Oncolytic Virotherapy versus Cancer Stem Cells: A Review of Approaches and Mechanisms

Abstract
:1. Introduction
2. Identification of Cancer Stem Cells
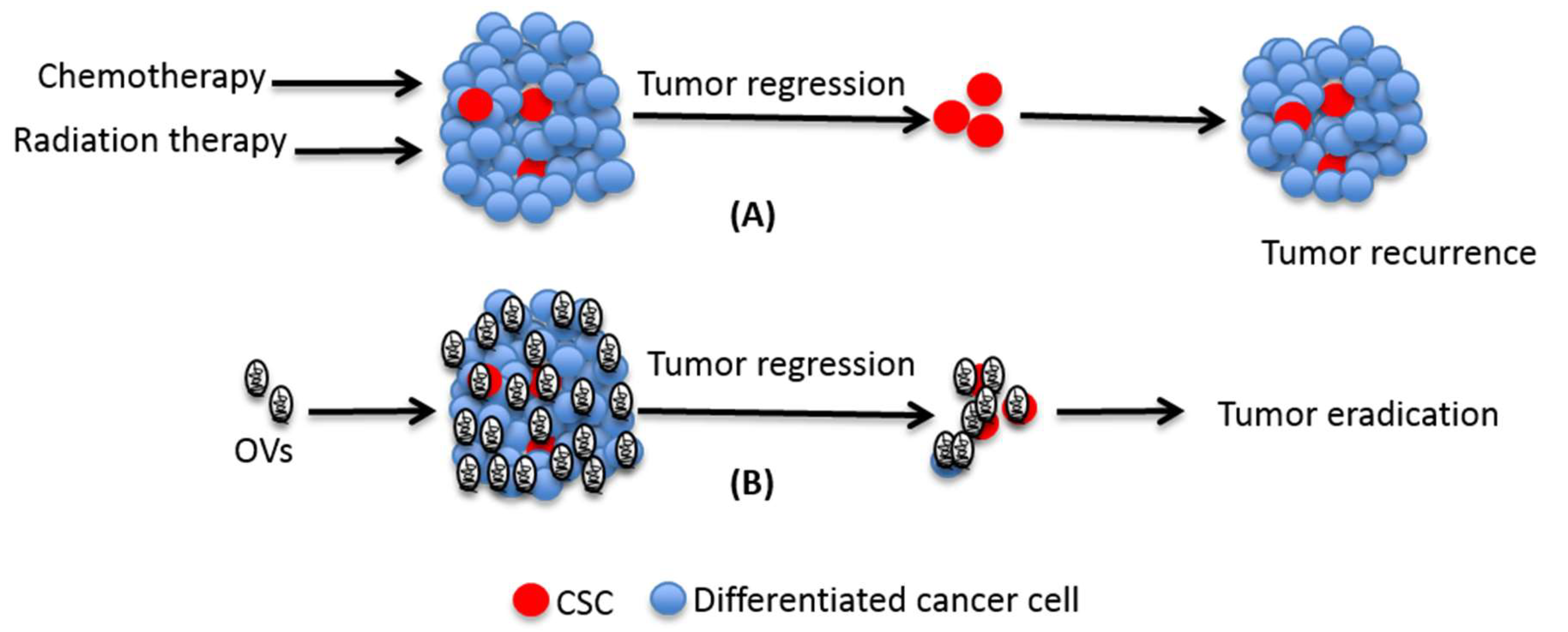
3. Conventional Cancer Therapy and CSCs
4. Oncolytic Viruses and Mechanisms of Cancer Cell Death
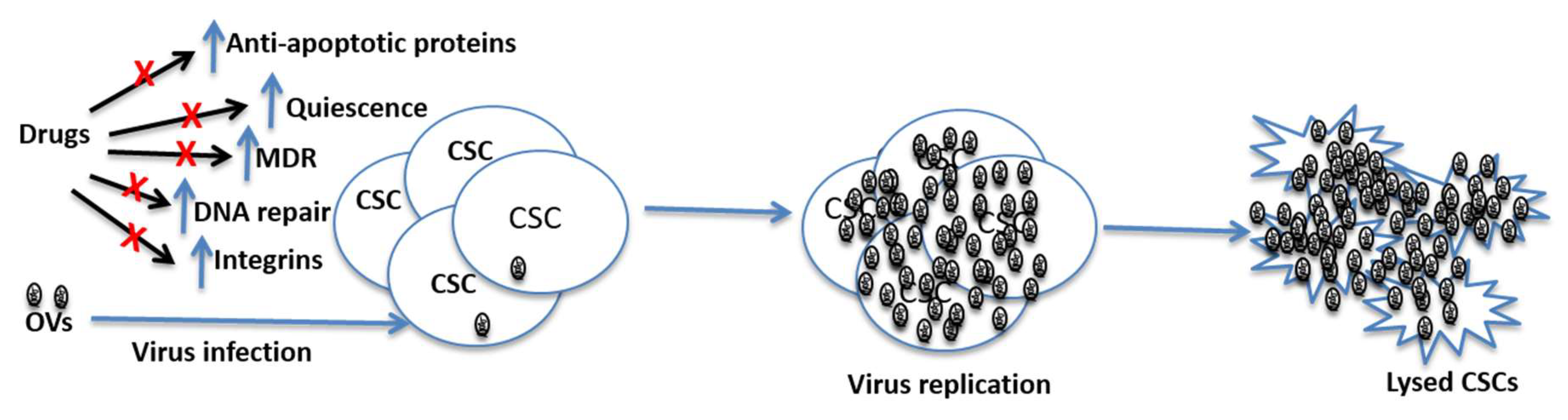
5. Mechanisms of Resistance to Chemotherapy and Radiation Therapies and the Prospect of Oncolytic Viruses in Killing CSCs
6. Oncolytic Viruses Targeting CSCs
6.1. Leukemia
6.2. Breast Cancer
6.3. Glioblastoma
6.4. Colorectal Cancer
6.5. Liver Cancer
6.6. Lung Cancer
6.7. Gastric Cancer
7. Discussions and Conclusions
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.L.; Polyak, K. Breast tumor heterogeneity: Cancer stem cells or clonal evolution? Cell Cycle 2007, 6, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Klonisch, T.; Wiechec, E.; Hombach-Klonisch, S.; Ande, S.R.; Wesselborg, S.; Schulze-Osthoff, K.; Los, M. Cancer stem cell markers in common cancers—Therapeutic implications. Trends Mol. Med. 2008, 14, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Ito, S.; Nakamura, H. Side population in MDA-MB-231 human breast cancer cells exhibits cancer stem cell-like properties without higher bone-metastatic potential. Oncol. Rep. 2011, 25, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Xiong, B.; Ma, L.; Hu, X.; Zhang, C.; Cheng, Y. Characterization of side population cells isolated from the colon cancer cell line SW480. Int. J. Oncol. 2014, 45, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Ebben, J.D.; Treisman, D.M.; Zorniak, M.; Kutty, R.G.; Clark, P.A.; Kuo, J.S. The cancer stem cell paradigm: A new understanding of tumor development and treatment. Expert Opin. Ther. Targets 2010, 14, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D. Replication-selective oncolytic adenoviruses: Virotherapy aimed at genetic targets in cancer. Oncogene 2000, 19, 6660–6669. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Target for cancer therapy: Proliferating cells or stem cells. Leukemia 2006, 20, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Kruger, G.M.; Morrison, S.J. Brain repair by endogenous progenitors. Cell 2002, 110, 399–402. [Google Scholar] [CrossRef]
- Lou, H.; Dean, M. Targeted therapy for cancer stem cells: The patched pathway and ABC transporters. Oncogene 2007, 26, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- McDermott, S.P.; Wicha, M.S. Targeting breast cancer stem cells. Mol. Oncol. 2010, 4, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Everts, B.; van der Poel, H.G. Replication-selective oncolytic viruses in the treatment of cancer. Cancer Gene Ther. 2005, 12, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Evgin, L.; Vaha-Koskela, M.; Rintoul, J.; Falls, T.; Le Boeuf, F.; Barrett, J.W.; Bell, J.C.; Stanford, M.M. Potent oncolytic activity of raccoonpox virus in the absence of natural pathogenicity. Mol. Ther. 2010, 18, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.A.; Watkins, G.; Jiang, W.G. The Coxsackie-adenovirus receptor has elevated expression in human breast cancer. Clin. Exp. Med. 2005, 5, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, X.; Fernandez, P.L.; Miquel, R.; Munoz, J.; Castronovo, V.; Menard, S.; Palacin, A.; Cardesa, A.; Campo, E. Overexpression of the 67-kD laminin receptor correlates with tumour progression in human colorectal carcinoma. J. Pathol. 1996, 179, 376–380. [Google Scholar] [CrossRef]
