BRCA1-Dependent Transcriptional Regulation: Implication in Tissue-Specific Tumor Suppression


{kind=link}
{kind=link}
{kind=link}
Abstract
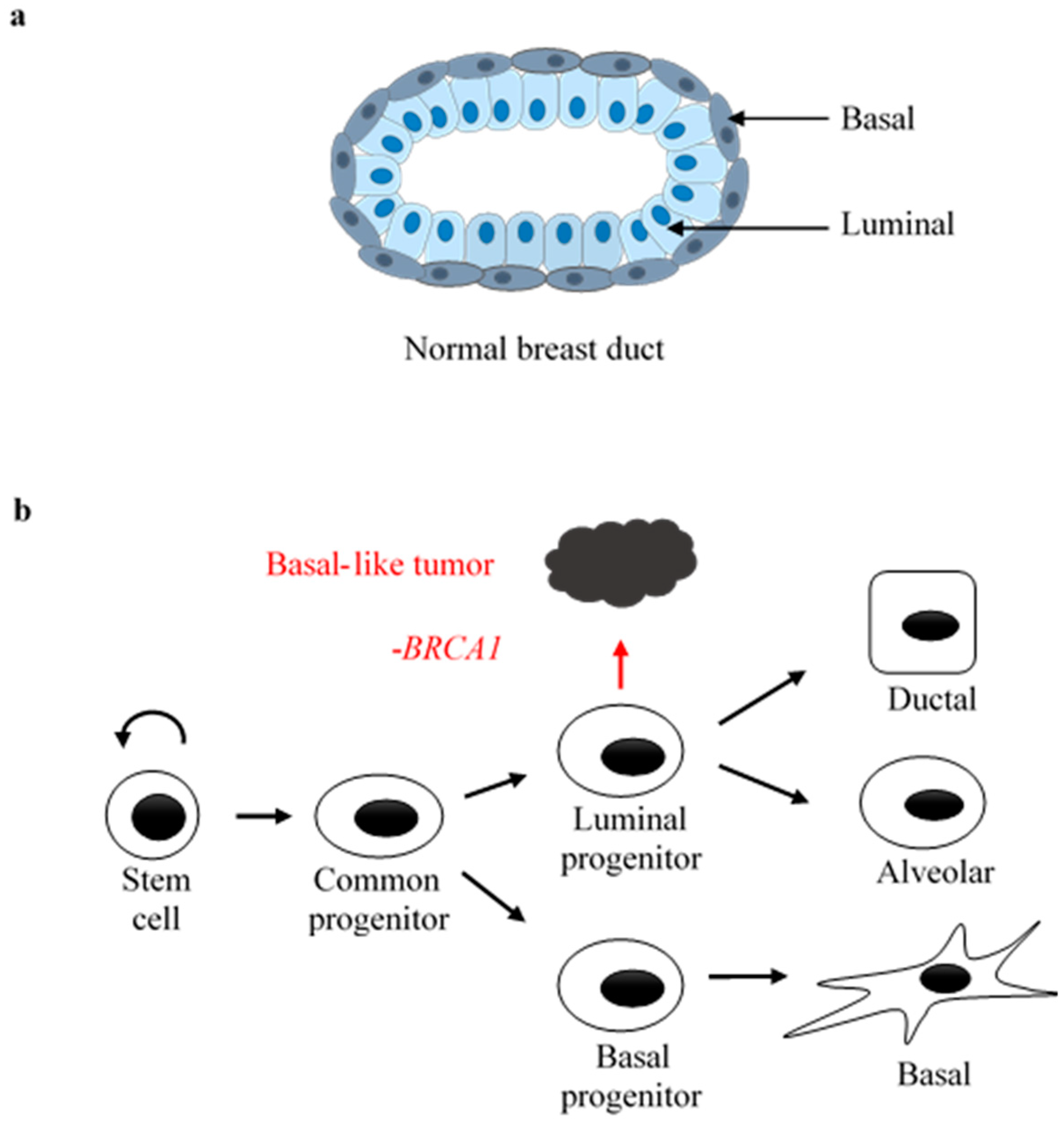
:1. Introduction
2. Functional Interaction between BRCA1 and Transcription Factors
2.1. BRCA1 with p53
2.2. BRCA1 with ERα
2.3. BRCA1 with COBRA1/NELF-B
3. The Roles of BRCA1 in Epigenetic Regulation
3.1. BRCA1 in DNA Methylation
3.2. BRCA1 in Histone Acetylation
3.3. BRCA1 in Histone Ubiquitination
4. BRCA1 in Chromatin Reorganization
5. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Whittemore, A.S.; Gong, G.; John, E.M.; McGuire, V.; Li, F.P.; Ostrow, K.L.; DiCioccio, R.; Felberg, A.; West, D.W. Prevalence of BRCA1 mutation carriers among US non-hispanic whites. Cancer Epidemiol. Biomark. 2004, 13, 2078–2083. [Google Scholar]
- John, E.M.; Miron, A.; Gong, G.; Phipps, A.I.; Felberg, A.; Li, F.P.; West, D.W.; Whittemore, A.S. Prevalence of pathogenic BRCA1 mutation carriers in 5 US racial/ethnic groups. JAMA 2007, 298, 2869–2876. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.; Pharoah, P.D.P.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.N.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.C.; Domchek, S.; Parmigiani, G.; Chen, S. Breast cancer risk among male BRCA1 and BRCA2 mutation carriers. J. Natl. Cancer Inst. 2007, 99, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- Phillips, K.A.; Nichol, K.; Ozcelik, H.; Knight, J.; Done, S.J.; Goodwin, P.J.; Andrulis, I.L. Frequency of p53 mutations in breast carcinomas from Ashkenazi Jewish carriers of BRCA1 mutations. J. Natl. Cancer Inst. 1999, 91, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.D.; Crossland, S.; Parker, G.; Osin, P.; Brooks, L.; Waller, J.; Philp, E.; Crompton, M.R.; Gusterson, B.A.; Allday, M.J.; et al. Novel p53 mutants selected in BRCA-associated tumours which dissociate transformation suppression from other wild-type p53 functions. Oncogene 1999, 18, 2451–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atchley, D.P.; Albarracin, C.T.; Lopez, A.; Valero, V.; Amos, C.I.; Gonzalez-Angulo, A.M.; Hortobagyi, G.N.; Arun, B.K. Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J. Clin. Oncol. 2008, 26, 4282–4288. [Google Scholar] [CrossRef] [PubMed]
- Brekelmans, C.T.M.; Tilanus-Linthorst, M.M.A.; Seynaeve, C.; Van der Ouweland, A.; Menke-Pluymers, M.B.E.; Bartels, C.C.M.; Kriege, M.; van Geel, A.; Burger, C.W.; Eggermont, A.M.M.; et al. Tumour characteristics, survival and prognostic factors of hereditary breast cancer from BRCA2-, BRCA1- and non-BRCA1/2 families as compared to sporadic breast cancer cases. Eur. J. Cancer 2007, 43, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, S.R.; Reis-Filho, J.S.; Fulford, L.; Penault-Llorca, F.; van der Vjiver, M.; Parry, S.; Bishop, T.; Benitez, J.; Rivas, C.; Bignon, Y.J.; et al. Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin. Cancer Res. 2005, 11, 5175–5180. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Stefansson, I.M.; Chappuis, P.O.; Begin, L.R.; Goffin, J.R.; Wong, N.; Trudel, M.; Akslen, L.A. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 2003, 95, 1482–1485. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M.; Loman, N.; Borg, A.; Isola, J. Cytokeratin 5/14-positive breast cancer: True basal phenotype confined to BRCA1 tumors. Mod. Pathol. 2005, 18, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: Results from the consortium of investigators of modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. 