CRISPR-Mediated Reactivation of DKK3 Expression Attenuates TGF-β Signaling in Prostate Cancer

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
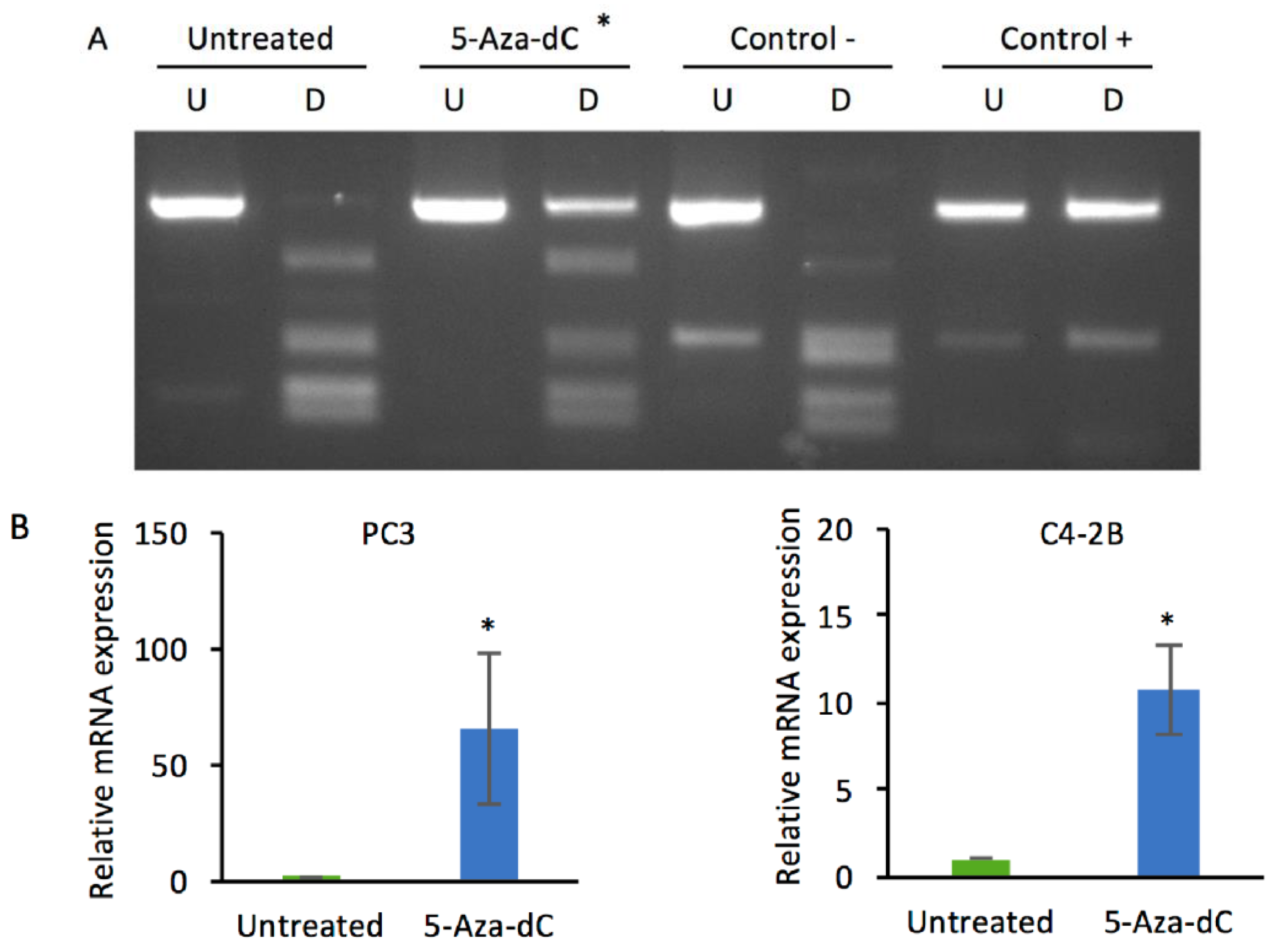
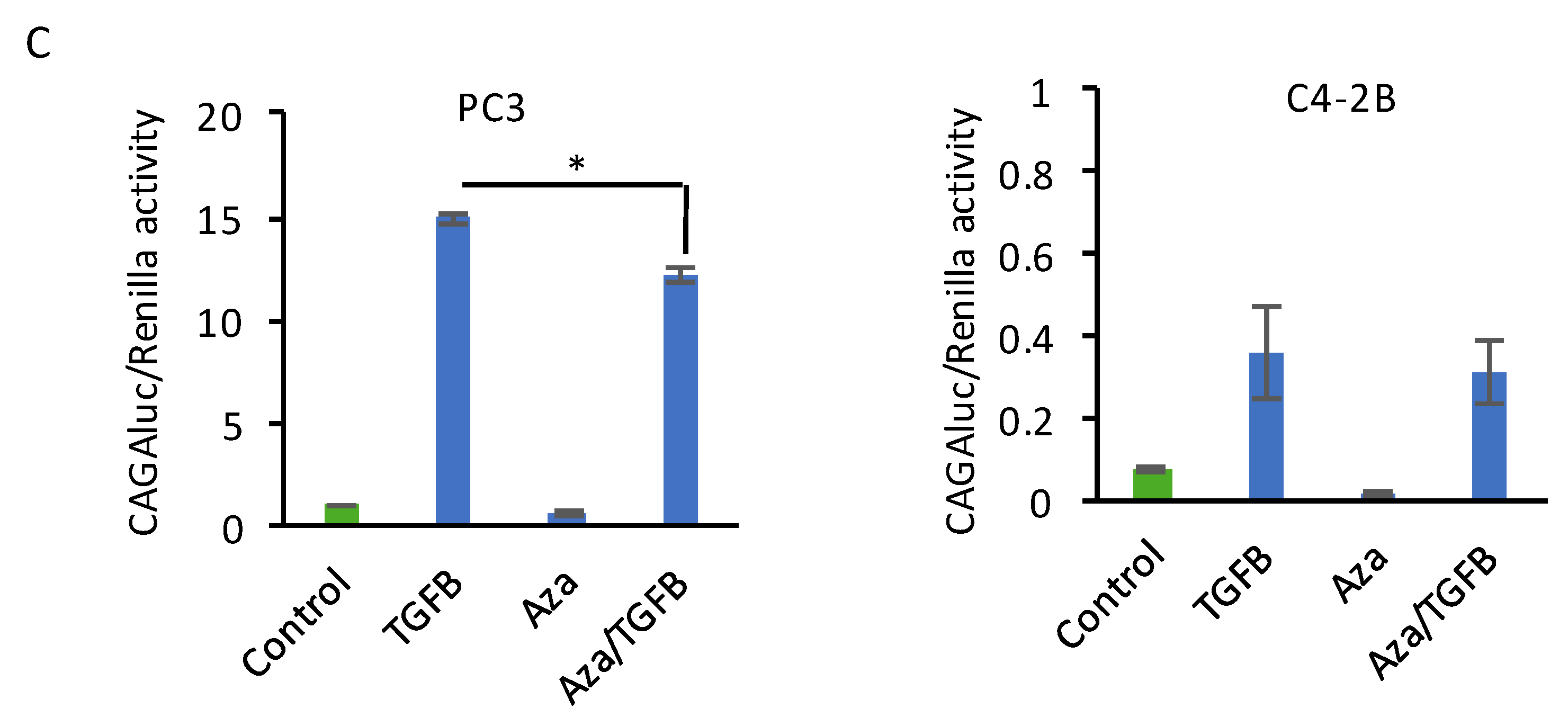
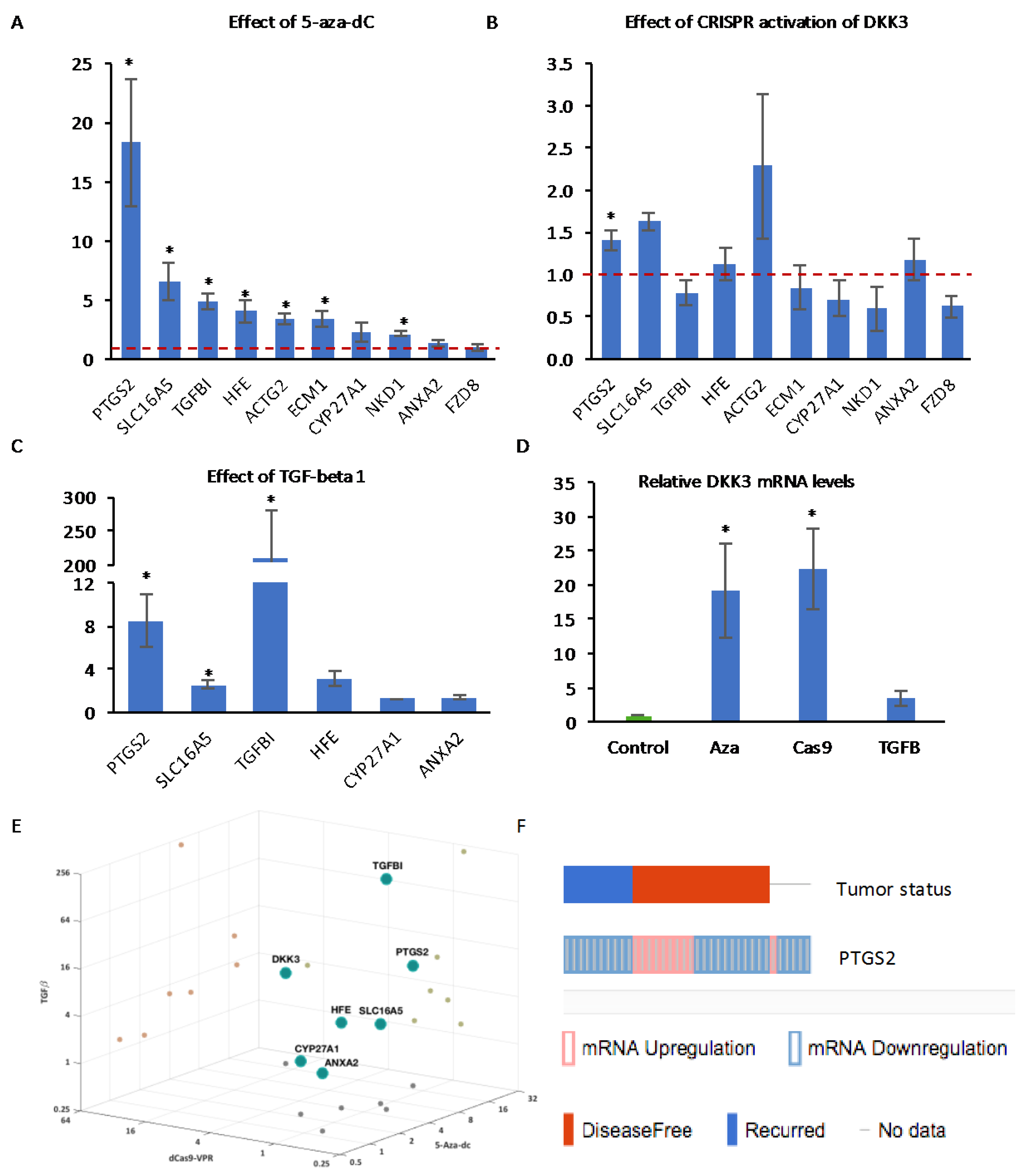
2.1. Decitabine Treatment of PC3 Cells Increases DKK3 Expression and Inhibits TGF-β-Dependent Gene Reporter Activity
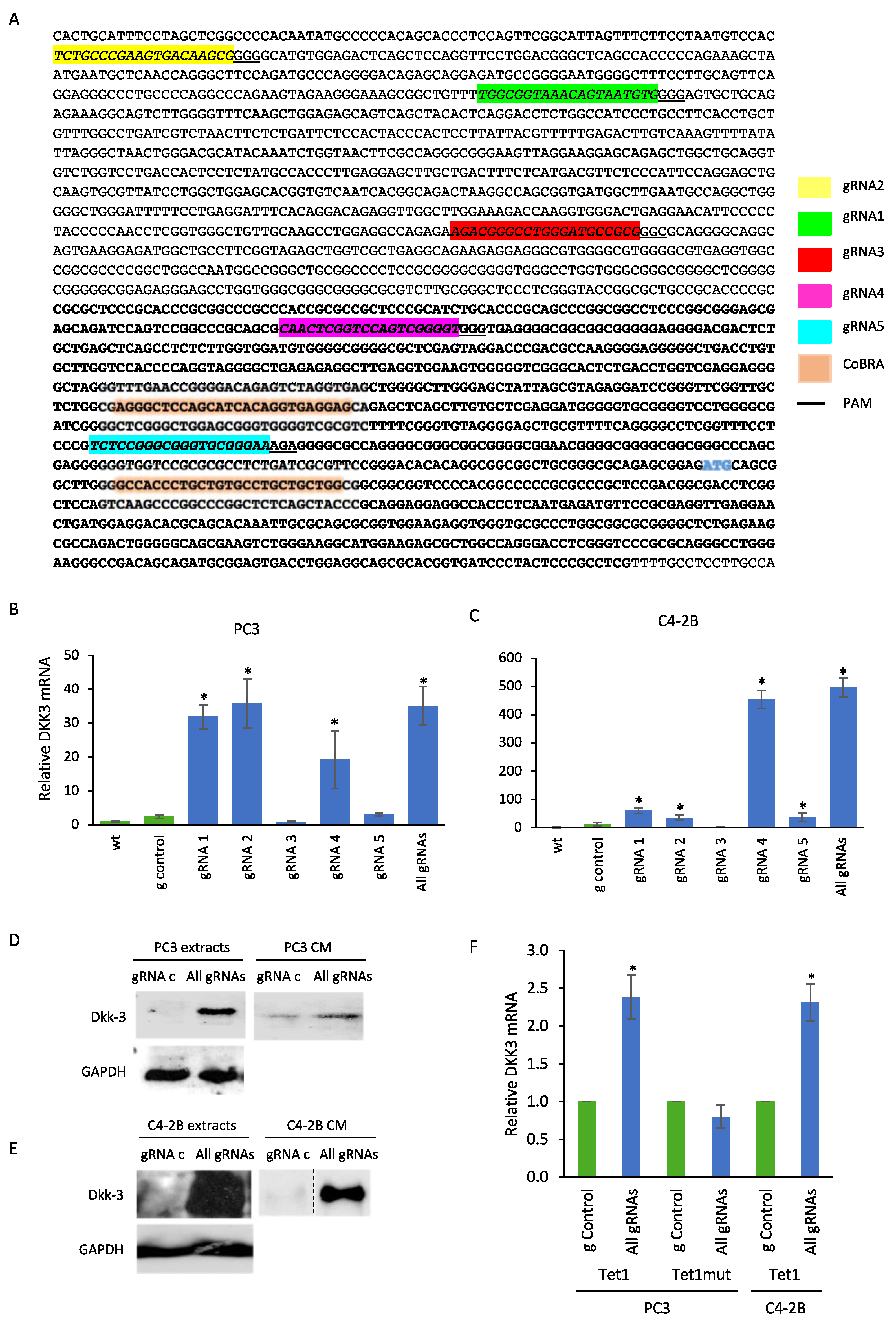
2.2. CRISPR-Mediated Activation of the DKK3 Promoter Increases DKK3 mRNA and Dkk-3 Protein Levels
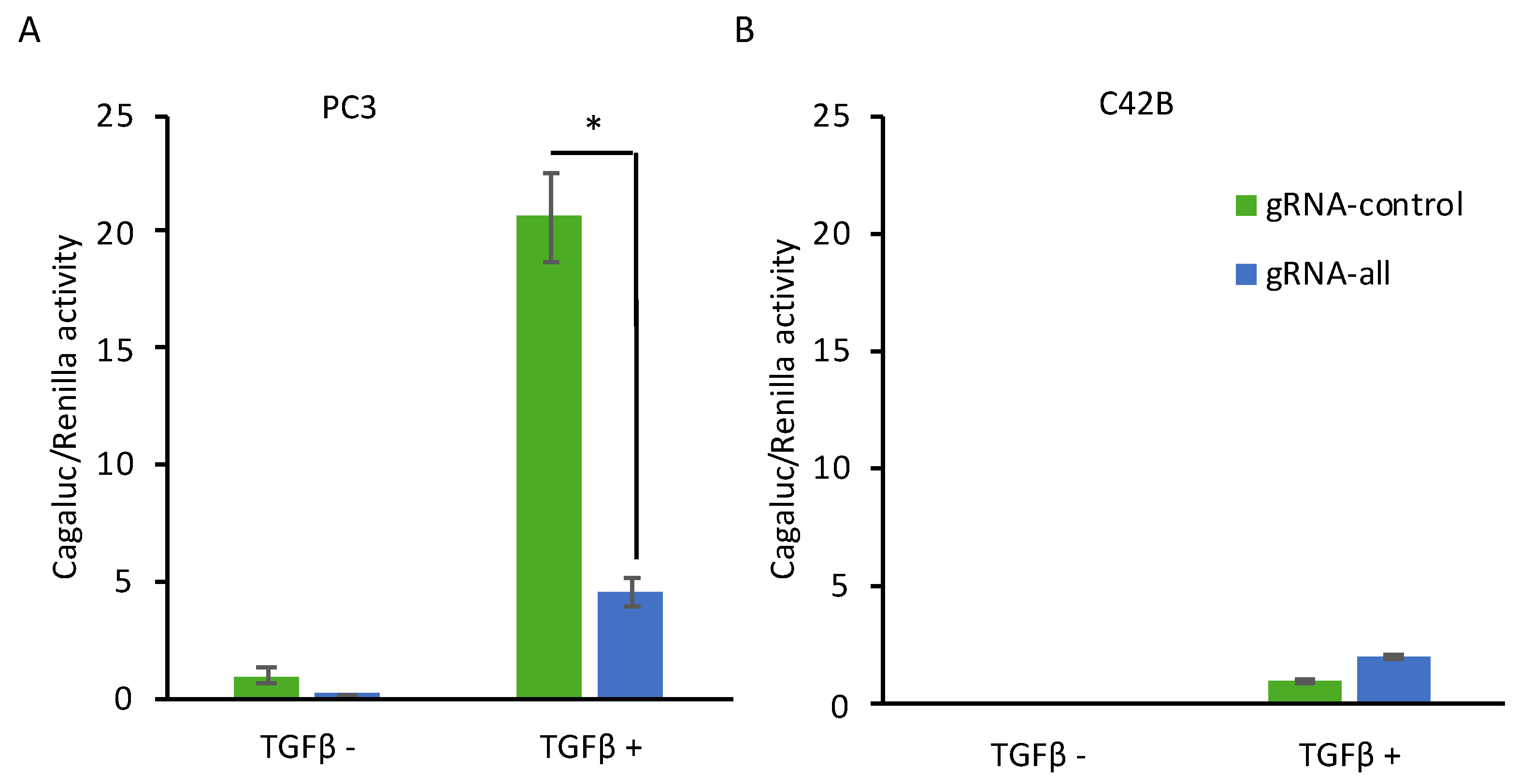
2.3. CRISPR-Mediated Induction of Dkk-3 Inhibits TGF-β Signaling in PC3 Cells