- Masson, D.; Jarry, A.; Baury, B.; Blanchardie, P.; Laboisse, C.; Lustenberger, P.; Denis, M.G. Overexpression of the CD155 gene in human colorectal carcinoma. Gut 2001, 49, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.D.; Nakamura, T.; Russell, S.J.; Peng, K.W. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004, 64, 4919–4926. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, S.H.; Cho, Y.S.; Choi, J.J.; Kim, Y.H.; Lee, J.H. Enhancement of the adenoviral sensitivity of human ovarian cancer cells by transient expression of coxsackievirus and adenovirus receptor (CAR). Gynecol. Oncol. 2002, 85, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.C.; Levin, B.; Hirano, T.; Yee, H.; Pampeno, C.; Meruelo, D. In vivo antitumor activity of Sindbis viral vectors. J. Natl. Cancer Inst. 2002, 94, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Ohka, S.; Matsuda, N.; Tohyama, K.; Oda, T.; Morikawa, M.; Kuge, S.; Nomoto, A. Receptor (CD155)-dependent endocytosis of poliovirus and retrograde axonal transport of the endosome. J. Virol. 2004, 78, 7186–7198. [Google Scholar] [CrossRef] [PubMed]
- Dorig, R.E.; Marcil, A.; Chopra, A.; Richardson, C.D. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 1993, 75, 295–305. [Google Scholar] [CrossRef]
- Mullen, J.T.; Tanabe, K.K. Viral oncolysis. Oncologist 2002, 7, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.C.; Thorne, S.H.; Kirn, D.H. Oncolytic adenoviruses for cancer gene therapy. Methods Mol. Biol. 2008, 433, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, S.; Hew, P.; Crosley, P.; Sharon, D.; Potts, K.; Agopsowicz, K.; Long, M.; Shi, C.; Hitt, M.M. Breast cancer gene therapy using an adenovirus encoding human IL-2 under control of mammaglobin promoter/enhancer sequences. Cancer Gene Ther. 2016, 23, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Andreansky, S.; He, B.; van Cott, J.; McGhee, J.; Markert, J.M.; Gillespie, G.Y.; Roizman, B.; Whitley, R.J. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther. 1998, 5, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oh, J.Y.; Park, B.H.; Lee, D.E.; Kim, J.S.; Park, H.E.; Roh, M.S.; Je, J.E.; Yoon, J.H.; Thorne, S.H.; et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol. Ther. 2006, 14, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.Q.; Assenberg, M.; Liu, B.L.; Wang, Y.B.; Simpson, G.; Thomas, S.; Coffin, R.S. Development of a second-generation oncolytic Herpes simplex virus expressing TNFalpha for cancer therapy. J. Gene Med. 2007, 9, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Heiber, J.F.; Barber, G.N. Vesicular stomatitis virus expressing tumor suppressor p53 is a highly attenuated, potent oncolytic agent. J. Virol. 2011, 85, 10440–10450. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.L.; Yu, Y.H.; Tian, H.; Ren, G.P.; Wang, H.; Zhou, B.; Han, X.H.; Yu, Q.Z.; Li, D.S. Genetically engineered Newcastle disease virus expressing interleukin-2 and TNF-related apoptosis-inducing ligand for cancer therapy. Cancer Biol. Ther. 2014, 15, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Arulanandam, R.; De Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.H.; et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013, 73, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Angarita, F.A.; Acuna, S.A.; Ottolino-Perry, K.; Zerhouni, S.; McCart, J.A. Mounting a strategic offense: Fighting tumor vasculature with oncolytic viruses. Trends Mol. Med. 2013, 19, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Vaha-Koskela, M.J.; Heikkila, J.E.; Hinkkanen, A.E. Oncolytic viruses in cancer therapy. Cancer Lett. 2007, 254, 178–216. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; You, L.; Liu, H.; Li, L.; Meng, H.; Qian, Q.; Qian, W. Potent antitumor activity of oncolytic adenovirus expressing Beclin-1 via induction of autophagic cell death in leukemia. Oncotarget 2013, 4, 860–874. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Madlambayan, G.J.; Rahman, M.M.; Smallwood, S.E.; Meacham, A.M.; Hosaka, K.; Scott, E.W.; Cogle, C.R.; McFadden, G. Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia 2009, 23, 2313–2317. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Guse, K.; Bauerschmitz, G.; Virkkunen, P.; Tarkkanen, M.; Tanner, M.; Hakkarainen, T.; Kanerva, A.; Desmond, R.A.; Pesonen, S.; et al. Oncolytic adenoviruses kill breast cancer initiating CD44+CD24−/low cells. Mol. Ther. 2007, 15, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Bauerschmitz, G.J.; Ranki, T.; Kangasniemi, L.; Ribacka, C.; Eriksson, M.; Porten, M.; Herrmann, I.; Ristimaki, A.; Virkkunen, P.; Tarkkanen, M.; et al. Tissue-specific promoters active in CD44+CD24-/low breast cancer cells. Cancer Res. 2008, 68, 5533–5539. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zeng, W.; Huang, Y.; Zhang, Q.; Hu, P.; Rabkin, S.D.; Liu, R. Treatment of breast cancer stem cells with oncolytic herpes simplex virus. Cancer Gene Ther. 2012, 19, 707–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Oncolytic reovirus effectively targets breast cancer stem cells. Mol. Ther. 2009, 17, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Warner, S.G.; Haddad, D.; Au, J.; Carson, J.S.; O’Leary, M.P.; Lewis, C.; Monette, S.; Fong, Y. Oncolytic herpes simplex virus kills stem-like tumor-initiating colon cancer cells. Mol. Ther. Oncolytics 2016, 3, 16013. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Gomez-Manzano, C.; Aoki, H.; Alonso, M.M.; Kondo, S.; McCormick, F.; Xu, J.; Kondo, Y.; Bekele, B.N.; Colman, H.; et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: Role of autophagic cell death. J. Natl. Cancer Inst. 2007, 99, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Kesari, S.; Farrell, C.J.; Curry, W.T., Jr.; Zaupa, C.; Aghi, M.; Kuroda, T.; Stemmer-Rachamimov, A.; Shah, K.; Liu, T.C.; et al. Human glioblastoma-derived cancer stem cells: Establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009, 69, 3472–3481. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Su, W.; Jin, G.; Xu, F.; Hao, S.; Guan, F.; Jia, W.; Liu, F. Glioma stem cells targeted by oncolytic virus carrying endostatin-angiostatin fusion gene and the expression of its exogenous gene in vitro. Brain Res. 2011, 1390, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Meng, S.; Zhang, R.; Ma, B.; Liu, T.; Yang, Y.; Xie, W.; Liu, X.; Huang, F.; Liu, T.; et al. GP73-regulated oncolytic adenoviruses possess potent killing effect on human liver cancer stem-like cells. Oncotarget 2016, 7, 29346–29358. [Google Scholar] [CrossRef] [PubMed]
