator of epidermal growth factor receptor ( EGFR ) signaling in breast cancer. Oncotarget 2017, 8, 9885–9898. [Google Scholar] [CrossRef]
- Su, Y.; Loos, M.; Giese, N.; Hines, O.J.; Diebold, I.; Görlach, A.; Metzen, E.; Pastorekova, S.; Friess, H.; Büchler, P. PHD3 regulates differentiation, tumour growth and angiogenesis in pancreatic cancer. Br. J. Cancer 2010, 1–9. [Google Scholar] [CrossRef]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential Function of the Prolyl Hydroxylases PHD1, PHD2, and PHD3 in the Regulation of Hypoxia-inducible Factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [Green Version]
- Hopfer, U.; Hopfer, A.; Jablonski, K.; Stahl, R.A.; Wolf, G. The Novel WD-repeat Protein Morg1 Acts as a Molecular Scaffold for Hypoxia-inducible Factor Prolyl Hydroxylase 3 (PHD3). J. Biol. Chem. 2006, 281, 8645–8655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koditz, J.; Nesper, J.; Wottawa, M.; Stiehl, D.P.; Camenisch, G.; Franke, C.; Myllyharju, J.; Wenger, R.H.; Katschinski, D.M. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood 2007, 110, 3610–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Sakota, M.; Nakayama, K. Human PRP19 interacts with prolyl-hydroxylase PHD3 and inhibits cell death in hypoxia. Exp. Cell Res. 2010, 316, 2871–2882. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Jiao, S.; Zhang, H.; Lv, D.; Xue, J.; Fan, L.; Wu, G.; Fang, J. Prolyl hydroxylase domain protein 3 targets Pax2 for destruction. Biochem. Biophys. Res. Commun. 2011, 409, 315–320. [Google Scholar] [CrossRef]
- German, N.J.; Yoon, H.; Yusuf, R.Z.; Murphy, J.P.; Finley, L.W.S.; Laurent, G.; Haas, W.; Satterstrom, F.K.; Guarnerio, J.; Zaganjor, E.; et al. PHD3 Loss in Cancer Enables Metabolic Reliance on Fatty Acid Oxidation via Deactivation of ACC2. Mol. Cell 2016, 63, 1006–1020. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Torigoe, T.; Hirohashi, Y.; Kitamura, H.; Tanaka, T.; Honma, I.; Asanuma, H.; Harada, K.; Takasu, H.; Masumori, N.; et al. Identification of an immunogenic CTL epitope of HIFPH3 for immunotherapy of renal cell carcinoma. Clin. Cancer Res. 2008, 14, 6916–6923. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Li, X.; Jiao, S.; Wei, Y.; Wu, G.; Fang, J. Prolyl hydroxylase-3 is down-regulated in colorectal cancer cells and inhibits IKKbeta independent of hydroxylase activity. Gastroenterology 2010, 138, 606–615. [Google Scholar] [CrossRef]
- Andersen, S.; Donnem, T.; Stenvold, H.; Al-Saad, S.; Al-Shibli, K.; Busund, L.T.; Bremnes, R.M. Overexpression of the hif hydroxylases phd1, phd2, phd3 and fih are individually and collectively unfavorable prognosticators for nsclc survival. PLoS ONE 2011, 6, e23847. [Google Scholar] [CrossRef] [Green Version]
- Miikkulainen, P.; Högel, H.; Rantanen, K.; Suomi, T.; Kouvonen, P.; Elo, L.L.; Jaakkola, P.M. HIF prolyl hydroxylase PHD3 regulates translational machinery and glucose metabolism in clear cell renal cell carcinoma. Cancer Metab. 2017, 5, 5. [Google Scholar] [CrossRef]
- Klotzsche-von Ameln, A.; Muschter, A.; Mamlouk, S.; Kalucka, J.; Prade, I.; Franke, K.; Rezaei, M.; Poitz, D.M.; Breier, G.; Wielockx, B. Inhibition of HIF prolyl hydroxylase-2 blocks tumor growth in mice through the antiproliferative activity of TGFβ. Cancer Res. 2011, 71, 3306–3316. [Google Scholar] [CrossRef]
