Obesity-Linked Cancers: Current Knowledge, Challenges and Limitations in Mechanistic Studies and Rodent Models



{kind=link}
{kind=link}
Abstract
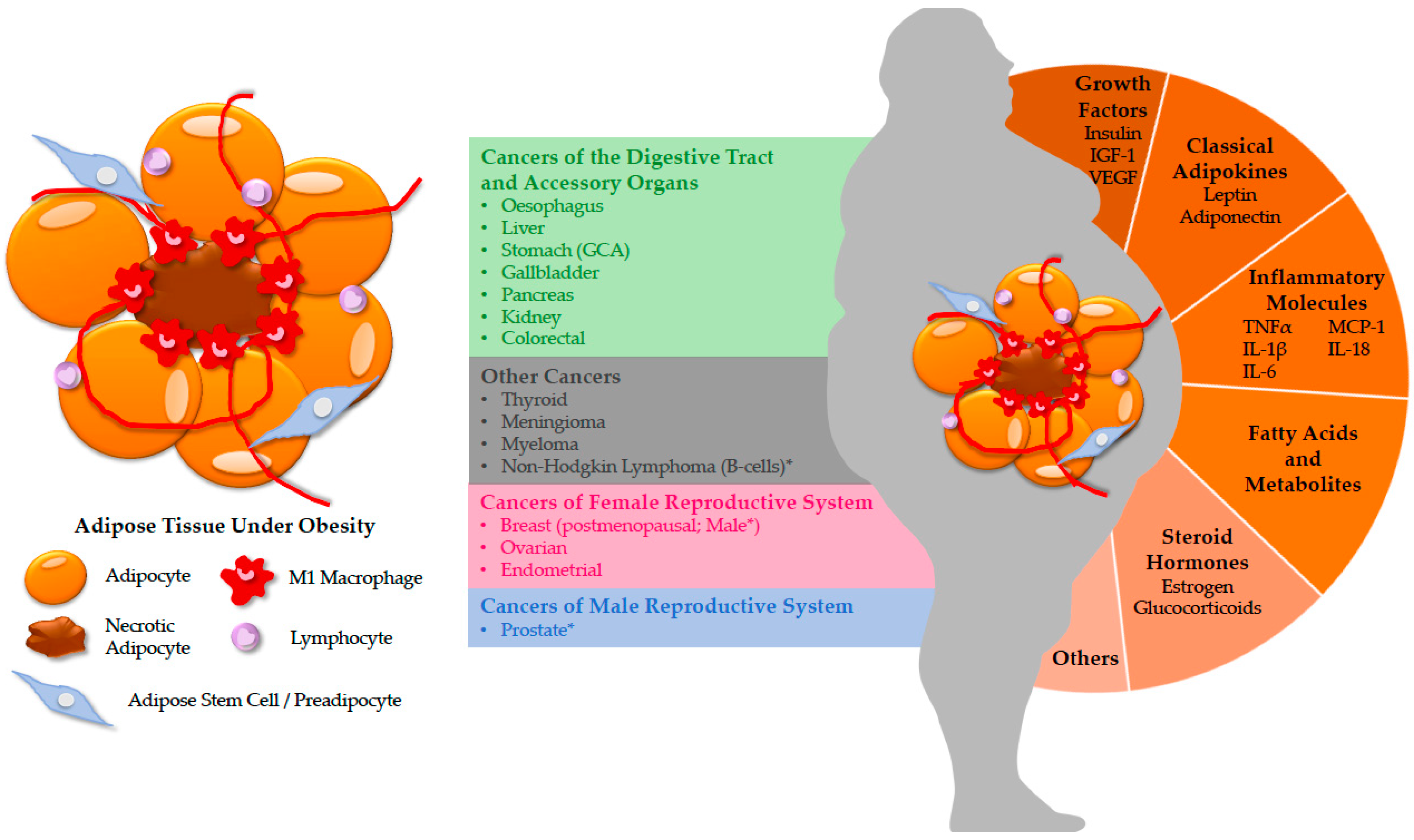
:1. Introduction
2. Cancers in the Digestive Tract and Accessory Organs
2.1. Gastric Cancer
2.2. Liver Cancer
2.3. Pancreatic Cancer
3. Cancers in the Female Reproductive System
Breast Cancer
4. Cancers in the Male Reproductive System
Prostate Cancer
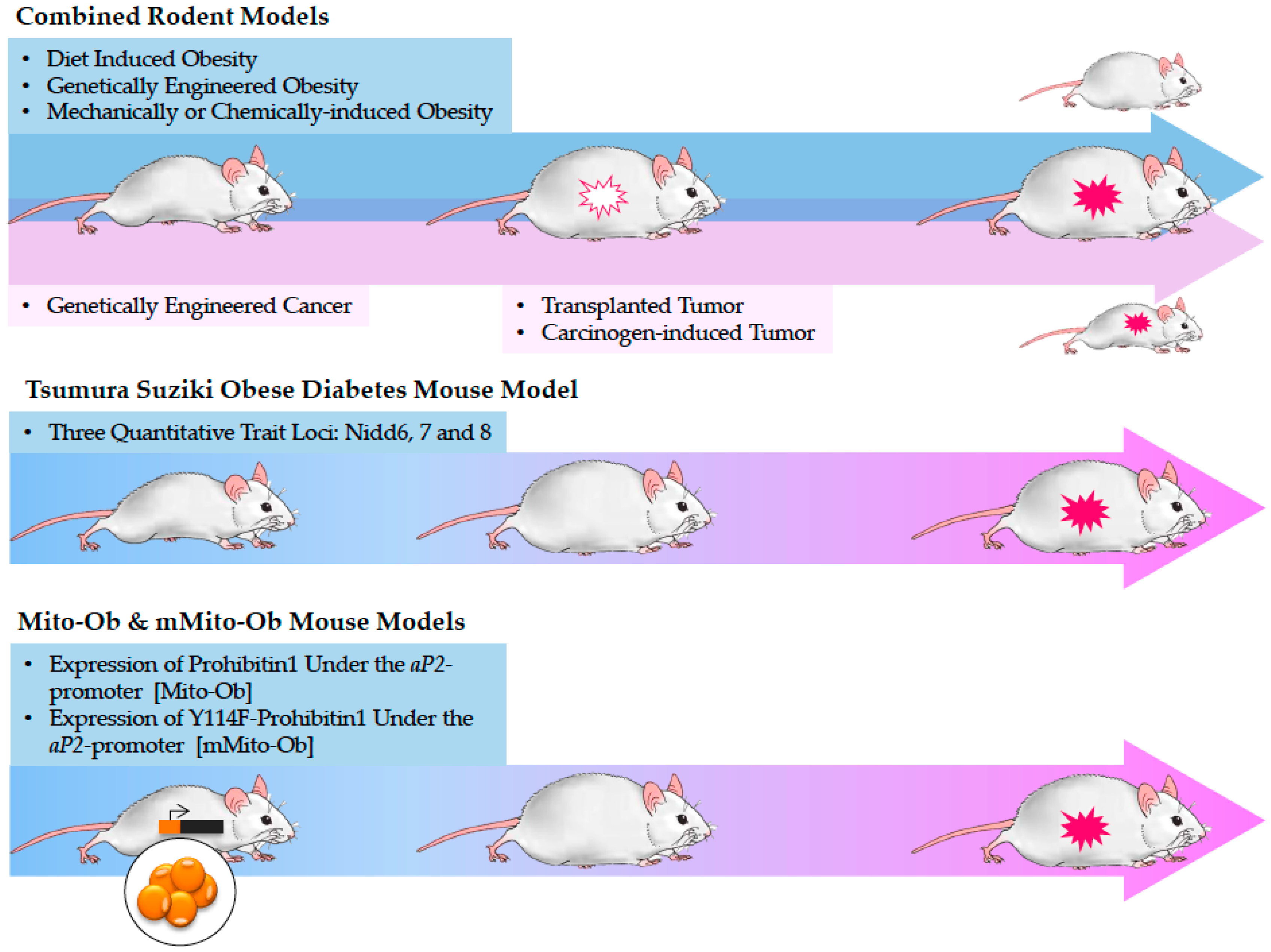
5. Current Approaches to Study Obesity-Linked Cancers and Their Limitations
5.1. Tsumura Suziki Obese Diabetes (TSOD) Mouse Model
5.2. Mito-Ob and mMito-Ob Mouse Models
6. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, P.A.G.; Stevens, G.A.; Ezzati, P.M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R.; et al. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36–46. [Google Scholar] [CrossRef]
- Vos, T.; Abajobir, A.A.; Abate, K.H.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abdulkader, R.S.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Lond. Engl. 2017, 390, 1211–1259. [Google Scholar] [CrossRef]
- Frayn, K.N. Obesity and metabolic disease: Is adipose tissue the culprit? Proc. Nutr. Soc. 2005, 64, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Basen-Engquist, K.; Chang, M. Obesity and Cancer Risk: Recent Review and Evidence. Curr. Oncol. Rep. 2011, 13, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. International Agency for Research on Cancer Handbook Working Group. Body fatness and cancer—Viewpoint of the IARC working group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.B.; Thomas, C.C.; Henley, S.J.; Massetti, G.M.; Galuska, D.A.; Agurs-Collins, T.; Puckett, M.; Richardson, L.C. Vital Signs: Trends in Incidence of Cancers Associated with Overweight and Obesity—United States, 2005–2014. MMWR Morbidity Mortality Wkly. Rep. 2017, 66, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; McPherson, K.; Marsh, T.; Gortmaker, S.L.; Brown, M. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet 2011, 378, 815–825. [Google Scholar] [CrossRef]
- Office of the Surgeon General (US); Office of Disease Prevention and Health Promotion (US); Centers for Disease Control and Prevention (US); National Institutes of Health (US). The Surgeon General’s Call To Action To Prevent and Decrease Overweight and Obesity; Publications and Reports of the Surgeon General; Office of the Surgeon General (US): Rockville, MD, USA, 2001.
- Castaner, O.; Goday, A.; Park, Y.-M.; Lee, S.-H.; Magkos, F.; Shiow, S.-A.T.E.; Schröder, H. The Gut Microbiome Profile in Obesity: A Systematic Review. Int. J. Endocrinol. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Patel, K.V.; Lavie, C.J. Obesity, Central Adiposity, and Fitness: Understanding the Obesity Paradox in the Context of Other Cardiometabolic Parameters. Mayo Clin. Proc. 2018, 93, 676–678. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Chang, H.-W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [PubMed]