- Bach, P.; Abel, T.; Hoffmann, C.; Gal, Z.; Braun, G.; Voelker, I.; Ball, C.R.; Johnston, I.C.; Lauer, U.M.; Herold-Mende, C.; et al. Specific elimination of CD133+ tumor cells with targeted oncolytic measles virus. Cancer Res. 2013, 73, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, H.; Huang, W.; Ding, M.; Xiao, J.; Yang, D.; Li, H.; Liu, X.Y.; Chu, L. Targeting lung cancer stem-like cells with TRAIL gene armed oncolytic adenovirus. J. Cell. Mol. Med. 2015, 19, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Tazawa, H.; Hashimoto, Y.; Shirakawa, Y.; Kuroda, S.; Nishizaki, M.; Kishimoto, H.; Uno, F.; Nagasaka, T.; Urata, Y.; et al. A genetically engineered oncolytic adenovirus decoys and lethally traps quiescent cancer stem-like cells in S/G2/M phases. Clin. Cancer Res. 2013, 19, 6495–6505. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and cancer: Regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.; Wei, X. Cancer stem cells and drug resistance: The potential of nanomedicine. Nanomedicine 2012, 7, 597–615. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, H.; Shen, J.; Yang, Y.; Wu, S.; Xiao, J.; Xu, Y.; Liu, X.Y.; Chu, L. RGD-modifided oncolytic adenovirus exhibited potent cytotoxic effect on CAR-negative bladder cancer-initiating cells. Cell Death Dis. 2015, 6, e1760. [Google Scholar] [CrossRef] [PubMed]
- Ammayappan, A.; Peng, K.W.; Russell, S.J. Characteristics of oncolytic vesicular stomatitis virus displaying tumor-targeting ligands. J. Virol. 2013, 87, 13543–13555. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bao, Q.; Renner, A.; Camaj, P.; Eichhorn, M.; Ischenko, I.; Angele, M.; Kleespies, A.; Jauch, K.W.; Bruns, C. Cancer stem cells and angiogenesis. Int. J. Dev. Biol. 2011, 55, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Adini, A.; Adini, I.; Ghosh, K.; Benny, O.; Pravda, E.; Hu, R.; Luyindula, D.; D’Amato, R.J. The stem cell marker prominin-1/CD133 interacts with vascular endothelial growth factor and potentiates its action. Angiogenesis 2013, 16, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.Y.; Schwartz, B.E.; Hsu, M.Y. CD133+ melanoma subpopulations contribute to perivascular niche morphogenesis and tumorigenicity through vasculogenic mimicry. Cancer Res. 2012, 72, 5111–5118. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.B.; Schnegg, C.; Lai, C.Y.; Ghosh, S.; Yang, M.H.; Moffat, J.; Hsu, M.Y. CD133-targeted niche-dependent therapy in cancer: A multipronged approach. Am. J. Pathol. 2014, 184, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Folkins, C.; Man, S.; Xu, P.; Shaked, Y.; Hicklin, D.J.; Kerbel, R.S. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007, 67, 3560–3564. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Chen, H.; Rojas, J.; Sampath, P.; Thorne, S.H. Oncolytic vaccinia virus demonstrates antiangiogenic effects mediated by targeting of VEGF. Int. J. Cancer 2014, 135, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Joo, K.M.; Jin, J.; Nam, D.H. Cancer stem cells and their mechanism of chemo-radiation resistance. Int. J. Stem Cells 2009, 2, 109–114. [Google Scholar] [CrossRef] [PubMed]