- Berra, E.; Benizri, E.; Ginouves, A.; Volmat, V.; Roux, D.; Pouyssegur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, J.; Li, X.; Luo, X.; Fang, J.; Chen, H. The expression of prolyl hydroxylase domain enzymes are up-regulated and negatively correlated with Bcl-2 in non-small cell lung cancer. Mol. Cell. Biochem. 2011, 358, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Henze, A.-T.; Riedel, J.; Diem, T.; Wenner, J.; Flamme, I.; Pouyseggur, J.; Plate, K.H.; Acker, T. Prolyl hydroxylases 2 and 3 act in gliomas as protective negative feedback regulators of hypoxia-inducible factors. Cancer Res. 2009, 70, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Tennant, D.A.; Gottlieb, E. HIF prolyl hydroxylase-3 mediates alpha-ketoglutarate-induced apoptosis and tumor suppression. J. Mol. Med. 2010, 88, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Zwerner, J.P.; May, W. a PDGF-C is an EWS/FLI induced transforming growth factor in Ewing family tumors. Oncogene 2001, 20, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Velez, G.; Hovland, P.; Hirose, T.; Kazlauskas, A. Plasmin is the major protease responsible for processing PDGF-C in the vitreous of patients with proliferative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2008, 49, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef]
- Kuzmanov, A.; Wielockx, B.; Rezaei, M.; Kettelhake, A.; Breier, G. Overexpression of factor inhibiting HIF-1 enhances vessel maturation and tumor growth via platelet-derived growth factor-C. Int. J. Cancer 2012, 131. [Google Scholar] [CrossRef] [Green Version]
- Manzat Saplacan, R.M.; Balacescu, L.; Gherman, C.; Chira, R.I.; Craiu, A.; Mircea, P.A.; Lisencu, C.; Balacescu, O. The Role of PDGFs and PDGFRs in Colorectal Cancer. Mediators Inflamm. 2017, 2017. [Google Scholar] [CrossRef]
- Klotzsche-von Ameln, A.; Muschter, A.; Heimesaat, M.M.; Breier, G.; Wielockx, B. HIF prolyl hydroxylase-2 inhibition diminishes tumor growth through matrix metalloproteinase-induced TGFbeta activation. Cancer Biol. Ther. 2012, 13, 216–223. [Google Scholar] [CrossRef]
- Sciorra, V.A.; Sanchez, M.A.; Kunibe, A.; Wurmser, A.E. Suppression of Glioma progression by Egln3. PLoS ONE 2012, 7, e40053. [Google Scholar] [CrossRef] [PubMed]
- Henze, A.T.; Garvalov, B.K.; Seidel, S.; Cuesta, A.M.; Ritter, M.; Filatova, A.; Foss, F.; Dopeso, H.; Essmann, C.L.; Maxwell, P.H.; et al. Loss of PHD3 allows tumours to overcome hypoxic growth inhibition and sustain proliferation through EGFR. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.-J.; Jiang, X.-T.; Jiang, S.-B.; He, X.-J.; Luo, J.-G.; Liu, Z.-C.; Wang, L.; Tao, H.-Q.; Chen, J.-Z. PHD3 affects gastric cancer progression by negatively regulating HIF1A. Mol. Med. Rep. 2017, 16, 6882–6889. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, L.E.; Hemo, I.; Keshet, E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 1998, 125, 1591–1598. [Google Scholar] [PubMed]
- Morikawa, S.; Baluk, P.; Kaidoh, T.; Haskell, A.; Jain, R.K.; McDonald, D.M. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2002, 160, 985–1000. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S.; Meyer-Morse, N.; Bergsland, E.; Hanahan, D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J. Clin. Invest. 2003, 111, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.-J.; You, W.-K.; Bonaldo, P.; Seyfried, T.N.; Pasquale, E.B.; Stallcup, W.B. Pericyte deficiencies lead to aberrant tumor vascularizaton in the brain of the NG2 null mouse. Dev. Biol. 2010, 344, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef]
- Di Tomaso, E.; London, N.; Fuja, D.; Logie, J.; Tyrrell, J.A.; Kamoun, W.; Munn, L.L.; Jain, R.K. PDGF-C induces maturation of blood vessels in a model of glioblastoma and attenuates the response to anti-VEGF treatment. PLoS ONE 2009, 4, e5123. [Google Scholar] [CrossRef]
