Precision Immuno-Oncology: Prospects of Individualized Immunotherapy for Pancreatic Cancer



{kind=link}
{kind=link}
Abstract
:1. Introduction
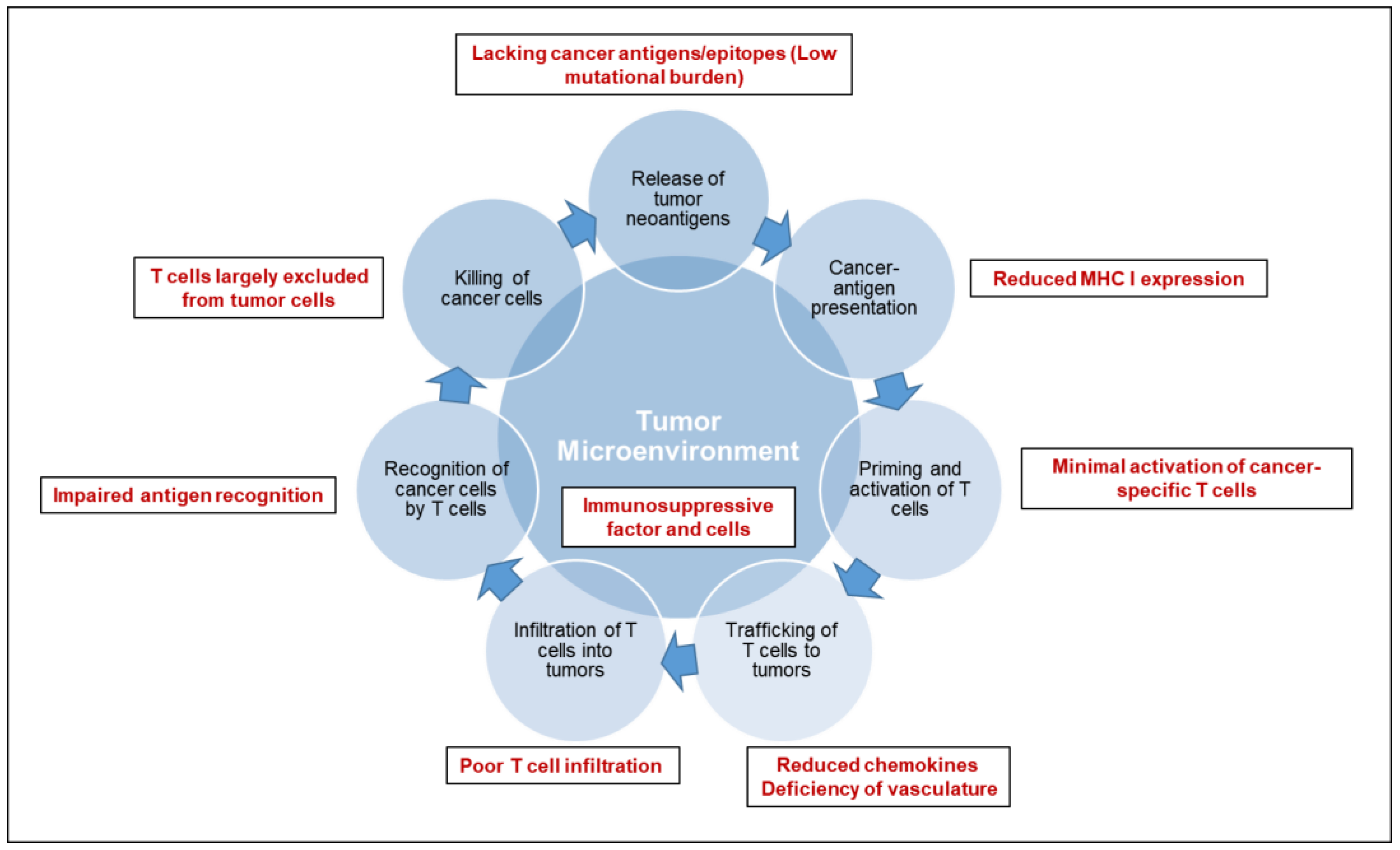
2. Overview of Immune-Biology of Pancreatic Cancer
3. Biomarkers Identification
3.1. Tumor Microenvironment
3.1.1. PD-L1 Expression
3.1.2. Tumor Infiltrating Lymphocytes
3.2. Biomarkers in the Genome
3.2.1. Mutations and Mutation Burden
3.2.2. Chromosomal Chaos
4. Immunotherapeutic Targets for Pancreatic Cancer
4.1. Immune Checkpoints
4.2. Activating or Introducing Cancer Antigen-Specific T Cells
4.2.1. Tumor Vaccines
4.2.2. Neoantigens and Neoepitopes
4.3. Targeting the Microenvironment
4.3.1. Stromal Matrix
4.3.2. CD40/CD40L Pathway
4.3.3. RAS/MAPK Activation
4.3.4. Focal Adhesion Kinase
4.4. Targeting DNA Repair Mechanisms
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Seufferlein, T.; Mayerle, J. Pancreatic cancer in 2015: Precision medicine in pancreatic cancer—Fact or fiction? Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 74–75. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer—Immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Predina, J.; Eruslanov, E.; Judy, B.; Kapoor, V.; Cheng, G.; Wang, L.-C.; Sun, J.; Moon, E.K.; Fridlender, Z.G.; Albelda, S.; et al. Changes in the local tumor microenvironment in recurrent cancers may explain the failure of vaccines after surgery. Proc. Natl. Acad. Sci. USA 2013, 110, E415–E424. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-H. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jonesa, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Eder, J.P.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Bellmunt, J.; Burris, H.A.; Petrylak, D.P.; Teng, S.L.; et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014, 515, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Soares, K.C.; Rucki, A.A.; Wu, A.A.; Olino, K.; Xiao, Q.; Chai, Y.; Wamwea, A.; Bigelow, E.; Lutz, E.; Liu, L.; et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J. Immunother. 2015, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy Converts Nonimmunogenic Pancreatic Tumors into Immunogenic Foci of Immune Regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef] [PubMed]