- Dalby, M.J.; Ross, A.W.; Walker, A.W.; Morgan, P.J. Dietary Uncoupling of Gut Microbiota and Energy Harvesting from Obesity and Glucose Tolerance in Mice. Cell Rep. 2017, 21, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.-S.; Lee, Y.-R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Valencak, T.G.; Osterrieder, A.; Schulz, T.J. Sex matters: The effects of biological sex on adipose tissue biology and energy metabolism. Redox Biol. 2017, 12, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, J.; Champagne, C.; de Jonge, L.; Xie, H.; Smith, S. Increased visceral fat and decreased energy expenditure during the menopausal transition. Int. J. Obes. 2008, 32, 949–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorak, M.T.; Karpuzoglu, E. Gender differences in cancer susceptibility: An inadequately addressed issue. Front. Genet. 2012, 3, 268. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Liu, L.; Chen, H.; Wang, Y.; Xu, Y.; Mao, H.; Li, J.; Mills, G.B.; Shu, Y.; Li, L.; et al. Comprehensive Characterization of Molecular Differences in Cancer between Male and Female Patients. Cancer Cell 2016, 29, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Stomach Cancer Risk Factors. Available online: https://www.cancer.org/cancer/stomach-cancer/causes-risks-prevention/risk-factors.html (accessed on 10 December 2018).
- Chen, Y.; Liu, L.; Wang, X.; Wang, J.; Yan, Z.; Cheng, J.; Gong, G.; Li, G. Body Mass Index and Risk of Gastric Cancer: A Meta-analysis of a Population with More Than Ten Million from 24 Prospective Studies. Cancer Epidemiol. Prev. Biomark. 2013, 22, 1395–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Zhang, J.; Zhou, Y.; Qiao, L. Obesity and gastric cancer. Front. Biosci. Landmark Ed. 2012, 17, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Olefson, S.; Moss, S.F. Obesity and related risk factors in gastric cardia adenocarcinoma. Gastric Cancer 2015, 18, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Nomoto-Kojima, N.; Aoki, S.; Uchihashi, K.; Matsunobu, A.; Koike, E.; Ootani, A.; Yonemitsu, N.; Fujimoto, K.; Toda, S. Interaction between adipose tissue stromal cells and gastric cancer cells in vitro. Cell Tissue Res. 2011, 344, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.-C.; Zhao, B.; Han, J.-G.; Ma, H.-C.; Wang, Z.-J. Adipose-derived stem cells promote gastric cancer cell growth, migration and invasion through SDF-1/CXCR4 axis. Hepatogastroenterology 2010, 57, 1382–1389. [Google Scholar] [PubMed]
- Dong, Z.; Fu, S.; Xu, X.; Yang, Y.; Du, L.; Li, W.; Kan, S.; Li, Z.; Zhang, X.; Wang, L.; et al. Leptin-mediated regulation of ICAM-1 is Rho/ROCK dependent and enhances gastric cancer cell migration. Br. J. Cancer 2014, 110, 1801–1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, R.; Lin, C.; Tran, T.; Tarnawski, A. Leptin activates STAT and ERK2 pathways and induces gastric cancer cell proliferation. Biochem. Biophys. Res. Commun. 2005, 331, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Shida, D.; Kitayama, J.; Mori, K.; Watanabe, T.; Nagawa, H. Transactivation of Epidermal Growth Factor Receptor Is Involved in Leptin-Induced Activation of Janus-Activated Kinase 2 and Extracellular Signal–Regulated Kinase 1/2 in Human Gastric Cancer Cells. Cancer Res. 2005, 65, 9159–9163. [Google Scholar] [CrossRef] [PubMed]
- Cammisotto, P.G.; Levy, É.; Bukowiecki, L.J.; Bendayan, M. Cross-talk between adipose and gastric leptins for the control of food intake and energy metabolism. Prog. Histochem. Cytochem. 2010, 45, 143–200. [Google Scholar] [CrossRef] [PubMed]
- Xiang, F.; Wu, K.; Liu, Y.; Shi, L.; Wang, D.; Li, G.; Tao, K.; Wang, G. Omental adipocytes enhance the invasiveness of gastric cancer cells by oleic acid-induced activation of the PI3K-Akt signaling pathway. Int. J. Biochem. Cell Biol. 2017, 84, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Gislette, T.; Chen, J. The Possible Role of IL-17 in Obesity-Associated Cancer. Available online: https://www.hindawi.com/journals/tswj/2010/982036/abs/ (accessed on 10 December 2018).