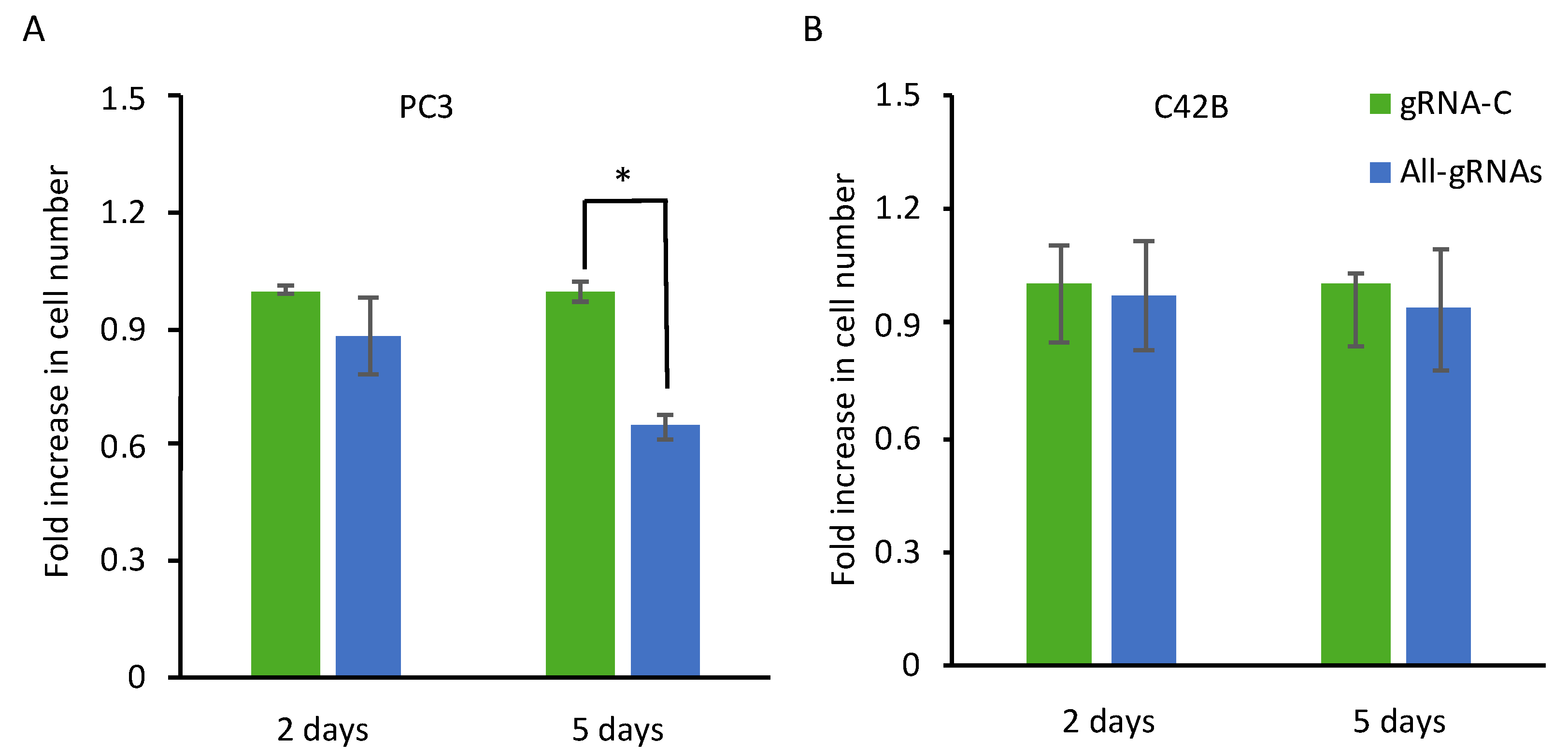
2.4. CRISPR-Mediated Induction of Dkk-3 Reduces PC3 Cell Number
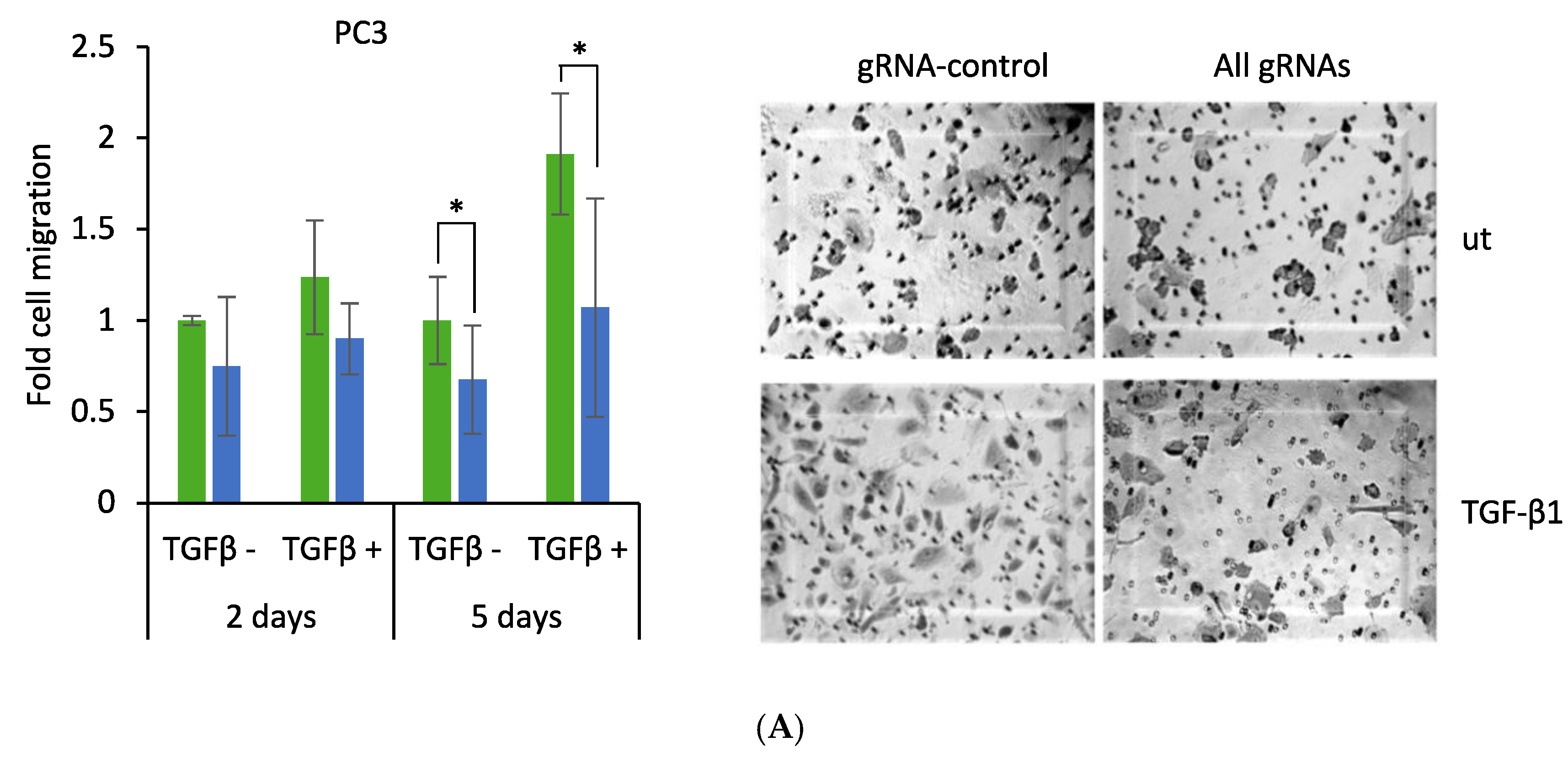
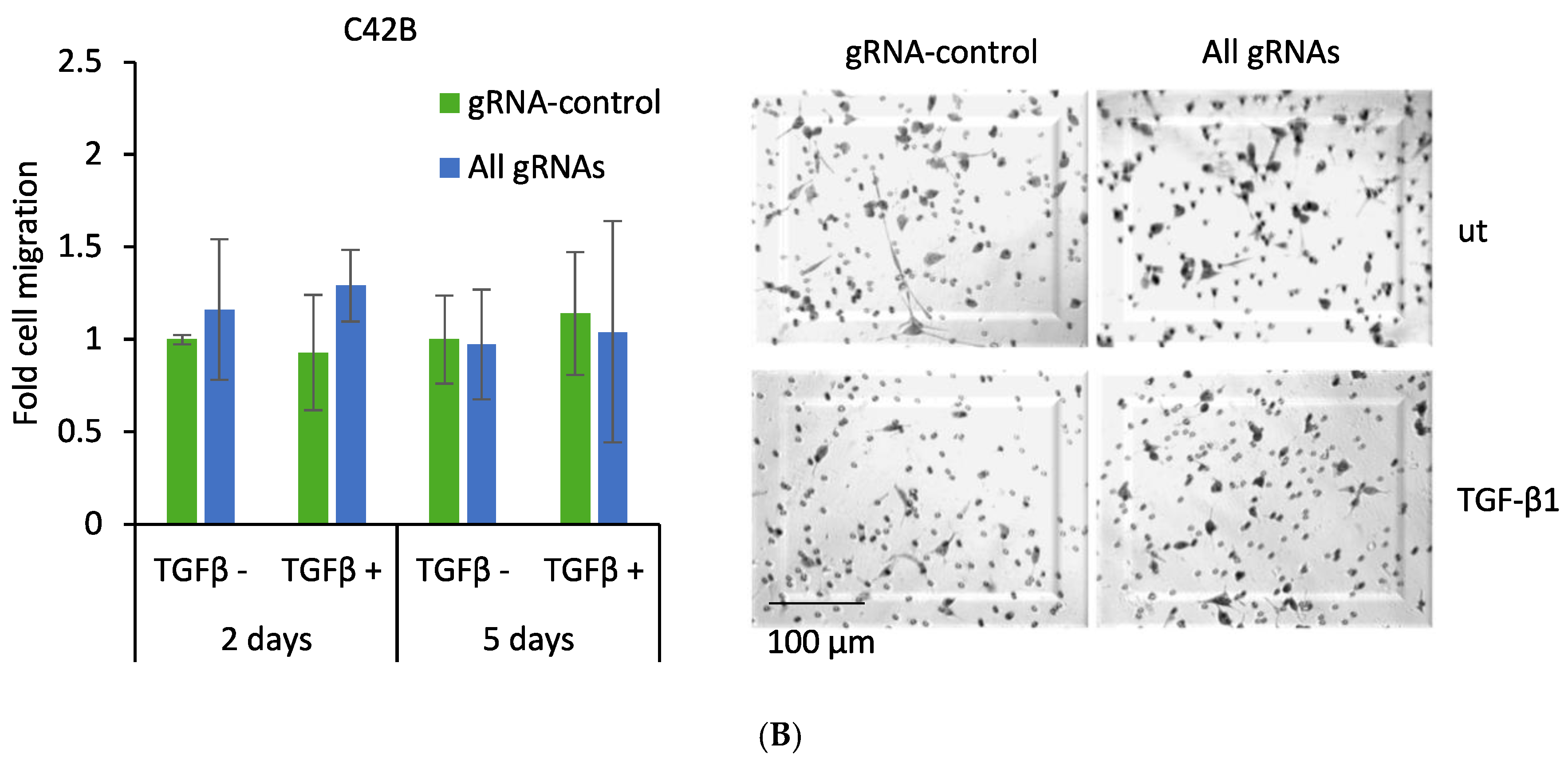
2.5. CRISPR-Mediated Induction of Dkk-3 Reduces PC3 Cell Migration
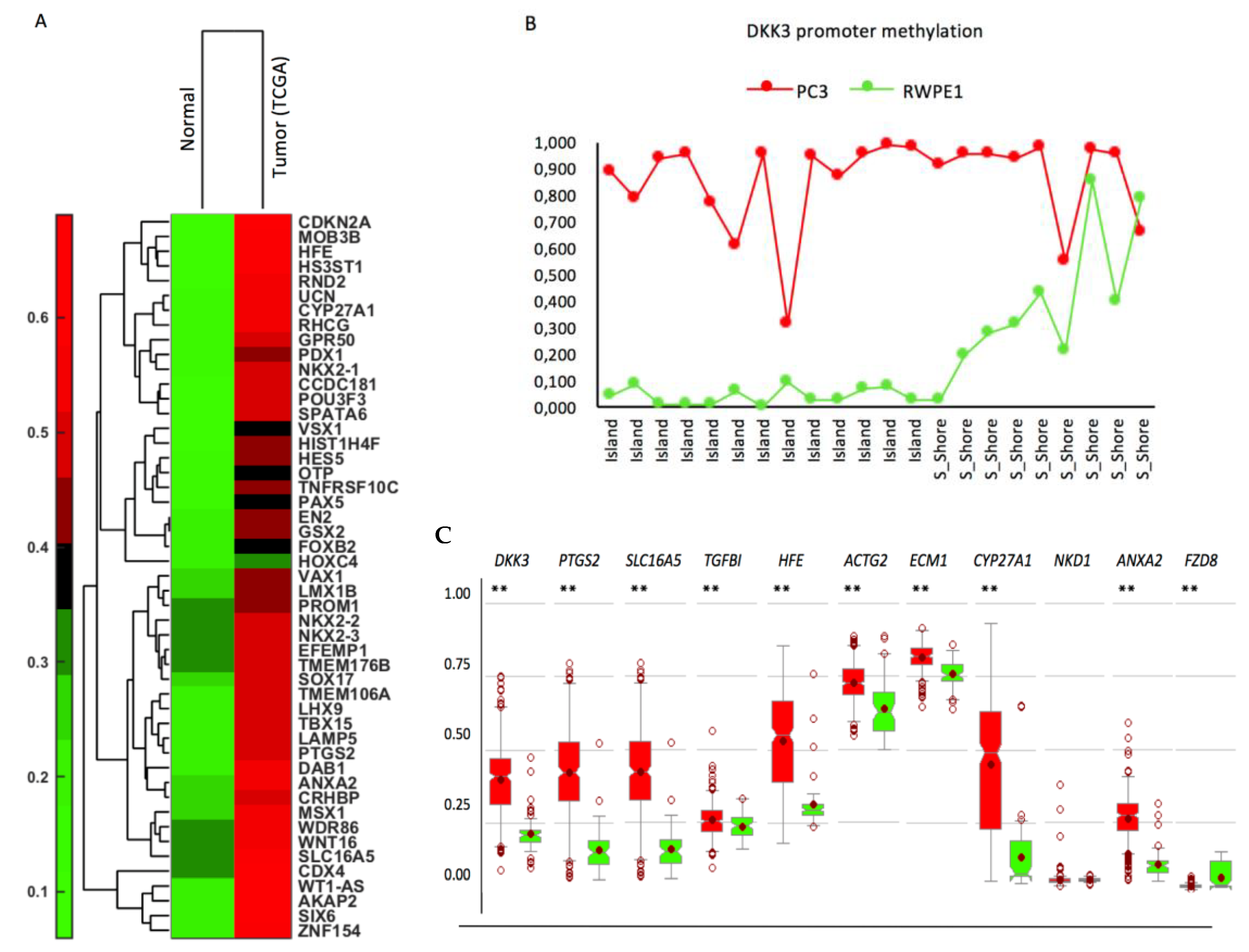
2.6. Effect of Activation of Endogenous DKK3 on Gene Expression
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Plasmid Vectors and gRNA Design
4.3. Transient Transfection
4.4. RNA Extraction, cDNA Synthesis, and qPCR
4.5. Demethylation Using 5-Aza-2′Deoxycytidine (5-Aza-dC)
4.6. Genomic DNA Extraction, Bisulfite Conversion, and Combined Bisulfite Restriction Analysis (CoBRA)