- McAuliffe, S.M.; Morgan, S.L.; Wyant, G.A.; Tran, L.T.; Muto, K.W.; Chen, Y.S.; Chin, K.T.; Partridge, J.C.; Poole, B.B.; Cheng, K.H.; et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E2939–E2948. [Google Scholar] [CrossRef] [PubMed]
- Mato-Berciano, A.; Raimondi, G.; Maliandi, M.V.; Alemany, R.; Montoliu, L.; Fillat, C. A NOTCH-sensitive uPAR-regulated oncolytic adenovirus effectively suppresses pancreatic tumor growth and triggers synergistic anticancer effects with gemcitabine and nab-paclitaxel. Oncotarget 2017, 8, 22700–22715. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.; Majumder, B.; Dhawan, A.; Ravi, S.; Goldman, D.; Kohandel, M.; Majumder, P.K.; Sengupta, S. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat. Commun. 2015, 6, 6139. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien-Ball, C.; Biddle, A. Reprogramming to developmental plasticity in cancer stem cells. Dev. Biol. 2017, 430, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Deheeger, M.; Lesniak, M.S.; Ahmed, A.U. Cellular plasticity regulated cancer stem cell niche: A possible new mechanism of chemoresistance. Cancer Cell. Microenviron. 2014, 1. [Google Scholar] [CrossRef]
- Blick, T.; Hugo, H.; Widodo, E.; Waltham, M.; Pinto, C.; Mani, S.A.; Weinberg, R.A.; Neve, R.M.; Lenburg, M.E.; Thompson, E.W. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi)/CD24 (lo)/-stem cell phenotype in human breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Chen, W.Y.; Lin, S.F.; Wong, R.J. Epithelial-mesenchymal transition enhances response to oncolytic herpesviral therapy through nectin-1. Hum. Gene Ther. 2014, 25, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Trumpp, A.; Wiestler, O.D. Mechanisms of Disease: Cancer stem cells--targeting the evil twin. Nat. Clin. Pract. Oncol. 2008, 5, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Rich, J.N. Survival of the fittest: Cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 2008, 26, 2839–2845. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Cherubini, G.; Lemoine, N.R.; Hallden, G. The E1B19K-deleted oncolytic adenovirus mutant AdDelta19K sensitizes pancreatic cancer cells to drug-induced DNA-damage by down-regulating Claspin and Mre11. Oncotarget 2016, 7, 15703–15724. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, G.; Kallin, C.; Mozetic, A.; Hammaren-Busch, K.; Muller, H.; Lemoine, N.R.; Hallden, G. The oncolytic adenovirus AdDeltaDelta enhances selective cancer cell killing in combination with DNA-damaging drugs in pancreatic cancer models. Gene Ther. 2011, 18, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Aghi, M.; Rabkin, S.; Martuza, R.L. Effect of chemotherapy-induced DNA repair on oncolytic herpes simplex viral replication. J. Natl. Cancer Inst. 2006, 98, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Scully, R. Hijacking the DNA damage response to enhance viral replication: Gamma-herpesvirus 68 orf36 phosphorylates histone H2AX. Mol. Cell 2007, 27, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin®) as treatment of recurrent glioblastoma multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.C.; Farrell, A.T.; Madabushi, R.; Booth, B.; Chattopadhyay, S.; Sridhara, R.; Justice, R.; Pazdur, R. Sorafenib for the treatment of unresectable hepatocellular carcinoma. Oncologist 2009, 14, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Waxman, D.J. Combination of antiangiogenesis with chemotherapy for more effective cancer treatment. Mol. Cancer Ther. 2008, 7, 3670–3684. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, S.; Dobrucki, I.T.; Kim, E.Y.; Marrison, S.T.; Vu, V.T. Hypoxia and radiation therapy: Past history, ongoing research, and future promise. Curr. Mol. Med. 2009, 9, 442–458. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Hypoxia and drug resistance. Cancer Metastasis Rev. 1994, 13, 139–168. [Google Scholar] [CrossRef] [PubMed]
- Benencia, F.; Courreges, M.C.; Conejo-Garcia, J.R.; Buckanovich, R.J.; Zhang, L.; Carroll, R.H.; Morgan, M.A.; Coukos, G. Oncolytic HSV exerts direct antiangiogenic activity in ovarian carcinoma. Hum. Gene Ther. 2005, 16, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Sunamura, M.; Motoi, F.; Abe, H.; Egawa, S.; Duda, D.G.; Hoshida, T.; Fukuyama, S.; Hamada, H.; Matsuno, S. Oncolytic replication-competent adenovirus suppresses tumor angiogenesis through preserved E1A region. Cancer Gene Ther. 2006, 13, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Paterson, J.M.; Lemay, C.G.; Falls, T.J.; McGuire, A.; Parato, K.A.; Stojdl, D.F.; Daneshmand, M.; Speth, K.; Kirn, D.; et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol. Ther. 2007, 15, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Fasullo, M.; Burch, A.D.; Britton, A. Hypoxia enhances the replication of oncolytic herpes simplex virus in p53- breast cancer cells. Cell Cycle 2009, 8, 2194–2197. [Google Scholar] [CrossRef] [PubMed]
- Hay, J.G. The potential impact of hypoxia on the success of oncolytic virotherapy. Curr. Opin. Mol. Ther. 2005, 7, 353–358. [Google Scholar] [PubMed]
- Hiley, C.T.; Yuan, M.; Lemoine, N.R.; Wang, Y. Lister strain vaccinia virus, a potential therapeutic vector targeting hypoxic tumours. Gene Ther. 2010, 17, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Reinblatt, M.; Pin, R.H.; Federoff, H.J.; Fong, Y. Utilizing tumor hypoxia to enhance oncolytic viral therapy in colorectal metastases. Ann. Surg. 2004, 239, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Pajonk, F.; Vlashi, E.; McBride, W.H. Radiation resistance of cancer stem cells: The 4 R’s of radiobiology revisited. Stem Cells 2010, 28, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Holtz, M.; Forman, S.J.; Bhatia, R. Growth factor stimulation reduces residual quiescent chronic myelogenous leukemia progenitors remaining after imatinib treatment. Cancer Res. 2007, 67, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Lechman, E.R.; Gentner, B.; Ng, S.W.K.; Schoof, E.M.; van Galen, P.; Kennedy, J.A.; Nucera, S.; Ciceri, F.; Kaufmann, K.B.; Takayama, N.; et al. miR-126 Regulates Distinct Self-Renewal Outcomes in Normal and Malignant Hematopoietic Stem Cells. Cancer Cell 2016, 29, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, N.G.; Minev, B.R.; Szalay, A.A. Oncolytic vaccinia virus GLV-1h68 strain shows enhanced replication in human breast cancer stem-like cells in comparison to breast cancer cells. J. Transl. Med. 2012, 10, 167. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, S.; Favis, N.; Umer, B.; Potts, K.; Noyce, R.; Irwin, C.; Evans, D.H.; Hitt, M.M. F4L-deleted Vaccinia Virus Exhibits Oncolytic Activity in Breast Cancer Models. Sci. Rep. (under revision).