2012, 21, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Bitler, B.G.; Watson, Z.L.; Wheeler, L.J.; Behbakht, K. PARP inhibitors: Clinical utility and possibilities of overcoming resistance. Gynecol. Oncol. 2017, 147, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Jhan, J.R.; Andrechek, E.R. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics 2017, 18, 1595–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godet, I.; Gilkes, D.M. BRCA1 and BRCA2 mutations and treatment strategies for breast cancer. Integr. Cancer Sci. Ther. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E.; Stingl, J. Mammary stem cells and the differentiation hierarchy: Current status and perspectives. Genes Dev. 2014, 28, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Brisken, C.; O'Malley, B. Hormone action in the mammary gland. Cold Spring Harb. Perspect. Biol. 2010, 2, a003178. [Google Scholar] [CrossRef]
- Macias, H.; Hinck, L. Mammary gland development. Wiley Interdiscip. Rev. Dev. Boil. 2012, 1, 533–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Proia, T.A.; Keller, P.J.; Gupta, P.B.; Klebba, I.; Jones, A.D.; Sedic, M.; Gilmore, H.; Tung, N.; Naber, S.P.; Schnitt, S.; et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 2011, 8, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.J.; Zhang, X.W.; Chiang, H.C.; Jahid, M.J.; Wang, Y.; Garza, P.; April, C.; Salathia, N.; Banerjee, T.; Alenazi, F.S.; et al. Genetic suppression reveals DNA repair-independent antagonism between BRCA1 and COBRA1 in mammary gland development. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Nolan, E.; Vaillant, F.; Branstetter, D.; Pal, B.; Giner, G.; Whitehead, L.; Lok, S.W.; Mann, G.B.; Rohrbach, K.; Huang, L.Y.; et al. RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat. Med. 2016, 22, 933. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, G.; Geyer, F.C.; Magnay, F.A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; Mackay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010, 7, 403–417. [Google Scholar] [CrossRef]
- Powell, S.N.; Kachnic, L.A. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 2003, 22, 5784–5791. [Google Scholar] [CrossRef] [Green Version]
- Silver, D.P.; Livingston, D.M. Mechanisms of BRCA1 tumor suppression. Cancer Discov. 2012, 2, 679–684. [Google Scholar] [CrossRef]
- Walsh, C.S. Two decades beyond BRCA1/2: Homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol. Oncol. 2015, 137, 343–350. [Google Scholar] [CrossRef]
- Mullan, P.B.; Quinn, J.E.; Harkin, D.P. The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene 2006, 25, 5854–5863. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.X. Brca1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Bade, S.; Le Guillou, M.; Burke, K.; Reed, R.; Bowman-Colin, C.; Su, Y.; Ting, D.T.; Polyak, K.; Richardson, A.L.; et al. BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat. Commun. 2014, 5, 5496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, G.M.; Janknecht, R.; Ruffner, H.; Hunter, T.; Verma, I.M. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc. Natl. Acad. Sci. USA 2000, 97, 1020–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Hu, Y.F.; Zhong, H.; Nye, A.C.; Belmont, A.S.; Li, R. BRCA1-induced large-scale chromatin unfolding and allele-specific effects of cancer-predisposing mutations. J. Cell Boil. 2001, 155, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.F.; Schlegel, B.P.; Nakajima, T.; Wolpin, E.S.; Parvin, J.D. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat Genet. 1998, 19, 254–256. [Google Scholar] [CrossRef]