- Li, X.; Kumar, A.; Zhang, F.; Lee, C.; Li, Y.; Tang, Z. VEGF-independent angiogenic pathways induced by PDGF-C. Oncotarget 2010, 1, 309–314. [Google Scholar] [CrossRef]
- Li, X.; Tjwa, M.; Moons, L.; Fons, P.; Noel, A.; Ny, A.; Zhou, J.M.; Lennartsson, J.; Li, H.; Luttun, A.; et al. Revascularization of ischemic tissues by PDGF-CC via effects on endothelial cells and their progenitors. J. Clin. Invest. 2005, 115, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, D.G.; Duff, M.E.; West, J.W.; Kelly, J.D.; Sheppard, P.O.; Hofstrand, P.D.; Gao, Z.; Shoemaker, K.; Bukowski, T.R.; Moore, M.; et al. Platelet-derived growth factor C (PDGF-C), a novel growth factor that binds to PDGF alpha and beta receptor. J. Biol. Chem. 2001, 276, 27406–27414. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.S.; Johnson, M.M.; Bauer, R.L.; Hudkins, K.L.; Gilbertson, D.G.; Riehle, K.J.; Yeh, M.M.; Alpers, C.E.; Fausto, N. Targeting stromal cells for the treatment of platelet-derived growth factor C-induced hepatocellular carcinogenesis. Differentiation 2007, 75, 843–852. [Google Scholar] [CrossRef]
- Li, X.; Ponten, A.; Aase, K.; Karlsson, L.; Abramsson, A.; Uutela, M.; Backstrom, G.; Hellstrom, M.; Bostrom, H.; Li, H.; et al. PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nat. Cell Biol. 2000, 2, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef]
- Lei, H.; Hovland, P.; Velez, G.; Haran, A.; Gilbertson, D.; Hirose, T.; Kazlauskas, A. A potential role for PDGF-C in experimental and clinical proliferative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2007, 48, 2335–2342. [Google Scholar] [CrossRef]
- Lei, H.; Kazlauskas, A. Focus on Molecules: Platelet-derived Growth Factor C, PDGF-C. Exp. Eye Res. 2008, 86, 711–712. [Google Scholar] [CrossRef]
- Hurst, N.J.; Najy, A.J.; Ustach, C.V.; Movilla, L.; Kim, H.-R.C. Platelet-derived growth factor-C (PDGF-C) activation by serine proteases: Implications for breast cancer progression. Biochem. J. 2012, 441, 909–918. [Google Scholar] [CrossRef]
- Schafer, A.I.; Rodriguez, R.; Loscalzo, J.; Gimbrone, M.A.J. Inhibition of Vascular Endothelial Cell Prostacyclin Synthesis by Plasmin. Blood 1989, 74, 1015–1020. [Google Scholar]
- Kwaan, H.C.; Wang, J.; Weiss, I. Expression of receptors for plasminogen activators on endothelial cell surface depends on their origin. J. Thromb. Haemost. 2004, 2, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Eklund, L.; Olsen, B.R. Tie receptors and their angiopoietin ligands are context-dependent regulators of vascular remodeling. Exp. Cell Res. 2006, 312, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Arjunan, P.; Lee, C.; Li, Y.; Kumar, A.; Hou, X.; Wang, B.; Wardega, P.; Zhang, F.; Dong, L.; et al. Survival effect of PDGF-CC rescues neurons from apoptosis in both brain and retina by regulating GSK3β phosphorylation. J. Exp. Med. 2010, 207, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Chai, H.; Yu, Z.; Ge, W.; Kang, N.; Xia, W.; Che, Y. HIF-1α effects on angiogenic potential in human small cell lung carcinoma. J. Exp. Clin. Cancer Res. 2011, 30. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Schnittler, H.J.; Aust, D.E.; Friedrich, K.; Baretton, G.; Vestweber, D.; Breier, G. Vascular endothelial cadherin promotes breast cancer progression via transforming growth factor beta signaling. Cancer Res. 2008, 68, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, M.; Friedrich, K.; Wielockx, B.; Kuzmanov, A.; Kettelhake, A.; Labelle, M.; Schnittler, H.; Baretton, G.; Breier, G. Interplay between neural-cadherin and vascular endothelial-cadherin in breast cancer progression. Breast Cancer Res. 2012, 14, R154. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clone | shRNA Sequence |