- Levine, P.H.; Steinhorn, S.C.; Ries, L.G.; Aron, J.L. Inflammatory breast cancer: The experience of the surveillance, epidemiology, and end results (SEER) program. J. Natl. Cancer Inst. 1985, 74, 291–297. [Google Scholar] [PubMed]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeyer, M.R.; Wall, K.M.; Dharmawardhane, S.F. In vitro analysis of the invasive phenotype of SUM 149, an inflammatory breast cancer cell line. Cancer Cell Int. 2005, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.C.; Ehrlich, S.B. Positional influences on job satisfaction and job performance: A multivariate, predictive approach. Psychol. Rep. 1991, 69, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Edlund, K.; Bergenheim, A.T.; Wadell, G. Adenoviruses 16 and CV23 efficiently transduce human low-passage brain tumor and cancer stem cells. Mol. Ther. 2007, 15, 2140–2145. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.Y.; Bang, S.Y.; Jeong, S.N.; Kang, D.H.; Heo, J. A cancer-favoring oncolytic vaccinia virus shows enhanced suppression of stem-cell like colon cancer. Oncotarget 2016, 7, 16479–16489. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.K.; Wo, J.Y.; Ng, I.O.; Zheng, B.J.; Guan, X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef] [PubMed]
- Sagara, M.; Inoue, H.; Miyamoto, S.; Sakamoto, C.; Nakano, Y.; Takayama, K.; Shimizu, H.; Nakanishi, Y.; Tani, K. CVB3 Infection Elicits Potent Oncolytic Activity Against Lung Cancer Stem Cells. Mol. Ther. 2013, 21, S170. [Google Scholar] [CrossRef]
- Chaurasiya, S.; Chen, N.G.; Fong, Y.; City of Hope National Medical Center, Duarte, CA 91010, USA. Personal observation, 2017.
- Zhang, X.; Komaki, R.; Wang, L.; Fang, B.; Chang, J.Y. Treatment of radioresistant stem-like esophageal cancer cells by an apoptotic gene-armed, telomerase-specific oncolytic adenovirus. Clin. Cancer Res. 2008, 14, 2813–2823. [Google Scholar] [CrossRef] [PubMed]
- Schatton, T.; Frank, M.H. The in vitro spheroid melanoma cell culture assay: Cues on tumor initiation? J. Investig. Dermatol. 2010, 130, 1769–1771. [Google Scholar] [CrossRef] [PubMed]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig. 2010, 120, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Stiles, B.M.; Chan, M.K.; Chou, T.C.; Wong, R.J.; Rusch, V.W.; Fong, Y. Radiation therapy potentiates effective oncolytic viral therapy in the treatment of lung cancer. Ann. Thorac. Surg. 2005, 80, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.H.; Zamarin, D.; Gao, S.P.; Chou, T.C.; Gonzalez, L.; Lin, S.F.; Fong, Y. Synergistic action of oncolytic herpes simplex virus and radiotherapy in pancreatic cancer cell lines. Br. J. Surg 2010, 97, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Sikorski, R.; Kirn, D.H.; Thorne, S.H. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the IFN response and HMGB1. Gene Ther. 2011, 18, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, L.; Simpson, G.R.; Boxall, A.; Kottke, T.; Relph, K.L.; Vile, R.; Melcher, A.; Prestwich, R.; Harrington, K.J.; Morgan, R.; et al. Synergistic effects of oncolytic reovirus and docetaxel chemotherapy in prostate cancer. BMC Cancer 2011, 11, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Cancer Type | OV | CSC Source | CSCs Susceptible to OV? | OV Replicates in CSC? | Comments | Reference |
|---|---|---|---|---|---|---|