- Yarden, R.I.; Brody, L.C. BRCA1 interacts with components of the histone deacetylase complex. Proc. Natl. Acad. Sci. USA 1999, 96, 4983–4988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Somasundaram, K.; Peng, Y.; Tian, H.; Zhang, H.; Bi, D.; Weber, B.L.; El-Deiry, W.S. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene 1998, 16, 1713–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starita, L.M.; Parvin, J.D. The multiple nuclear functions of BRCA1: Transcription, ubiquitination and DNA repair. Curr. Opin. Cell Biol. 2003, 15, 345–350. [Google Scholar] [CrossRef]
- Monteiro, A.N.; August, A.; Hanafusa, H. Evidence for a transcriptional activation function of BRCA1 c-terminal region. Proc. Natl. Acad. Sci. USA 1996, 93, 13595–13599. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.F.; Miyake, T.; Ye, Q.; Li, R. Characterization of a novel trans-activation domain of BRCA1 that functions in concert with the BRCA1 c-terminal (BRCT) domain. J. Boil. Chem. 2000, 275, 40910–40915. [Google Scholar] [CrossRef] [PubMed]
- Haile, D.T.; Parvin, J.D. Activation of transcription in vitro by the BRCA1 carboxyl-terminal domain. J. Boil. Chem. 1999, 274, 2113–2117. [Google Scholar] [CrossRef]
- Scully, R.; Anderson, S.F.; Chao, D.M.; Wei, W.; Ye, L.; Young, R.A.; Livingston, D.M.; Parvin, J.D. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 5605–5610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouchi, T.; Monteiro, A.N.; August, A.; Aaronson, S.A.; Hanafusa, H. BRCA1 regulates p53-dependent gene expression. Proc. Natl. Acad. Sci. USA 1998, 95, 2302–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.; Ma, Y.X.; Wang, C.; Yuan, R.Q.; Meng, Q.; Wang, J.A.; Erdos, M.; Goldberg, I.D.; Webb, P.; Kushner, P.J.; et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene 2001, 20, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhang, H.; Kajino, K.; Greene, M.I. BRCA1 binds c-Myc and inhibits its transcriptional and transforming activity in cells. Oncogene 1998, 17, 1939–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Pan, H.; Li, S.; Flesken-Nikitin, A.; Chen, P.L.; Boyer, T.G.; Lee, W.H. Sequence-specific transcriptional corepressor function for BRCA1 through a novel zinc finger protein, zbrk1. Mol. Cell 2000, 6, 757–768. [Google Scholar] [CrossRef]
- Tkocz, D.; Crawford, N.T.; Buckley, N.E.; Berry, F.B.; Kennedy, R.D.; Gorski, J.J.; Harkin, D.P.; Mullan, P.B. BRCA1 and GATA3 corepress FOXC1 to inhibit the pathogenesis of basal-like breast cancers. Oncogene 2012, 31, 3667–3678. [Google Scholar] [CrossRef]
- Ouchi, T.; Lee, S.W.; Ouchi, M.; Aaronson, S.A.; Horvath, C.M. Collaboration of signal transducer and activator of transcription 1 (STAT1) and BRCA1 in differential regulation of IFN-gamma target genes. Proc. Natl. Acad. Sci. USA 2000, 97, 5208–5213. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y. BRCA1, hormone, and tissue-specific tumor suppression. Int. J. Boil. Sci. 2009, 5, 20–27. [Google Scholar] [CrossRef]
- Rosen, E.M.; Fan, S.; Ma, Y. BRCA1 regulation of transcription. Cancer Lett. 2006, 236, 175–185. [Google Scholar] [CrossRef]