- Ericksen, R.E.; Rose, S.; Westphalen, C.B.; Shibata, W.; Muthupalani, S.; Tailor, Y.; Friedman, R.A.; Han, W.; Fox, J.G.; Ferrante, A.W.; et al. Obesity accelerates Helicobacter felis-induced gastric carcinogenesis by enhancing immature myeloid cell trafficking and TH17 response. Gut 2014, 63, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Liver Cancer Risk Factors. Available online: https://www.cancer.org/cancer/liver-cancer/causes-risks-prevention/risk-factors.html (accessed on 10 December 2018).
- Aleksandrova, K.; Stelmach-Mardas, M.; Schlesinger, S. Obesity and Liver Cancer. Recent Res. Cancer Res. Fortsch. Krebsforschung Progres Dans Rech. Cancer 2016, 208, 177–198. [Google Scholar]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Turati, F.; Talamini, R.; Pelucchi, C.; Polesel, J.; Franceschi, S.; Crispo, A.; Izzo, F.; La Vecchia, C.; Boffetta, P.; Montella, M. Metabolic syndrome and hepatocellular carcinoma risk. Br. J. Cancer 2013, 108, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.L.; Freedman, N.D.; Demuth, J.; Yang, B.; Van Den Eeden, S.K.; Engel, L.S.; McGlynn, K.A. Obesity, diabetes, serum glucose, and risk of primary liver cancer by birth cohort, race/ethnicity, and sex: Multiphasic health checkup study. Cancer Epidemiol. 2016, 42, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Moschen, A.R. Mechanisms behind the link between obesity and gastrointestinal cancers. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.-Y.; Ma, T.; Lock, E.-J.; Hao, Q.; Kristiansen, K.; Frøyland, L.; Madsen, L. Depot-Dependent Effects of Adipose Tissue Explants on Co-Cultured Hepatocytes. PLoS ONE 2011, 6, e20917. [Google Scholar] [CrossRef] [PubMed]
- Nov, O.; Kohl, A.; Lewis, E.C.; Bashan, N.; Dvir, I.; Ben-Shlomo, S.; Fishman, S.; Wueest, S.; Konrad, D.; Rudich, A. Interleukin-1β May Mediate Insulin Resistance in Liver-Derived Cells in Response to Adipocyte Inflammation. Endocrinology 2010, 151, 4247–4256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.-R.; Liu, J.; Plumeri, D.; Cao, Y.-B.; He, T.; Lin, L.; Li, Y.; Jiang, Y.-Y.; Li, J.; Shang, J. Lipotoxicity in HepG2 cells triggered by free fatty acids. Am. J. Transl. Res. 2011, 3, 284–291. [Google Scholar] [PubMed]
- Asrih, M.; Montessuit, C.; Philippe, J.; Jornayvaz, F.R. Free Fatty Acids Impair FGF21 Action in HepG2 Cells. Cell. Physiol. Biochem. 2015, 37, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Donato, M.T. Potential Impact of Steatosis on Cytochrome P450 Enzymes of Human Hepatocytes Isolated from Fatty Liver Grafts. Drug Metab. Dispos. 2006, 34, 1556–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free Fatty Acids Induce JNK-dependent Hepatocyte Lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.A.; Hebbard, L.W. Molecular cross-talk between the liver and white adipose tissue links excessive noURIshment to hepatocellular carcinoma. Transl. Cancer Res. 2016, 5, S1222–S1226. [Google Scholar] [CrossRef]
- Buechler, C.; Haberl, E.M.; Rein-Fischboeck, L.; Aslanidis, C. Adipokines in Liver Cirrhosis. Int. J. Mol. Sci. 2017, 18, 1392. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Li, J.; Shao, D.; Pan, Y.; Chen, Y.; Li, S.; Yao, X.; Li, H.; Liu, W.; Zhang, M.; et al. Adipose tissue-secreted miR-27a promotes liver cancer by targeting FOXO1 in obese individuals. OncoTargets Ther. 2015, 8, 735–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Xu, M.; Li, X.; Su, X.; Xiao, X.; Keating, A.; Zhao, R.C. Exosomes released by hepatocarcinoma cells endow adipocytes with tumor-promoting properties. J. Hematol. Oncol. 2018, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Andersen, D.K.; Korc, M.; Petersen, G.M.; Eibl, G.; Li, D.; Rickels, M.R.; Chari, S.T.; Abbruzzese, J.L. Diabetes, Pancreatogenic Diabetes, and Pancreatic Cancer. Diabetes 2017, 66, 1103–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebours, V.; Gaujoux, S.; d’Assignies, G.; Sauvanet, A.; Ruszniewski, P.; Levy, P.; Paradis, V.; Bedossa, P.; Couvelard, A. Obesity and Fatty Pancreatic Infiltration Are Risk Factors for Pancreatic Precancerous Lesions (PanIN). Clin. Cancer Res. 2015, 21, 3522–3528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malka, D.; Hammel, P.; Maire, F.; Rufat, P.; Madeira, I.; Pessione, F.; Lévy, P.; Ruszniewski, P. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut 2002, 51, 849–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowenfels, A.B.; Maisonneuve, P.; Cavallini, G.; Ammann, R.W.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andren-Sandberg, A.; Domellof, L.; the International Pancreatitis Study Group. Pancreatitis and the Risk of Pancreatic Cancer. N. Engl. J. Med. 1993, 328, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Barbacid, M. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol. Oncol. 2013, 7, 232–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, P.B.; True, E.M.; Ziegler, K.M.; Wang, S.S.; Swartz-Basile, D.A.; Pitt, H.A.; Zyromski, N.J. Insulin, leptin, and tumoral adipocytes promote murine pancreatic cancer growth. J. Gastrointest. Surg. 2010, 14, 1888–1894. [Google Scholar] [CrossRef] [PubMed]