4.7. Protein Extraction, Western Blotting, and ELISAs
4.8. Gene Reporter, Proliferation, and Migration Assays
4.9. Bioinformatics Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, N.; Dahut, W.L.; Steinberg, S.M.; Figg, W.D.; Tarassoff, C.; Arlen, P.; Gulle, J.L. A retrospective study of the time to clinical endpoints for advanced prostate cancer. BJU Int. 2005, 96, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Mills, I.G.; Lynch, A.G. The importance of DNA methylation in prostate cancer development. J. Steroid Biochem. Mol. Biol. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005, 2, S4–S11. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Herman, J.G. Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J. Pathol. 2002, 196, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rhee, I.; Bachman, K.E.; Park, B.H.; Jair, K.-W.; Yen, R.-W.C.; Schuebel, K.E.; Cui, H.; Feinberg, A.P.; Lengauer, C.; Kinzler, K.W.; Baylin, S.B.; Vogelstein, B. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002, 416, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases DNMT3A and DNMT3B are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Robert, M.-F.; Morin, S.; Beaulieu, N.; Gauthier, F.; Chute, I.C.; Barsalou, A.; MacLeod, A.R. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat. Genet. 2003, 33, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Smeets, E.; Lynch, A.G.; Prekovic, S.; Van den Broeck, T.; Moris, L.; Helsen, C.; Joniau, S.; Claessens, F.; Massie, C.E. The role of TET-mediated DNA hydroxymethylation in prostate cancer. Mol. Cell. Endocrinol. 2018, 462, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Abarzua, F. Adenovirus-Mediated Overexpression of REIC/Dkk-3 Selectively Induces Apoptosis in Human Prostate Cancer Cells through Activation of c-Jun-NH2-Kinase. Cancer Res. 2005, 65, 9617–9622. [Google Scholar] [CrossRef] [PubMed]