- Chai, Y.L.; Cui, J.; Shao, N.; Shyam, E.; Reddy, P.; Rao, V.N. The second BRCT domain of BRCA1 proteins interacts with p53 and stimulates transcription from the p21WAF1/CIP1 promoter. Oncogene 1999, 18, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLachlan, T.K.; Takimoto, R.; El-Deiry, W.S. BRCA1 directs a selective p53-dependent transcriptional response towards growth arrest and DNA repair targets. Mol. Cell. Boil. 2002, 22, 4280–4292. [Google Scholar] [CrossRef]
- Ongusaha, P.P.; Ouchi, T.; Kim, K.T.; Nytko, E.; Kwak, J.C.; Duda, R.B.; Deng, C.X.; Lee, S.W. BRCA1 shifts p53-mediated cellular outcomes towards irreversible growth arrest. Oncogene 2003, 22, 3749–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somasundaram, K.; MacLachlan, T.K.; Burns, T.F.; Sgagias, M.; Cowan, K.H.; Weber, B.L.; el-Deiry, W.S. BRCA1 signals ARF-dependent stabilization and coactivation of p53. Oncogene 1999, 18, 6605–6614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arizti, P.; Fang, L.; Park, I.; Yin, Y.; Solomon, E.; Ouchi, T.; Aaronson, S.A.; Lee, S.W. Tumor suppressor p53 is required to modulate BRCA1 expression. Mol. Cell. Boil. 2000, 20, 7450–7459. [Google Scholar] [CrossRef]
- MacLachlan, T.K.; Dash, B.C.; Dicker, D.T.; El-Deiry, W.S. Repression of BRCA1 through a feedback loop involving p53. J. Boil. Chem. 2000, 275, 31869–31875. [Google Scholar] [CrossRef] [PubMed]
- Hakem, R.; delaPompa, J.L.; Sirard, C.; Mo, R.; Woo, M.; Hakem, A.; Wakeham, A.; Potter, J.; Reitmair, A.; Billia, F.; et al. The tumor suppressor gene BRCA1 is required for embryonic cellular proliferation in the mouse. Cell 1996, 85, 1009–1023. [Google Scholar] [CrossRef]
- Hakem, R.; de la Pompa, J.L.; Elia, A.; Potter, J.; Mak, T.W. Partial rescue of BRCA1 (5-6) early embryonic lethality by p53 or p21 null mutation. Nat. Genet. 1997, 16, 298–302. [Google Scholar] [CrossRef]
- Xu, X.; Qiao, W.; Linke, S.P.; Cao, L.; Li, W.M.; Furth, P.A.; Harris, C.C.; Deng, C.X. Genetic interactions between tumor suppressors BRCA1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat. Genet. 2001, 28, 266–271. [Google Scholar] [CrossRef]
- Ludwig, T.; Chapman, D.L.; Papaioannou, V.E.; Efstratiadis, A. Targeted mutations of breast cancer susceptibility gene homologs in mice: phenotypes of BRCA1, BRCA2, BRCA1/BRCA2, BRCA1/p53, and BRCA2/p53 nullizygous embryos. Genes Dev. 1997, 11, 1226–1241. [Google Scholar] [CrossRef]
- Cao, L.; Li, W.; Kim, S.; Brodie, S.G.; Deng, C.X. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the BRCA1 full-length isoform. Genes Dev. 2003, 17, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.L.; Wagner, K.U.; Larson, D.; Weaver, Z.; Li, C.L.; Ried, T.; Hennighausen, L.; Wynshaw-Boris, A.; Deng, C.X. Conditional mutation of BRCA1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat. Genet. 1999, 22, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Holstege, H.; van der Gulden, H.; Treur-Mulder, M.; Zevenhoven, J.; Velds, A.; Kerkhoven, R.M.; van Vliet, M.H.; Wessels, L.F.; Peterse, J.L.; et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12111–12116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenblatt, M.S.; Chappuis, P.O.; Bond, J.P.; Hamel, N.; Foulkes, W.D. TP53 mutations in breast cancer associated with BRCA1 or BRCA2 germ-line mutations: Distinctive spectrum and structural distribution. Cancer Res. 2001, 61, 4092–4097. [Google Scholar] [PubMed]