- Feygenzon, V.; Loewenstein, S.; Lubezky, N.; Pasmanic-Chor, M.; Sher, O.; Klausner, J.M.; Lahat, G. Unique cellular interactions between pancreatic cancer cells and the omentum. PLoS ONE 2017, 12, e0179862. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.A.; Neeley, C.K.; Baker, N.A.; Washabaugh, A.R.; Flesher, C.G.; Nelson, B.S.; Frankel, T.L.; Lumeng, C.N.; Lyssiotis, C.A.; Wynn, M.L.; et al. Adipocytes promote pancreatic cancer cell proliferation via glutamine transfer. Biochem. Biophys. Rep. 2016, 7, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Okumura, T.; Ohuchida, K.; Sada, M.; Abe, T.; Endo, S.; Koikawa, K.; Iwamoto, C.; Miura, D.; Mizuuchi, Y.; Moriyama, T.; et al. Extra-pancreatic invasion induces lipolytic and fibrotic changes in the adipose microenvironment, with released fatty acids enhancing the invasiveness of pancreatic cancer cells. Oncotarget 2017, 8, 18280–18295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Daniluk, J.; Liu, Y.; Chu, J.; Li, Z.; Ji, B.; Logsdon, C.D. Oncogenic K-Ras requires activation for enhanced activity. Oncogene 2014, 33, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Philip, B.; Roland, C.L.; Daniluk, J.; Liu, Y.; Chatterjee, D.; Gomez, S.B.; Ji, B.; Huang, H.; Wang, H.; Fleming, J.B.; et al. A High-Fat Diet Activates Oncogenic Kras and COX2 to Induce Development of Pancreatic Ductal Adenocarcinoma in Mice. Gastroenterology 2013, 145, 1449–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delitto, D.; Black, B.S.; Sorenson, H.L.; Knowlton, A.E.; Thomas, R.M.; Sarosi, G.A.; Moldawer, L.L.; Behrns, K.E.; Liu, C.; George, T.J.; et al. The inflammatory milieu within the pancreatic cancer microenvironment correlates with clinicopathologic parameters, chemoresistance and survival. BMC Cancer 2015, 15, 783. [Google Scholar] [CrossRef] [PubMed]
- Incio, J.; Liu, H.; Suboj, P.; Chin, S.M.; Chen, I.X.; Pinter, M.; Ng, M.R.; Nia, H.T.; Grahovac, J.; Kao, S.; et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discov. 2016. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.C.; Wu, Y.; Nordbeck, E.B.; Musovic, S.; Olofsson, C.S.; Rorsman, P.; Wernstedt, A. Pancreatic Adipose Tissue in Diet-Induced Type 2 Diabetes. Diabetes 2018, 67 (Suppl. 1). [Google Scholar] [CrossRef]
- Danai, L.V.; Babic, A.; Rosenthal, M.H.; Dennstedt, E.A.; Muir, A.; Lien, E.C.; Mayers, J.R.; Tai, K.; Lau, A.N.; Jones-Sali, P.; et al. Altered exocrine function can drive adipose wasting in early pancreatic cancer. Nature 2018, 558, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Webb, P.M. Obesity and gynecologic cancer etiology and survival. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Carmina, E.; Lobo, R.A. Polycystic Ovary Syndrome (PCOS): Arguably the Most Common Endocrinopathy Is Associated with Significant Morbidity in Women. J. Clin. Endocrinol. Metab. 1999, 84, 1897–1899. [Google Scholar] [CrossRef] [PubMed]
- Serhat, E.; Cogendez, E.; Selcuk, S.; Asoglu, M.R.; Arioglu, P.F.; Eren, S. Is there a relationship between endometrial polyps and obesity, diabetes mellitus, hypertension? Arch. Gynecol. Obstet. 2014, 290, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Sommer, E.M.; Balkwill, A.; Reeves, G.; Green, J.; Beral, D.V.; Coffey, K. Effects of obesity and hormone therapy on surgically-confirmed fibroids in postmenopausal women. Eur. J. Epidemiol. 2015, 30, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Dunaif, A. Insulin resistance and the polycystic ovary syndrome: Mechanism and implications for pathogenesis. Endocr. Rev. 1997, 18, 774–800. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Falconer, H.; Yin, L.; Xu, L.; Ye, W. Association Between Polycystic Ovary Syndrome and Cancer Risk. JAMA Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Zeitoun, K.; Sasano, H.; Simpson, E.R. Aromatase in aging women. Semin. Reprod. Endocrinol. 1999, 17, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.R. Sources of estrogen and their importance. J. Steroid Biochem. Mol. Biol. 2003, 86, 225–230. [Google Scholar] [CrossRef]
- Cleary, M.P.; Grossmann, M.E. Obesity and Breast Cancer: The Estrogen Connection. Endocrinology 2009, 150, 2537–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simó, R.; Sáez-López, C.; Barbosa-Desongles, A.; Hernández, C.; Selva, D.M. Novel insights in SHBG regulation and clinical implications. Trends Endocrinol. Metab. 2015, 26, 376–383. [Google Scholar] [CrossRef] [PubMed]
- In BRCA Mutation Carriers, Obesity is Linked with Increased DNA Damage in Normal Breast Gland Cells | Endocrine Society. Available online: https://www.endocrine.org/news-room/2018/in-brca-mutation-carriers-obesity-is-linked-with-increased-dna-damage-in-normal-breast-gland-cells (accessed on 10 December 2018).
- Canadian Cancer Statistics Publication—Canadian Cancer Society. Available online: http://www.cancer.ca/en/cancer-information/cancer-101/canadian-cancer-statistics-publication/?region=on (accessed on 10 December 2018).