- Loos, M.; Giese, N.A.; Kleeff, J.; Giese, T.; Gaida, M.M.; Bergmann, F.; Laschinger, M.; Büchler, M.W.; Friess, H.; et al. Clinical significance and regulation of the costimulatory molecule B7-H1 in pancreatic cancer. Cancer Lett. 2008, 268, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Paschall, A.V.; Shi, H.; Savage, N.; Waller, J.L.; Sabbatini, M.E.; Oberlies, N.H.; Pearce, C.; Liu, K.; et al. The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L. PD-L1 Expression in Pancreatic Cancer. J. Natl. Cancer Inst. 2017, 109, djw304. [Google Scholar] [CrossRef] [PubMed]
- Church, D.N.; Stelloo, E.; Nout, R.A.; Valtcheva, N.; Depreeuw, J.; ter Haar, N.; Noske, A.; Amant, F.; Tomlinson, I.P.; Wild, P.J.; et al. Prognostic significance of POLE proofreading mutations in endometrial cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L.; et al. Colocalization of Inflammatory Response with B7-H1 Expression in Human Melanocytic Lesions Supports an Adaptive Resistance Mechanism of Immune Escape. Sci. Transl. Med. 2012, 4, 127ra37. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, T.; Kumar, S.; Borkar, R.N.; Azimi, V.; Thibault, G.; Chang, Y.H.; Balter, A.; Kawashima, R.; Choe, G.; Sauer, D.; et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017, 19, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Irving, B.A.; Hodi, F.S. Molecular pathways: Next-generation immunotherapy-inhibiting programmed death-ligand 1 and programmed death-1. Clin. Cancer Res. 2012, 18, 6580–6587. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Yao, Q.; Chen, Z.; Xiang, J.; William, F.E.; Gibbs, R.A.; Chen, C. Genetic and molecular alterations in pancreatic cancer: Implications for personalized medicine. Med. Sci. Monit. 2013, 19, 916–926. [Google Scholar] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Haraldsdottir, S.; Hampel, H.; Tomsic, J.; Frankel, W.L.; Pearlman, R.; De La Chapelle, A.; Pritchard, C.C. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology 2014, 147, 1308–1316.e1. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Freeman-Mills, L.; Rayner, E.; Glaire, M.; Briggs, S.; Vermeulen, L.; Fessler, E.; Medema, J.P.; Boot, A.; Morreau, H.; et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroenterol. Hepatol. 2016, 1, 207–216. [Google Scholar] [CrossRef]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.A. The Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Johanns, T.M.; Miller, C.A.; Dorward, I.G.; Tsien, C.; Chang, E.; Perry, A.; Uppaluri, R.; Ferguson, C.; Schmidt, R.E.; Dahiya, S.; et al. Immunogenomics of hypermutated glioblastoma: A patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov. 2016, 6, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- cBioPortal. Available online: http://www.cbioportal.org/ (accessed on 1 December 2017).