- Hosey, A.M.; Gorski, J.J.; Murray, M.M.; Quinn, J.E.; Chung, W.Y.; Stewart, G.E.; James, C.R.; Farragher, S.M.; Mulligan, J.M.; Scott, A.N.; et al. Molecular basis for estrogen receptor alpha deficiency in BRCA1-linked breast cancer. J. Natl. Cancer Inst. 2007, 99, 1683–1694. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Wang, J.; Yuan, R.; Ma, Y.; Meng, Q.; Erdos, M.R.; Pestell, R.G.; Yuan, F.; Auborn, K.J.; Goldberg, I.D.; et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science 1999, 284, 1354–1356. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Annab, L.A.; Afshari, C.A.; Lee, W.H.; Boyer, T.G. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 9587–9592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanstein, B.; Eckner, R.; DiRenzo, J.; Halachmi, S.; Liu, H.; Searcy, B.; Kurokawa, R.; Brown, M. p300 is a component of an estrogen receptor coactivator complex. Proc. Natl. Acad. Sci. USA 1996, 93, 11540–11545. [Google Scholar] [CrossRef] [PubMed]
- Vo, N.; Goodman, R.H. CREB-binding protein and p300 in transcriptional regulation. J. Boil. Chem. 2001, 276, 13505–13508. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Ma, Y.X.; Wang, C.; Yuan, R.Q.; Meng, Q.; Wang, J.A.; Erdos, M.; Goldberg, I.D.; Webb, P.; Kushner, P.J.; et al. p300 modulates the BRCA1 inhibition of estrogen receptor activity. Cancer Res. 2002, 62, 141–151. [Google Scholar]
- Ma, Y.; Fan, S.; Hu, C.; Meng, Q.; Fuqua, S.A.; Pestell, R.G.; Tomita, Y.A.; Rosen, E.M. BRCA1 regulates acetylation and ubiquitination of estrogen receptor-alpha. Mol. Endocrinol. 2010, 24, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Fan, S.; Rosen, E.M. Regulation of the estrogen-inducible gene expression profile by the breast cancer susceptibility gene BRCA1. Endocrinology 2005, 146, 2031–2047. [Google Scholar] [CrossRef] [PubMed]
- Marquis, S.T.; Rajan, J.V.; Wynshaw-Boris, A.; Xu, J.; Yin, G.Y.; Abel, K.J.; Weber, B.L.; Chodosh, L.A. The developmental pattern of BRCA1 expression implies a role in differentiation of the breast and other tissues. Nat. Genet. 1995, 11, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Spillman, M.A.; Bowcock, A.M. BRCA1 and BRCA2 mRNA levels are coordinately elevated in human breast cancer cells in response to estrogen. Oncogene 1996, 13, 1639–1645. [Google Scholar] [PubMed]
- Liu, S.; Ginestier, C.; Charafe-Jauffret, E.; Foco, H.; Kleer, C.G.; Merajver, S.D.; Dontu, G.; Wicha, M.S. BRCA1 regulates human mammary stem/progenitor cell fate. Proc. Natl. Acad. Sci. USA 2008, 105, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Shehata, M.; Teschendorff, A.; Sharp, G.; Novcic, N.; Russell, I.A.; Avril, S.; Prater, M.; Eirew, P.; Caldas, C.; Watson, C.J.; et al. Phenotypic and functional characterisation of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. BCR 2012, 14, R134. [Google Scholar] [CrossRef] [PubMed]
- Giraddi, R.R.; Shehata, M.; Gallardo, M.; Blasco, M.A.; Simons, B.D.; Stingl, J. Stem and progenitor cell division kinetics during postnatal mouse mammary gland development. Nat. Commun. 2015, 6, 8487. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zurrer-Hardi, U.; Bell, G.; et al. Slug and sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef]
- Amleh, A.; Nair, S.J.; Sun, J.; Sutherland, A.; Hasty, P.; Li, R. Mouse cofactor of BRCA1 (Cobra1) is required for early embryogenesis. PLoS ONE 2009, 4, e5034. [Google Scholar] [CrossRef]
- Chiang, H.C.; Zhang, X.W.; Zhao, X.Y.; Zhang, C.; Chen, J.; Garza, P.; Smith, S.; Ludwig, T.; Baer, R.J.; Li, R.; et al. Gene-specific genetic complementation between Brca1 and Cobra1 during mouse mammary gland development. Sci. Rep. UK 2018, 8. [Google Scholar] [CrossRef]