- Daling, J.R.; Malone, K.E.; Doody, D.R.; Johnson, L.G.; Gralow, J.R.; Porter, P.L. Relation of body mass index to tumor markers and survival among young women with invasive ductal breast carcinoma. Cancer 2001, 92, 720–729. [Google Scholar] [CrossRef] [Green Version]
- De Azambuja, E.; McCaskill-Stevens, W.; Francis, P.; Quinaux, E.; Crown, J.P.A.; Vicente, M.; Giuliani, R.; Nordenskjöld, B.; Gutiérez, J.; Andersson, M.; et al. The effect of body mass index on overall and disease-free survival in node-positive breast cancer patients treated with docetaxel and doxorubicin-containing adjuvant chemotherapy: the experience of the BIG 02-98 trial. Breast Cancer Res. Treat. 2010, 119, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, C.B.; Kjær, S.K.; Ejlertsen, B.; Andersson, M.; Jensen, M.-B.; Christensen, J.; Langballe, R.; Mellemkjær, L. Incidence of metachronous contralateral breast cancer in Denmark 1978–2009. Int. J. Epidemiol. 2014, 43, 1855–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantarero-Villanueva, I.; Galiano-Castillo, N.; Fernández-Lao, C.; Diaz-Rodríguez, L.; Fernández-Pérez, A.M.; Sánchez, M.J.; Arroyo-Morales, M. The influence of body mass index on survival in breast cancer patients. Clin. Breast Cancer 2015, 15, e117–e123. [Google Scholar] [CrossRef] [PubMed]
- Copson, E.R.; Cutress, R.I.; Maishman, T.; Eccles, B.K.; Gerty, S.; Stanton, L.; Altman, D.G.; Durcan, L.; Wong, C.; Simmonds, P.D.; et al. POSH Study Steering Group Obesity and the outcome of young breast cancer patients in the UK: The POSH study. Ann. Oncol. 2015, 26, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Schautz, B.; Later, W.; Heller, M.; Müller, M.J.; Bosy-Westphal, A. Associations between breast adipose tissue, body fat distribution and cardiometabolic risk in women: Cross-sectional data and weight-loss intervention. Eur. J. Clin. Nutr. 2011, 65, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, A.; Tamimi, R.M. Breast fat and breast cancer. Breast Cancer Res. Treat. 2012, 135, 321–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gérard, C.; Brown, K.A. Obesity and breast cancer—Role of estrogens and the molecular underpinnings of aromatase regulation in breast adipose tissue. Mol. Cell. Endocrinol. 2018, 466, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Milne, R.L.; Friedlander, M.L.; McCredie, M.R.E.; Giles, G.G.; Hopper, J.L.; Phillips, K.-A. Obesity and outcomes in premenopausal and postmenopausal breast cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1686–1691. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, M.; Decorby, K.; Choi, B.C.K. The Association between Obesity and Cancer Risk: A Meta-Analysis of Observational Studies from 1985 to 2011. ISRN Prev. Med. 2013, 2013, 680536. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, L.; Zhou, Q.; Imam, M.U.; Cai, J.; Wang, Y.; Qi, M.; Sun, P.; Ping, Z.; Fu, X. Body mass index had different effects on premenopausal and postmenopausal breast cancer risks: A dose-response meta-analysis with 3,318,796 subjects from 31 cohort studies. BMC Public Health 2017, 17, 936. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, L.; Cui, S.; Tian, F.; Fan, Z.; Geng, C.; Cao, X.; Yang, Z.; Wang, X.; Liang, H.; et al. Distinct Effects of Body Mass Index and Waist/Hip Ratio on Risk of Breast Cancer by Joint Estrogen and Progestogen Receptor Status: Results from a Case-Control Study in Northern and Eastern China and Implications for Chemoprevention. Oncologist 2017, 22, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Savolainen-Peltonen, H.; Vihma, V.; Leidenius, M.; Wang, F.; Turpeinen, U.; Hämäläinen, E.; Tikkanen, M.J.; Mikkola, T.S. Breast Adipose Tissue Estrogen Metabolism in Postmenopausal Women with or Without Breast Cancer. J. Clin. Endocrinol. Metab. 2014, 99, E2661–E2667. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Samad, F.; Mueller, B.M. Local adipocytes enable estrogen-dependent breast cancer growth. Adipocyte 2013, 2, 165–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyengar, N.M.; Hudis, C.A.; Dannenberg, A.J. Obesity and inflammation: New insights into breast cancer development and progression. Am. Soc. Clin. Oncol. Educ. Book 2013, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Péqueux, C.; Raymond-Letron, I.; Blacher, S.; Boudou, F.; Adlanmerini, M.; Fouque, M.-J.; Rochaix, P.; Noël, A.; Foidart, J.-M.; Krust, A.; et al. Stromal Estrogen Receptor-α Promotes Tumor Growth by Normalizing an Increased Angiogenesis. Cancer Res. 2012, 72, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- Maehle, B.O.; Tretli, S.; Thorsen, T. The associations of obesity, lymph node status and prognosis in breast cancer patients: Dependence on estrogen and progesterone receptor status. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2004, 112, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Komninou, D.; Stephenson, G.D. Obesity, adipocytokines, and insulin resistance in breast cancer. Obes. Rev. 2004, 5, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, B.Y.; Wilkens, L.R.; LeMarchand, L.; Horio, D.; Chong, C.D.; Loo, L.W. Difference in IGF-axis protein expression and survival among multiethnic breast cancer patients. Cancer Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, C.; Gramigano, G.; Floris, C.; Muenu, G.; Sollari, G.; Maccio, A. Role of inflammation and oxidative stress in post-menopausal oestrogen-dependent breast cancer. J. Cell. Mol. Med. 2014, 18, 2519–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Flanagan, C.H.; Rossi, E.L.; McDonell, S.B.; Chen, X.; Tsai, Y.-H.; Parker, J.S.; Usary, J.; Perou, C.M.; Hursting, S.D. Metabolic reprogramming underlies metastatic potential in an obesity-responsive murine model of metastatic triple negative breast cancer. NPJ Breast Cancer. 2017, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Blücher, C.; Stadler, S.C. Obesity and Breast Cancer: Current Insights on the Role of Fatty Acids and Lipid Metabolism in Promoting Breast Cancer Growth and Progression. Front. Endocrinol. 2017, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Himbert, C.; Delphan, M.; Scherer, D.; Bowers, L.W.; Hursting, S.; Ulrich, C.M. Signals from the Adipose Microenvironment and the Obesity-Cancer Link—A Systematic Review. Cancer Prev. Res. 2017, 10, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Hoy, A.J.; Balaban, S.; Saunders, D.N. Adipocyte–Tumor Cell Metabolic Crosstalk in Breast Cancer. Trends Mol. Med. 2017, 23, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Nickel, A.; Blücher, C.; Kadri, O.A.; Schwagarus, N.; Müller, S.; Schaab, M.; Thiery, J.; Burkhardt, R.; Stadler, S.C. Adipocytes induce distinct gene expression profiles in mammary tumor cells and enhance inflammatory signaling in invasive breast cancer cells. Sci. Rep. 2018, 8, 9482. [Google Scholar] [CrossRef] [PubMed]