|---|---|
| shPHD3#2 | Sense |
| 5’-CGCGTCCCGTCCAAGGCAATGGTGGCTTGTTCAAGAGACAAGCCAC CATTGCCTTGGACTTTTTGGAAAT-3’ | |
| Antisense | |
| 3’-AGGGGCAGGTTCCGTTACCACCGAACAAGTTCTCTGTTCGGTGGT AACGGAACCTGAAAAACCTTTAGC-5’ | |
| shPHD3#28 | Sense |
| 5’-CGCGTCCCGCAAATACTATGTCAAGGATTCAAGAGATCCTTGACATA GTATTTGCTTTTTGGAAAT-3’ | |
| Antisense | |
| 3’-AGGGGCGTTTATGATACAGTTCCTAAGTTCTCTAGGAACTGTATCA TAAACGAAAAACCTTTAGC-5’ | |
| scr#1, scr#2 | Sense |
| 5’-CGCGTCCCAGTCGCTTAGAAACGAGAATTCAAGAGATTCTCGTTTCT AAGCGACTTTTTTGGAAAT-3’ | |
| Antisense | |
| 3’-AGGGGTCAGCGAATCTTTGCTCTTAAGTTCTCTAAGAGCAAAGATT CGCTGAAAAAACCTTTAGC-5′ | |
| shPDGF-C#3, shPDGF-C#4 | Sense |
| 5′-CGCGTCCCAGTGGTGAATCTGAATCTCTTCAAGAGAGAGATTCAGAT TCACCACTTTTTTGGAAAT-3′ | |
| Antisense | |
| 3′-AGGGGTCACCACTTAGACTTAGAGAAGTTCTCTCTCTAAG TCTAAGTGGTGAAAAAACCTTTAGC-5′ |
| Primer | Sequence |
|---|---|
| Eef2 | 5′-atc ctc acc gac atc acc aag-3′ |
| 5′-ctg ctc tgg aca ctg gat ctc-3′ | |
| FIH | 5′-gta ctg gtg gca cca tat ag-3′ |
| 5′-cct ctc caa gca tct tct ca-3′ | |
| Glut-1 | 5′-gtc ggg ggc atg att ggt tcc tt-3′ |
| 5′-ctc ttg gcc cgg ttc tcc tcg tta-3′ | |
| Hmox | 5′-ttg tct gag gcc ttg aag ga-3′ |
| 5′-ctg ctt gtt gcg ctc tat ct-3′ | |
| PDGF-A | 5′-gag ata ccc cgg gag ttg at-3′ |
| 5′-aaa tga ccg tcc tgg tct tg-3′ | |
| PDGF-B | 5′-gat ctc tcg gaa cct cat cg-3′ |
| 5′-ggc ttc ttt cgc aca atc tc-3′ | |
| PDGF-C | 5′-gtg gag gaa att gtg cct gt-3′ |
| 5′-tcc aga gcc aca tca gtg ag-3′ | |
| PDGFR-α | 5′-cca cca gtg aag tct atg ag-3′ |
| 5′-acg cat tat cag agt cca cc-3′ | |
| PHD1 | 5′-tct acc cag gca atc tgg tc-3′ |
| 5′-gct agg ctg agg gag gaa gt-3′ | |
| PHD2 | 5′-cat acg cca caa ggt acg ca-3′ |
| 5′-aac tga gag gct gta ggt ga-3′ | |
| PHD3 | 5′-ggc cgc tgt atc acc tgt at-3′ |
| 5′-ttc tgc cct ttc ttc agc at-3′ | |
| PHD4 | 5′-acc tcc tgt cgc tac atg a-3′ |
| 5′-cac agt gcc ttc gag tgt cc-3′ | |
| Tbp | 5′-tct acc gtg aat ctt ggc tgt aaa-3′ |
| 5′-ttc tca tga tga ctg cag caa a-3′ | |
| VEGF-A | 5′-agt ccc atg aag tga tca agt tca-3′ |
| 5′-atc cgc atg atc tgc atg-3′ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egners, A.; Rezaei, M.; Kuzmanov, A.; Poitz, D.M.; Streichert, D.; Müller-Reichert, T.; Wielockx, B.; Breier, G. PHD3 Acts as Tumor Suppressor in Mouse Osteosarcoma and Influences Tumor Vascularization via PDGF-C Signaling. Cancers 2018, 10, 496. https://doi.org/10.3390/cancers10120496
Egners A, Rezaei M, Kuzmanov A, Poitz DM, Streichert D, Müller-Reichert T, Wielockx B, Breier G. PHD3 Acts as Tumor Suppressor in Mouse Osteosarcoma and Influences Tumor Vascularization via PDGF-C Signaling. Cancers. 2018; 10(12):496. https://doi.org/10.3390/cancers10120496
Chicago/Turabian StyleEgners, Antje, Maryam Rezaei, Aleksandar Kuzmanov, David M. Poitz, Doreen Streichert, Thomas Müller-Reichert, Ben Wielockx, and Georg Breier. 2018. "PHD3 Acts as Tumor Suppressor in Mouse Osteosarcoma and Influences Tumor Vascularization via PDGF-C Signaling" Cancers 10, no. 12: 496. https://doi.org/10.3390/cancers10120496
APA StyleEgners, A., Rezaei, M., Kuzmanov, A., Poitz, D. M., Streichert, D., Müller-Reichert, T., Wielockx, B., & Breier, G. (2018). PHD3 Acts as Tumor Suppressor in Mouse Osteosarcoma and Influences Tumor Vascularization via PDGF-C Signaling. Cancers, 10(12), 496. https://doi.org/10.3390/cancers10120496