- Pan, H.; Qin, K.; Guo, Z.; Ma, Y.; April, C.; Gao, X.; Andrews, T.G.; Bokov, A.; Zhang, J.; Chen, Y.; et al. Negative elongation factor controls energy homeostasis in cardiomyocytes. Cell Rep. 2014, 7, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Adelman, K.; Lis, J.T. Promoter-proximal pausing of RNA polymerase II: Emerging roles in metazoans. Nat. Rev. Genet. 2012, 13, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Aiyar, S.E.; Cho, H.; Lee, J.; Li, R. Concerted transcriptional regulation by BRCA1 and COBRA1 in breast cancer cells. Int. J. Boil. Sci. 2007, 3, 486–492. [Google Scholar] [CrossRef]
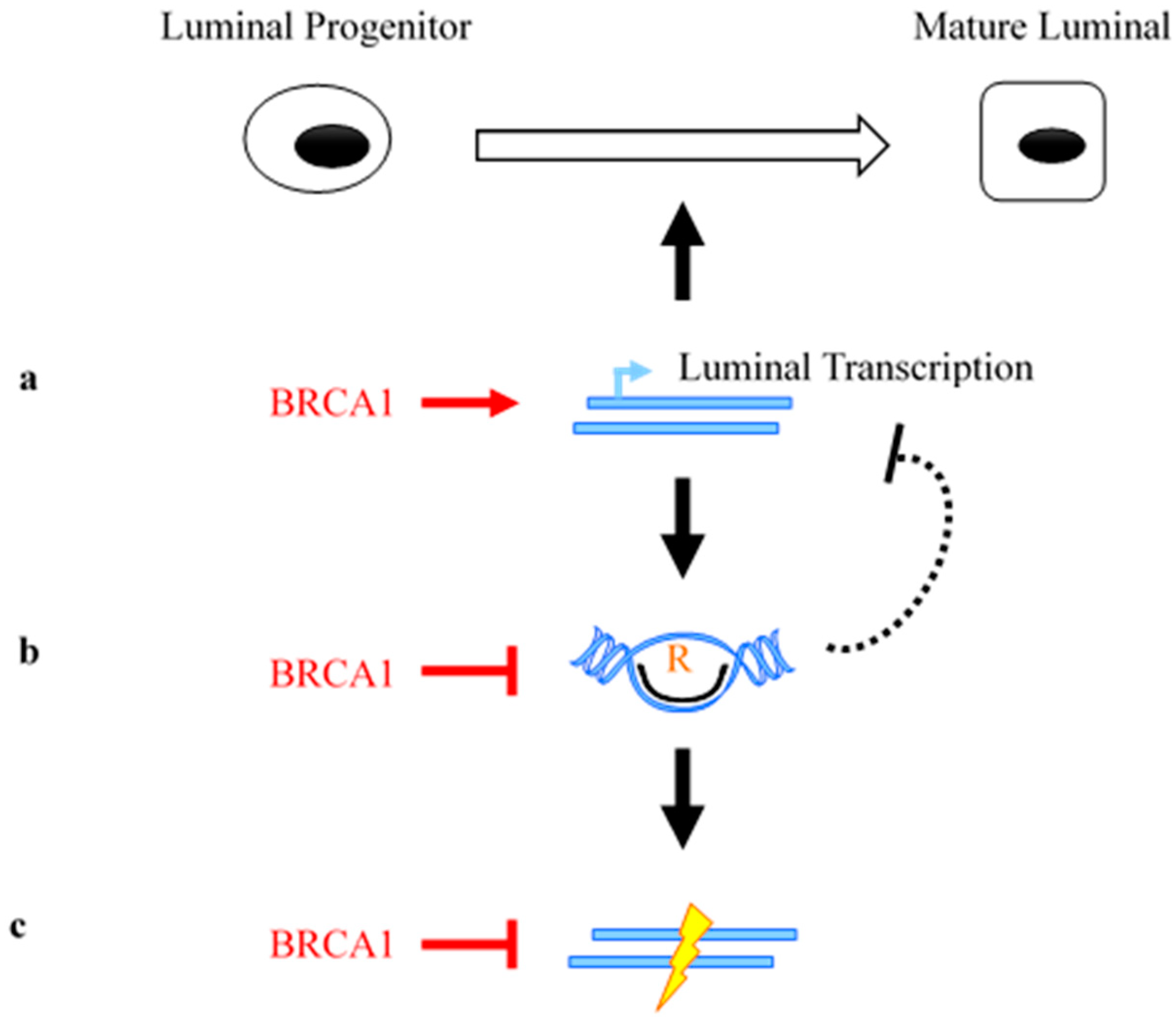
- Zhang, X.; Chiang, H.C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M.; et al. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017, 8, 15908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef] [Green Version]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Grewal, S.I.; Moazed, D. Heterochromatin and epigenetic control of gene expression. Science 2003, 301, 798–802. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics-UK 2009, 1, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.H.; Yazici, H.; Wu, H.C.; Terry, M.B.; Gonzalez, K.; Qu, M.X.; Dalay, N.; Santella, R.M. Aberrant promoter hypermethylation and genomic hypomethylation in tumor, adjacent normal tissues and blood from breast cancer patients. Anticancer Res. 2010, 30, 2489–2496. [Google Scholar]
- Torano, E.G.; Petrus, S.; Fernandez, A.F.; Fraga, M.F. Global DNA hypomethylation in cancer: Review of validated methods and clinical significance. Clin. Chem. Lab. Med. 2012, 50, 1733–1742. [Google Scholar] [CrossRef]
- Vasilatos, S.N.; Broadwater, G.; Barry, W.T.; Baker, J.C.; Lem, S.; Dietze, E.C.; Bean, G.R.; Bryson, A.D.; Pilie, P.G.; Goldenberg, V.; et al. CpG island tumor suppressor promoter methylation in non-BRCA-associated early mammary carcinogenesis. Cancer Epidemiol. Biomark. 2009, 18, 901–914. [Google Scholar] [CrossRef]
- Suijkerbuijk, K.P.M.; Fackler, M.J.; Sukumar, S.; van Gils, C.H.; van Laar, T.; van der Wall, E.; Vooijs, M.; van Diest, P.J. Methylation is less abundant in BRCA1-associated compared with sporadic breast cancer. Ann. Oncol. 2008, 19, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Shukla, V.; Coumoul, X.; Lahusen, T.; Wang, R.H.; Xu, X.L.; Vassilopoulos, A.; Xiao, C.Y.; Lee, M.H.; Man, Y.G.; Ouchi, M.; et al. BRCA1 affects global DNA methylation through regulation of DNMT1. Cell Res. 2010, 20, 1201–1215. [Google Scholar] [CrossRef] [Green Version]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and function of mammalian DNA methyltransferases. Chembiochem 2011, 12, 206–222. [Google Scholar] [CrossRef]
- Filipponi, D.; Muller, J.; Emelyanov, A.; Bulavin, D.V. Wip1 controls global heterochromatin silencing via ATM/BRCA1-dependent DNA methylation. Cancer Cell 2013, 24, 528–541. [Google Scholar] [CrossRef]
- Li, J.; Yang, Y.; Peng, Y.; Austin, R.J.; van Eyndhoven, G.; Nguyen, K.C.Q.; Gabriele, T.; McCurrach, M.E.; Marks, J.R.; Hoey, T.; et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat. Genet. 2002, 31, 133–134. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Demidov, O.N.; Saito, S.; Kauraniemi, P.; Phillips, C.; Amundson, S.A.; Ambrosino, C.; Sauter, G.; Nebreda, A.R.; Anderson, C.W.; et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat. Genet. 2002, 31, 210–215. [Google Scholar] [CrossRef]
- Beck, C.R.; Garcia-Perez, J.L.; Badge, R.M.; Moran, J.V. LINE-1 elements in structural variation and disease. Annu. Rev. Genom. Hum. Genet. 2011, 12, 187–215. [Google Scholar] [CrossRef]