- Bougaret, L.; Delort, L.; Billard, H.; Le Huede, C.; Boby, C.; De la Foye, A.; Rossary, A.; Mojallal, A.; Damour, O.; Auxenfans, C.; et al. Adipocyte/breast cancer cell crosstalk in obesity interferes with the anti-proliferative efficacy of tamoxifen. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Duong, M.N.; Cleret, A.; Matera, E.-L.; Chettab, K.; Mathé, D.; Valsesia-Wittmann, S.; Clémenceau, B.; Dumontet, C. Adipose cells promote resistance of breast cancer cells to trastuzumab-mediated antibody-dependent cellular cytotoxicity. Breast Cancer Res. 2015, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Mattei, E.; Velazquez-Torres, G.; Phan, L.; Zhang, F.; Chou, P.-C.; Shin, J.-H.; Choi, H.H.; Chen, J.-S.; Zhao, R.; Chen, J.; et al. Effects of obesity on transcriptomic changes and cancer hallmarks in estrogen receptor-positive breast cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Sabol, R.; Cote, A.; Bunnell, B. Adipose Stem Cells from Obese Individuals Promote Radioresistance of Estrogen Receptor Positive Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, E615–E616. [Google Scholar] [CrossRef]
- Strong, A.L.; Ohlstein, J.F.; Biagas, B.A.; Rhodes, L.V.; Pei, D.T.; Tucker, H.A.; Llamas, C.; Bowles, A.C.; Dutreil, M.F.; Zhang, S.; et al. Leptin produced by obese adipose stromal/stem cells enhances proliferation and metastasis of estrogen receptor positive breast cancers. Breast Cancer Res. 2015, 17, 112. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Miki, Y.; Takagi, K.; Suzuki, T.; Ishida, T.; Ohuchi, N.; Sasano, H. Interaction with adipocyte stromal cells induces breast cancer malignancy via S100A7 upregulation in breast cancer microenvironment. Breast Cancer Res. 2017, 19, 70. [Google Scholar] [CrossRef] [PubMed]
- Mullooly, M.; Yang, H.P.; Falk, R.T.; Nyante, S.J.; Cora, R.; Pfeiffer, R.M.; Radisky, D.C.; Visscher, D.W.; Hartmann, L.C.; Carter, J.M.; et al. Relationship between crown-like structures and sex-steroid hormones in breast adipose tissue and serum among postmenopausal breast cancer patients. Breast Cancer Res. 2017, 19, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, J.M.; Hoskin, T.L.; Pena, M.A.; Brahmbhatt, R.; Winham, S.J.; Frost, M.H.; Stallings-Mann, M.; Radisky, D.C.; Knutson, K.L.; Visscher, D.W.; et al. Macrophagic “Crown-like Structures” Are Associated with an Increased Risk of Breast Cancer in Benign Breast Disease. Cancer Prev. Res. 2018, 11, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Yan, F.; Zhang, Y.; Triplett, A.; Zhang, Y.; Schultz, D.A.; Sun, Y.; Zeng, J.; Silverstein, K.A.T.; Zheng, Q.; et al. Expression of adipocyte/macrophage fatty acid binding protein in tumor associated macrophages promotes breast cancer progression. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Sun, X.; Gupta, H.B.; Yuan, B.; Li, J.; Ge, F.; Chiang, H.-C.; Zhang, X.; Zhang, C.; Zhang, D.; et al. Adipose PD-L1 Modulates PD-1/PD-L1 Checkpoint Blockade Immunotherapy Efficacy in Breast Cancer. Oncoimmunology 2018, 7, e1500107. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Domchek, S.M.; Clark, A.S. Immunotherapy for Breast Cancer: What Are We Missing? Clin Cancer Res. 2017, 23, 2640–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svartberg, J.; von Mühlen, D.; Sundsfjord, J.; Jorde, R. Waist circumference and testosterone levels in community dwelling men. The Tromsø study. Eur. J. Epidemiol. 2004, 19, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.M.; Jones, T.H. Testosterone and obesity. Obes. Rev. 2015, 16, 581–606. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.G. Obesity in men: The hypogonadal-estrogen receptor relationship and its effect on glucose homeostasis. Med. Hypotheses 2008, 70, 358–360. [Google Scholar] [CrossRef] [PubMed]
- Ng Tang Fui, M.; Hoermann, R.; Prendergast, L.A.; Zajac, J.D.; Grossmann, M. Symptomatic response to testosterone treatment in dieting obese men with low testosterone levels in a randomized, placebo-controlled clinical trial. Int. J. Obes. 2017, 41, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.E.; Billups, K.L.; Partin, A.W. Testosterone and prostate cancer: An evidence-based review of pathogenesis and oncologic risk. Ther. Adv. Urol. 2015, 7, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P.; Koechlin, A.; Bota, M.; d’Onofrio, A.; Zaridze, D.G.; Perrin, P.; Fitzpatrick, J.; Burnett, A.L.; Boniol, M. Endogenous and exogenous testosterone and the risk of prostate cancer and increased prostate-specific antigen (PSA) level: A meta-analysis. BJU Int. 2016, 118, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Freedland, S.J.; Aronson, W.J. Examining the Relationship Between Obesity and Prostate Cancer. Rev. Urol. 2004, 6, 73–81. [Google Scholar] [PubMed]
- Campos-Silva, P.; Furriel, A.; Costa, W.S.; Sampaio, F.J.B.; Gregorio, B.M. Metabolic and testicular effects of the long-term administration of different high-fat diets in adult rats. Int. Braz. J. Urol. 2015, 41, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Xu, Y.; Liu, Y.; Zhang, Z.; Lu, L.; Ding, Z. Obesity or Overweight, a Chronic Inflammatory Status in Male Reproductive System, Leads to Mice and Human Subfertility. Front. Physiol. 2017, 8, 1117. [Google Scholar] [CrossRef] [PubMed]
- Barnes, K.T.; Smith, B.J.; Lynch, C.F.; Gupta, A. Obesity and invasive penile cancer. Eur. Urol. 2013, 63, 588–589. [Google Scholar] [CrossRef] [PubMed]
- Barnes, K.T.; McDowell, B.D.; Button, A.; Smith, B.J.; Lynch, C.F.; Gupta, A. Obesity is associated with increased risk of invasive penile cancer. BMC Urol. 2016, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Douglawi, A.; Masterson, T.A. Updates on the epidemiology and risk factors for penile cancer. Transl. Androl. Urol. 2017, 6, 785–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadra, G.; Photopoulos, C.; Loda, M. The fat side of prostate. Biochimica et Biophysica Acta 2013, 1831, 1518–1532. [Google Scholar] [CrossRef] [PubMed]