- Banville, N.; Geraghty, R.; Fox, E.; Leahy, D.T.; Green, A.; Keegan, D.; Geoghegan, J.; O’Donoghue, D.; Hyland, J.; Sheahan, K. Medullary carcinoma of the pancreas in a man with hereditary nonpolyposis colorectal cancer due to a mutation of the MSH2 mismatch repair gene. Hum. Pathol. 2006, 37, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Timms, L.; Kalimuthu, S.N.; Selander, I.; McPherson, T.; Wilson, G.W.; Chan-Seng-Yue, M.A.; Borozan, I.; et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2017, 3, 774. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Harders, C.; Wollscheid, U.; Staege, M.S.; Reske-Kunz, A.B.; Huber, C. Suppression of MHC class I antigens in oncogenic transformants: Association with decreased recognition by cytotoxic T lymphocytes. Exp. Hematol. 1996, 24, 1275–1279. [Google Scholar] [PubMed]
- Seliger, B.; Harders, C.; Lohmann, S.; Momburg, F.; Urlinger, S.; Tampé, R.; Huber, C. Down-regulation of the MHC class I antigen-processing machinery after oncogenic transformation of murine fibroblasts. Eur. J. Immunol. 1998, 28, 122–133. [Google Scholar] [CrossRef]
- Atkins, D.; Breuckmann, A.; Schmahl, G.E.; Binner, P.; Ferrone, S.; Krummenauer, F.; Störkel, S.; Seliger, B.; et al. MHC class I antigen processing pathway defects, ras mutations and disease stage in colorectal carcinoma. Int. J. Cancer 2004, 109, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Bradley, S.D.; Chen, Z.; Melendez, B.; Talukder, A.; Khalili, J.S.; Rodriguez-Cruz, T.; Liu, S.; Whittington, M.; Deng, W.; Li, F.; et al. BRAFV600E Co-opts a Conserved MHC Class I Internalization Pathway to Diminish Antigen Presentation and CD8+ T-cell Recognition of Melanoma. Cancer Immunol. Res. 2015, 3, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.J.; Bryant, K.L. KRAS mutant pancreatic cancer: No lone path to an effective treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M. Understanding the stroma of pancreatic cancer: Co-evolution of the microenvironment with epithelial carcinogenesis. J. Pathol. 2013, 231, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Redig, A.J.; Jänne, P.A. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J. Clin. Oncol. 2015, 33, 975–977. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Sciallero, S.; Giaretti, W.; Geido, E.; Bonelli, L.; Zhankui, L.; Saccomanno, S.; Zeraschi, E.; Pugliese, V. DNA aneuploidy is an independent factor of poor prognosis in pancreatic and peripancreatic cancer. Int. J. Pancreatol. 1993, 14, 21–28. [Google Scholar] [PubMed]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef] [PubMed]
- Foley, K.; Kim, V.; Jaffee, E.; Zheng, L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016, 381, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Jaffee, E.M. Next-generation cancer vaccine approaches: Integrating lessons learned from current successes with promising biotechnologic advances. J. Natl. Compr. Cancer Netw. 2013, 11, 766–772. [Google Scholar] [CrossRef]
- Jaffee, E.M.; Hruban, R.H.; Canto, M.; Kern, S.E. Focus on pancreas cancer. Cancer Cell 2002, 2, 25–28. [Google Scholar] [CrossRef]
- Keenan, B.P.; Saenger, Y.; Kafrouni, M.I.; Leubner, A.; Lauer, P.; Maitra, A.; Rucki, A.A.; Gunderson, A.J.; Coussens, L.M.; Brockstedt, D.G.; et al. A listeria vaccine and depletion of T-regulatory cells activate immunity against early stage pancreatic intraepithelial neoplasms and prolong survival of mice. Gastroenterology 2014, 146, 1784–1794.e6. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.R.; Kinkead, H.; Jaffee, E.M.; Zheng, L. Priming the pancreatic cancer tumor microenvironment for checkpoint-inhibitor immunotherapy. Oncoimmunology 2014, 3, e962401. [Google Scholar] [CrossRef] [PubMed]