- Le Guezennec, X.; Brichkina, A.; Huang, Y.F.; Kostromina, E.; Han, W.P.; Bulavin, D.V. Wip1-dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metab. 2012, 16, 68–80. [Google Scholar] [CrossRef]
- Shreeram, S.; Demidov, O.N.; Hee, W.K.; Yamaguchi, H.; Onishi, N.; Kek, C.; Timofeev, O.N.; Dudgeon, C.; Fornace, A.J.; Anderson, C.W.; et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol. Cell 2006, 23, 757–764. [Google Scholar] [CrossRef]
- Choi, J.D.; Park, M.A.; Lee, J.S. Suppression and recovery of BRCA1-mediated transcription by HP1gamma via modulation of promoter occupancy. Nucleic Acids Res. 2012, 40, 11321–11338. [Google Scholar] [CrossRef]
- Nabel, C.S.; Jia, H.; Ye, Y.; Shen, L.; Goldschmidt, H.L.; Stivers, J.T.; Zhang, Y.; Kohli, R.M. AID/APOBECdeaminases disfavor modified cytosines implicated in DNA demethylation. Nat. Chem. Boil. 2012, 8, 751–758. [Google Scholar] [CrossRef]
- Li, D.; Bi, F.F.; Cao, J.M.; Cao, C.; Liu, B.; Yang, Q. Regulation of DNA methyltransferase 1 transcription in BRCA1-mutated breast cancer: A novel crosstalk between E2F1 motif hypermethylation and loss of histone h3 lysine 9 acetylation. Mol. Cancer 2014, 13. [Google Scholar] [CrossRef]
- Gong, C.; Fujino, K.; Monteiro, L.J.; Gomes, A.R.; Drost, R.; Davidson-Smith, H.; Takeda, S.; Khoo, U.S.; Jonkers, J.; Sproul, D.; et al. FOXA1 repression is associated with loss of BRCA1 and increased promoter methylation and chromatin silencing in breast cancer. Oncogene 2015, 34, 5012–5024. [Google Scholar] [CrossRef]
- Gong, C.; Yao, S.; Gomes, A.R.; Man, E.P.S.; Lee, H.J.; Gong, G.; Chang, S.; Kim, S.B.; Fujino, K.; Kim, S.W.; et al. BRCA1 positively regulates FOXO3 expression by restricting FOXO3 gene methylation and epigenetic silencing through targeting EZH2 in breast cancer. Oncogenesis 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Wang, L.; Zeng, X.Z.; Chen, S.; Ding, L.Y.; Zhong, J.; Zhao, J.C.; Wang, L.G.; Sarver, A.; Koller, A.; Zhi, J.Z.; et al. BRCA1 is a negative modulator of the PRC2 complex. EMBO J. 2013, 32, 1584–1597. [Google Scholar] [CrossRef] [Green Version]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef]
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [Google Scholar] [CrossRef]
- Chang, S.; Wang, R.H.; Akagi, K.; Kim, K.A.; Martin, B.K.; Cavallone, L.; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab); Haines, D.C.; Basik, M.; Mai, P.; et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat. Med. 2011, 17, 1275–1282. [Google Scholar] [CrossRef] [Green Version]
- Lovelock, P.K.; Spurdle, A.B.; Mok, M.T.S.; Farrugia, D.J.; Lakhani, S.R.; Healey, S.; Arnold, S.; Buchanan, D.; Couch, F.J.; Henderson, B.R.; et al. Identification of BRCA1 missense substitutions that confer partial functional activity: moderate risk variants? Breast Cancer Res. 2007, 9. [Google Scholar] [CrossRef]
- Cao, J.; Yan, Q. Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Front. Oncol. 2012, 2, 26. [Google Scholar] [CrossRef]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, F.; Ohta, T. The ring heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Boil. Chem. 2001, 276, 14537–14540. [Google Scholar] [CrossRef] [PubMed]