- Bracarda, S.; Logothetis, C.; Sternberg, C.N.; Oudard, S. Current and emerging treatment modalities for metastatic castration-resistant prostate cancer. BJU Int. 2011, 107 (Suppl. 2), 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.A.; Lo, J.; Ascui, N.; Watt, M.J. Linking obesogenic dysregulation to prostate cancer progression. Endocr. Connect. 2015, 4, R68–R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gucalp, A.; Iyengar, N.M.; Zhou, X.K.; Giri, D.D.; Falcone, D.J.; Wang, H.; Williams, S.; Krasne, M.D.; Yaghnam, I.; Kunzel, B.; et al. Periprostatic adipose inflammation is associated with high-grade prostate cancer. Prostate Cancer Prostatic Dis. 2017, 20, 418–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, R.; Monteiro, C.; Catalán, V.; Hu, P.; Cunha, V.; Rodríguez, A.; Gómez-Ambrosi, J.; Fraga, A.; Príncipe, P.; Lobato, C.; et al. Obesity and prostate cancer: Gene expression signature of human periprostatic adipose tissue. BMC Med. 2012, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Laurent, V.; Guérard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y.; et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marzo, A.M.; Platz, E.A.; Sutcliffe, S.; Xu, J.; Grönberg, H.; Drake, C.G.; Nakai, Y.; Isaacs, W.B.; Nelson, W.G. Inflammation in prostate carcinogenesis. Nat. Rev. Cancer 2007, 7, 256–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sfanos, K.S.; De Marzo, A.M. Prostate cancer and inflammation: The evidence. Histopathology 2012, 60, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Duran, A.; Reina-Campos, M.; Valencia, T.; Castilla, E.A.; Müller, T.D.; Tschöp, M.H.; Moscat, J.; Diaz-Meco, M.T. Adipocyte p62/SQSTM1 Suppresses Tumorigenesis through Opposite Regulations of Metabolism in Adipose Tissue and Tumor. Cancer Cell 2018, 33, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Sagar, G.; Sah, R.P.; Javeed, N.; Dutta, S.K.; Smyrk, T.C.; Lau, J.S.; Giorgadze, N.; Tchkonia, T.; Kirkland, J.L.; Chari, S.T.; et al. Pathogenesis of pancreatic cancer exosome-induced lipolysis in adipose tissue. Gut 2016, 65, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Piro, G.; Gaianigo, N.; Ligorio, F.; Santoro, R.; Merz, V.; Simionato, F.; Zecchetto, C.; Falco, G.; Conti, G.; et al. Adipocytes sustain pancreatic cancer progression through a non-canonical WNT paracrine network inducing ROR2 nuclear shuttling. Int. J. Obes. 2018, 42, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.; Monteiro, C.; Cunha, V.; Oliveira, M.J.; Freitas, M.; Fraga, A.; Príncipe, P.; Lobato, C.; Lobo, F.; Morais, A.; et al. Human periprostatic adipose tissue promotes prostate cancer aggressiveness in vitro. J. Exp. Clin. Cancer Res. 2012, 31, 32. [Google Scholar] [CrossRef] [PubMed]
- Collier, A.; Ghosh, S.; Mcglynn, B.; Hollins, G. Prostate Cancer, Androgen Deprivation Therapy, Obesity, the Metabolic Syndrome, Type 2 Diabetes, and Cardiovascular Disease: A Review. Am. J. Clin. Oncol. 2012, 35, 504–509. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.W.; House, P.J.; Tomlinson, J.W. Understanding androgen action in adipose tissue. J. Steroid Biochem. Mol. Biol. 2014, 143, 277–284. [Google Scholar]
- Meyer, T.E.; Chu, L.W.; Li, Q.; Yu, K.; Rosenberg, P.S.; Menashe, I.; Chokkalingam, A.P.; Quraishi, S.M.; Huang, W.-Y.; Weiss, J.M.; et al. The association between inflammation-related genes and serum androgen levels in men: The Prostate, Lung, Colorectal, and Ovarian Study. Prostate 2012, 72, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Pitt, J.M.; Daillère, R.; Smyth, M.J.; Kroemer, G. Mouse models in oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Kleinert, M.; Clemmensen, C.; Hofmann, S.M.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; de Angelis, M.H.; Schürmann, A.; et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.K.; Sharma, D. Multifaceted Leptin network: The molecular connection between obesity and breast cancer. J. Mammary Gland Biol. Neoplasia 2013, 18, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Akase, T.; Shimada, T.; Harasawa, Y.; Akase, T.; Ikeya, Y.; Nagai, E.; Iizuka, S.; Nakagami, G.; Iizaka, S.; Sanada, H.; et al. Preventive Effects of Salacia reticulata on Obesity and Metabolic Disorders in TSOD Mice. Evid.-Based Complement. Altern. Med. 2011, 2011, 484590. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Deuschle, U.; Taira, S.; Nishida, T.; Fujimoto, M.; Hijikata, T.; Tsuneyama, K. Tsumura-Suzuki obese diabetic mice-derived hepatic tumors closely resemble human hepatocellular carcinomas in metabolism-related genes expression and bile acid accumulation. Hepatol. Int. 2018, 12, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, I.; Yi, Z.; Izumi, S.; Arai, I.; Suzuki, W.; Nagamachi, Y.; Kuwano, H.; Takeuchi, T.; Izumi, T. Genetic analysis of obese diabetes in the TSOD mouse. Diabetes 1999, 48, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Murotomi, K.; Umeno, A.; Yasunaga, M.; Shichiri, M.; Ishida, N.; Abe, H.; Yoshida, Y.; Nakajima, Y. Type 2 diabetes model TSOD mouse is exposed to oxidative stress at young age. J. Clin. Biochem. Nutr. 2014, 55, 216–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, T.; Tsuneyama, K.; Fujimoto, M.; Nomoto, K.; Hayashi, S.; Miwa, S.; Nakajima, T.; Nakanishi, Y.; Sasaki, Y.; Suzuki, W.; et al. Spontaneous onset of nonalcoholic steatohepatitis and hepatocellular carcinoma in a mouse model of metabolic syndrome. Lab. Invest. 2013, 93, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, S.; Suzuki, W.; Tabuchi, M.; Nagata, M.; Imamura, S.; Kobayashi, Y.; Kanitani, M.; Yanagisawa, T.; Kase, Y.; Takeda, S.; et al. Diabetic complications in a new animal model (TSOD mouse) of spontaneous NIDDM with obesity. Exp. Anim. 2005, 54, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nishida, T.; Baba, H.; Hatta, H.; Imura, J.; Sutoh, M.; Toyohara, S.; Hokao, R.; Watanabe, S.; Ogawa, H.; et al. Histopathological characteristics of glutamine synthetase-positive hepatic tumor lesions in a mouse model of spontaneous metabolic syndrome (TSOD mouse). Mol. Clin. Oncol. 2016, 5, 267–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuneyama, K.; Nishitsuji, K.; Matsumoto, M.; Kobayashi, T.; Morimoto, Y.; Tsunematsu, T.; Ogawa, H. Animal models for analyzing metabolic syndrome-associated liver diseases. Pathol. Int. 2017, 67, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Yamada, N.; Kwada, T. Possible involvement of hypothalamic nucleobindin-2 in hyperphagic feeding in Tsumara Suzuki Obese Diabetes mice. Biol. Pharm. Bull. 2012, 35, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Tabuchi, M.; Suzuki, W.; Iizuka, S.; Nagata, M.; Ikeya, Y.; Takeda, S.; Shimada, T.; Aburada, M. Insulin resistance and low sympathetic nerve activity in the Tsumura Suzuki obese diabetic mouse: A new model of spontaneous type 2 diabetes mellitus and obesity. Metabolism 2006, 55, 1664–1669. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, W.; Iizuka, S.; Tabuchi, M.; Funo, S.; Yanagisawa, T.; Kimura, M.; Sato, T.; Endo, T.; Kawamura, H. A new mouse model of spontaneous diabetes derived from ddY strain. Exp. Anim. 1999, 48, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Leiter, E.H. Selecting the “Right” Mouse Model for Metabolic Syndrome and Type 2 Diabetes Research. In Type 2 Diabetes: Methods and Protocols; Methods in Molecular Biology; Stocker, C., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 1–17. ISBN 978-1-59745-448-3. [Google Scholar]