| CML | Ad | Imatinib-resistant CML patients | Yes | NA | Inhibition of colony formation in vitro and elimination of xenografts in mice | [52] |
| AML | MYXV | AML patients | Yes | Yes | Prior infection of tumor cells with virus prevented engraftment in 90% of recipient mice compared to mock-infected cells | [53] |
| Breast | Ad | Pleural effusion from breast cancer patients | Yes | NA | Effective killing of CSCs in vitro; prior infection of CSCs prevented formation of xenografts; anti-tumor effect against CSCs derived tumors in mice | [54] |
| Breast | Ad | Pleural effusion from breast cancer patients | Yes | NA | Eradication of CSCs in vitro; anti-tumor effect against CSCs derived tumors in mice | [55] |
| Breast | HSV | Mammospheres generated from breast cancer cell lines | Yes | NA | Highly toxic to CSCs in vitro and effective against CSC-derived xenografts in mice | [56] |
| Breast | Reo | Human breast cancer patients | Yes | NA | Ras expression, a determinant of reovirus oncolysis, was similar in CSCs and non-CSCs; similar killing of CSC and non-CSCs both in vitro and in xenografts | [57] |
| Colon | HSV | Tumorspheres generated from HCT8 cells | Yes | Yes | Highly toxic to Akt overexpressing CSCs; effective against CSC-derived xenografts in mice | [58] |
| Glioblastoma | Ad | Glioblastoma patients | Yes | Yes | CSCs over-expressed Ad receptor (CAR) and were highly susceptible to the virus in vitro; significant anti-tumor effect against CSC-derived xenograft | [59] |
| Glioblastoma | HSV | Glioblastoma specimen from human patients | Yes | Yes | Effective killing of CSCs in vitro and significant anti-tumor effect against xenografts in mice | [60] |
| Glioblastoma | HSV | Glioblastoma specimen from human patients | Yes | NA | Effective oncolysis of CSC in vitro and anti-tumor effect against CSC-derived xenografts in mice | [61] |
| Liver | Ad | Liver cancer cell lines | Yes | NA | Highly toxic to CSCs both in vitro and in xenograft models | [62] |
| Liver | Measles | Surgical specimen from liver cancer patients | Yes | NA | CD133-targeted OV selectively killed CD133+ CSCs and prolonged survival of mice bearing orthotopic xenografts | [63] |
| Lung | Ad | Tumorspheres generated from A549 cells | Yes | NA | TRAIL-encoding OV was toxic to CSCs in vitro and showed significant anti-tumor effect against CSCs derived xenografts in mice | [64] |
| Gastric | Ad | Human castric cancer cell lines | Yes | Yes | The virus first-induced cell-cycle mobilization from G0-G1 to S/G2/M in CSCs and then killed them; OV also sensitized those cells to chemotherapies | [65] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaurasiya, S.; Chen, N.G.; Warner, S.G. Oncolytic Virotherapy versus Cancer Stem Cells: A Review of Approaches and Mechanisms. Cancers 2018, 10, 124. https://doi.org/10.3390/cancers10040124
Chaurasiya S, Chen NG, Warner SG. Oncolytic Virotherapy versus Cancer Stem Cells: A Review of Approaches and Mechanisms. Cancers. 2018; 10(4):124. https://doi.org/10.3390/cancers10040124
Chicago/Turabian StyleChaurasiya, Shyambabu, Nanhai G. Chen, and Susanne G. Warner. 2018. "Oncolytic Virotherapy versus Cancer Stem Cells: A Review of Approaches and Mechanisms" Cancers 10, no. 4: 124. https://doi.org/10.3390/cancers10040124
APA StyleChaurasiya, S., Chen, N. G., & Warner, S. G. (2018). Oncolytic Virotherapy versus Cancer Stem Cells: A Review of Approaches and Mechanisms. Cancers, 10(4), 124. https://doi.org/10.3390/cancers10040124