- Mallery, D.L.; Vandenberg, C.J.; Hiom, K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. Embo J. 2002, 21, 6755–6762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalb, R.; Mallery, D.L.; Larkin, C.; Huang, J.T.J.; Hiom, K. BRCA2 is a histone-H2A-specific ubiquitin ligase. Cell Rep. 2014, 8, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Thakar, A.; Parvin, J.D.; Zlatanova, J. BRCA1/BARD1 E3 ubiquitin ligase can modify histones H2A and H2B in the nucleosome particle. J. Biomol. Struct. Dyn. 2010, 27, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Boil. 2016, 23, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Chan, D.W.; Jung, S.Y.; Chen, Y.; Qin, J.; Wang, Y. The histone variant macroH2A1 is a BRCA1 ubiquitin ligase substrate. Cell Rep. 2017, 19, 1758–1766. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, A.A.; Affar, E.B.; Heine, G.F.; Shi, Y.; Parvin, J.D. A mechanism for transcriptional repression dependent on the BRCA1 E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2007, 104, 6614–6619. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Pao, G.M.; Huynh, A.M.; Suh, H.; Tonnu, N.; Nederlof, P.M.; Gage, F.H.; Verma, I.M. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 2011, 477, 179-U176. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Hu, Y.F.; Hao, Z.L.; Li, R. Chromatin remodeling and activation of chromosomal DNA replication by an acidic transcriptional activation domain from BRCA1. Genes Dev. 1999, 13, 637–642. [Google Scholar] [CrossRef] [Green Version]
- Miyake, T.; Hu, Y.F.; Yu, D.S.; Li, R. A functional comparison of BRCA1 c-terminal domains in transcription activation and chromatin remodeling. J. Boil. Chem. 2000, 275, 40169–40173. [Google Scholar] [CrossRef] [PubMed]
- Bochar, D.A.; Wang, L.; Beniya, H.; Kinev, A.; Xue, Y.; Lane, W.S.; Wang, W.; Kashanchi, F.; Shiekhattar, R. BRCA1 is associated with a human SWI/SNF-related complex: Linking chromatin remodeling to breast cancer. Cell 2000, 102, 257–265. [Google Scholar] [CrossRef]
- Kononenko, A.V.; Bansal, R.; Lee, N.C.; Grimes, B.R.; Masumoto, H.; Earnshaw, W.C.; Larionov, V.; Kouprina, N. A portable BRCA1-HAC (human artificial chromosome) module for analysis of BRCA1 tumor suppressor function. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [PubMed]
- Sollier, J.; Stork, C.T.; Garcia-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Gascard, P.; Bilenky, M.; Sigaroudinia, M.; Zhao, J.; Li, L.; Carles, A.; Delaney, A.; Tam, A.; Kamoh, B.; Cho, S.; et al. Epigenetic and transcriptional determinants of the human breast. Nat. Commun. 2015, 6, 6351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasanuma, H.; Tsuda, M.; Morimoto, S.; Saha, L.K.; Rahman, M.M.; Kiyooka, Y.; Fujiike, H.; Cherniack, A.D.; Itou, J.; Moreu, E.C.; et al. BRCA1 ensures genome integrity by eliminating estrogen-induced pathological topoisomerase II-DNA complexes. Proc. Natl. Acad. Sci. USA 2018, 115, E10642–E10651. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Li, R. BRCA1-Dependent Transcriptional Regulation: Implication in Tissue-Specific Tumor Suppression. Cancers 2018, 10, 513. https://doi.org/10.3390/cancers10120513
Zhang X, Li R. BRCA1-Dependent Transcriptional Regulation: Implication in Tissue-Specific Tumor Suppression. Cancers. 2018; 10(12):513. https://doi.org/10.3390/cancers10120513
Chicago/Turabian StyleZhang, Xiaowen, and Rong Li. 2018. "BRCA1-Dependent Transcriptional Regulation: Implication in Tissue-Specific Tumor Suppression" Cancers 10, no. 12: 513. https://doi.org/10.3390/cancers10120513
APA StyleZhang, X., & Li, R. (2018). BRCA1-Dependent Transcriptional Regulation: Implication in Tissue-Specific Tumor Suppression. Cancers, 10(12), 513. https://doi.org/10.3390/cancers10120513