- Merkwirth, C.; Langer, T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2009, 1793, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ande, S.R.; Gu, Y.; Nyomba, B.L.G.; Mishra, S. Insulin induced phosphorylation of prohibitin at tyrosine 114 recruits Shp1. Biochim. Biophys. Acta 2009, 1793, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Ande, S.R.; Nguyen, K.H.; Padilla-Meier, G.P.; Wahida, W.; Nyomba, B.L.G.; Mishra, S. Prohibitin overexpression in adipocytes induces mitochondrial biogenesis, leads to obesity development, and affects glucose homeostasis in a sex-specific manner. Diabetes 2014, 63, 3734–3741. [Google Scholar] [CrossRef] [PubMed]
- Ande, S.R.; Nguyen, K.H.; Grégoire Nyomba, B.L.; Mishra, S. Prohibitin-induced, obesity-associated insulin resistance and accompanying low-grade inflammation causes NASH and HCC. Sci. Rep. 2016, 6, 23608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ande, S.R.; Nguyen, K.H.; Padilla-Meier, G.P.; Nyomba, B.L.G.; Mishra, S. Expression of a mutant prohibitin from the aP2 gene promoter leads to obesity-linked tumor development in insulin resistance-dependent manner. Oncogene 2016, 35, 4459–4470. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Murphy, L.C.; Murphy, L.J. The Prohibitins: Emerging roles in diverse functions. J. Cell. Mol. Med. 2006, 10, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Rogers, P.C.; Meacham, L.R.; Oeffinger, K.C.; Henry, D.W.; Lange, B.J. Obesity in pediatric oncology. Pediatr. Blood Cancer 2005, 45, 881–891. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D.P.; Sarpong, K.; Jordan, L.; Ray, L.A.; Jain, S. Infant obesity: Are we ready to make this diagnosis? J. Pediatr. 2010, 157, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Sovio, U.; Kaakinen, M.; Tzoulaki, I.; Das, S.; Ruokonen, A.; Pouta, A.; Hartikainen, A.-L.; Molitor, J.; Järvelin, M.-R. How do changes in body mass index in infancy and childhood associate with cardiometabolic profile in adulthood? Findings from the Northern Finland Birth Cohort 1966 Study. Int. J. Obes. 2014, 38, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Berger, N.A. Young Adult Cancer: Influence of the Obesity Pandemic. Obesity (Silver Spring) 2018, 26, 641–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnea, D.; Raghunathan, N.; Friedman, D.N.; Tonorezos, E.S. Obesity and Metabolic Disease After Childhood Cancer. Oncology (Williston Park) 2015, 29, 849–855. [Google Scholar] [PubMed]
- Hursting, S.D.; Nunez, N.P.; Varticovski, L.; Vinson, C. The Obesity-Cancer Link: Lessons Learned from a Fatless Mouse. Cancer Res. 2007, 67, 2391–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberson, L.L.; Aneni, E.C.; Maziak, W.; Agatston, A.; Feldman, T.; Rouseff, M.; Tran, T.; Blaha, M.J.; Santos, R.D.; Sposito, A.; et al. Beyond BMI: The “Metabolically healthy obese” phenotype & its association with clinical/subclinical cardiovascular disease and all-cause mortality—A systematic review. BMC Public Health 2014, 14, 14. [Google Scholar]
- Mathew, H.; Farr, O.M.; Mantzoros, C.S. Metabolic Health and Weight: Understanding metabolically unhealthy normal weight or metabolically healthy obese patients. Metabolism 2016, 65, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.A.; Cao, Y.; Chan, A.T. Aspirin and colorectal cancer: The promise of precision chemoprevention. Nat. Rev. Cancer 2016, 16, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-H.; Moro, A.; Chou, C.E.N.; Dawson, D.W.; French, S.; Schmidt, A.I.; Sinnett-Smith, J.; Hao, F.; Hines, O.J.; Eibl, G.; et al. Metformin Decreases the Incidence of Pancreatic Ductal Adenocarcinoma Promoted by Diet-induced Obesity in the Conditional KrasG12D Mouse Model. Sci. Rep. 2018, 8, 5899. [Google Scholar] [CrossRef] [PubMed]
- Padwal, R.S.; Pajewski, N.M.; Allison, D.B.; Sharma, A.M. Using the Edmonton obesity staging system to predict mortality in a population-representative cohort of people with overweight and obesity. CMAJ 2011, 183, E1059–E1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalgaard, K.; Landgraf, K.; Heyne, S.; Lempradl, A.; Longinotto, J.; Gossens, K.; Ruf, M.; Orthofer, M.; Strogantsev, R.; Selvaraj, M.; et al. Trim28 Haploinsufficiency Triggers Bi-stable Epigenetic Obesity. Cell 2016, 164, 353–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Grimm, S.A.; Chrysovergis, K.; Kosak, J.; Wang, X.; Du, Y.; Burkholder, A.; Janardhan, K.; Mav, D.; Shah, R.; et al. Obesity, Rather Than Diet, Drives Epigenomic Alterations in Colonic Epithelium Resembling Cancer Progression. Cell Metab. 2014, 19, 702–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Grimm, S.A.; Mav, D.; Gu, H.; Djukovic, D.; Shah, R.; Merrick, B.A.; Raftery, D.; Wade, P.A. Transcriptome and DNA Methylome Analysis in a Mouse Model of Diet-Induced Obesity Predicts Increased Risk of Colorectal Cancer. Cell Rep. 2018, 22, 624–637. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.X.Z.; Mishra, S. Obesity-Linked Cancers: Current Knowledge, Challenges and Limitations in Mechanistic Studies and Rodent Models. Cancers 2018, 10, 523. https://doi.org/10.3390/cancers10120523
Xu YXZ, Mishra S. Obesity-Linked Cancers: Current Knowledge, Challenges and Limitations in Mechanistic Studies and Rodent Models. Cancers. 2018; 10(12):523. https://doi.org/10.3390/cancers10120523
Chicago/Turabian StyleXu, Yang Xin Zi, and Suresh Mishra. 2018. "Obesity-Linked Cancers: Current Knowledge, Challenges and Limitations in Mechanistic Studies and Rodent Models" Cancers 10, no. 12: 523. https://doi.org/10.3390/cancers10120523
APA StyleXu, Y. X. Z., & Mishra, S. (2018). Obesity-Linked Cancers: Current Knowledge, Challenges and Limitations in Mechanistic Studies and Rodent Models. Cancers, 10(12), 523. https://doi.org/10.3390/cancers10120523