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Chu, N.J.; Armstrong, T.D.; Jaffee, E.M. Nonviral oncogenic antigens and the inflammatory signals driving early cancer development as targets for cancer immunoprevention. Clin. Cancer Res. 2015, 21, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Laheru, D.; Lutz, E.; Burke, J.; Biedrzycki, B.; Solt, S.; Onners, B.; Tartakovsky, I.; Nemunaitis, J.; Le, D.; Sugar, E.; et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: A pilot study of safety, feasibility, and immune activation. Clin. Cancer Res. 2008, 14, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz Jr, L.A.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 9, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Mardis, E.R. Applications of Immunogenomics to Cancer. Cell 2017, 168, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Ruth, T.Y.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Bajor, D.L.; Winograd, R.; Evans, R.A.; Bayne, L.J.; Beatty, G.L. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar]
- Vonderheide, R.H.; Bajor, D.L.; Winograd, R.; Evans, R.A.; Bayne, L.J.; Beatty, G.L. CD40 immunotherapy for pancreatic cancer. Cancer Immunol. Immunother. 2013, 62, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef] [PubMed]
- Wilmott, J.S.; Long, G.V.; Howle, J.R.; Haydu, L.E.; Sharma, R.N.; Thompson, J.F.; Kefford, R.F.; Hersey, P.; Scolyer, R.A. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin. Cancer Res. 2012, 18, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.; Fagone, P.; Nicoletti, F.; Andersen, R.S.; Høgdall, E.; Straten, P.T.; Andersen, M.H.; Svane, I.M. BRAF inhibition improves tumor recognition by the immune system: Potential implications for combinatorial therapies against melanoma involving adoptive T-cell transfer. Oncoimmunology 2012, 1, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-tumor Immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.K.; Cheng, Y.; Cheng, M.L.; Yu, L.; Mu, M.; Li, H.; Liu, Y.; Zhang, B.; Yao, Y.; Guo, H.; et al. Focal Adhesion Kinase Regulates Fibroblast Migration via Integrin beta-1 and Plays a Central Role in Fibrosis. Sci. Rep. 2016, 6, 19276. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Durkacz, B.W.; Omidiji, O.; Gray, D.A.; Shall, S. (ADP-ribose)n participates in DNA excision repair. Nature 1980, 283, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, F.; Schreiber, V.; Niedergang, C.; Trucco, C.; Flatter, E.; De La Rubia, G.; Oliver, J.; Rolli, V.; Ménissier-de Murcia, J.; et al. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie 1999, 81, 69–75. [Google Scholar] [CrossRef]
- Shall, S.; de Murcia, G. Poly(ADP-ribose) polymerase-1: What have we learned from the deficient mouse model? Mutat. Res.-DNA Rep. 2000, 460, 1–15. [Google Scholar] [CrossRef]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Michels, J.; Vitale, I.; Saparbaev, M.; Castedo, M.; Kroemer, G. Predictive biomarkers for cancer therapy with PARP inhibitors. Oncogene 2013, 33, 3894–3907. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Wolfgang, C.L.; Zheng, L. Precision Immuno-Oncology: Prospects of Individualized Immunotherapy for Pancreatic Cancer. Cancers 2018, 10, 39. https://doi.org/10.3390/cancers10020039
Zhang J, Wolfgang CL, Zheng L. Precision Immuno-Oncology: Prospects of Individualized Immunotherapy for Pancreatic Cancer. Cancers. 2018; 10(2):39. https://doi.org/10.3390/cancers10020039
Chicago/Turabian StyleZhang, Jiajia, Christopher L. Wolfgang, and Lei Zheng. 2018. "Precision Immuno-Oncology: Prospects of Individualized Immunotherapy for Pancreatic Cancer" Cancers 10, no. 2: 39. https://doi.org/10.3390/cancers10020039
APA StyleZhang, J., Wolfgang, C. L., & Zheng, L. (2018). Precision Immuno-Oncology: Prospects of Individualized Immunotherapy for Pancreatic Cancer. Cancers, 10(2), 39. https://doi.org/10.3390/cancers10020039