- Lodygin, D.; Epanchintsev, A.; Menssen, A.; Diebold, J.; Hermeking, H. Functional epigenomics identifies genes frequently silenced in prostate cancer. Cancer Res. 2005, 65, 4218–4227. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Kitaoka, M.; Hamada, Y.; Walker, M.; Waxman, J.; Kypta, R. Regulation of prostate cell growth and morphogenesis by Dickkopf-3. Oncogene 2006, 25, 6528–6537. [Google Scholar] [CrossRef] [PubMed]
- Zenzmaier, C.; Untergasser, G.; Hermann, M.; Dirnhofer, S.; Sampson, N.; Berger, P. Dysregulation of Dkk-3 expression in benign and malignant prostatic tissue. Prostate 2008, 68, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Romero, D.; Kypta, R. Dickkopf-3 function in the prostate: implications for epithelial homeostasis and tumor progression. Bioarchitecture 2013, 3, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Veeck, J.; Dahl, E. Targeting the Wnt pathway in cancer: The emerging role of Dickkopf-3. Biochim. Biophys. Acta Rev. Cancer 2012, 1825, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Edamura, K.; Nasu, Y.; Takaishi, M.; Kobayashi, T.; Abarzua, F.; Sakaguchi, M.; Kashiwakura, Y.; Ebara, S.; Saika, T.; Watanabe, M.; et al. Adenovirus-mediated REIC/Dkk-3 gene transfer inhibits tumor growth and metastasis in an orthotopic prostate cancer model. Cancer Gene Ther. 2007, 14, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Romero, D.; Kawano, Y.; Bengoa, N.; Walker, M.M.; Maltry, N.; Niehrs, C.; Waxman, J.; Kypta, R. Downregulation of Dickkopf-3 disrupts prostate acinar morphogenesis through TGF-β/Smad signaling. J. Cell Sci. 2013, 6, 1208. [Google Scholar] [CrossRef]
- Kumon, H.; Sasaki, K.; Ariyoshi, Y.; Sadahira, T.; Ebara, S.; Hiraki, T.; Kanazawa, S.; Yanai, H.; Watanabe, M.; Nasu, Y. Ad-REIC gene therapy: Promising results in a patient with metastatic CRPC following chemotherapy. Clin. Med. Insights Oncol. 2015, 9, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Kumon, H.; Ariyoshi, Y.; Sasaki, K.; Sadahira, T.; Araki, M.; Ebara, S.; Yanai, H.; Watanabe, M.; Nasu, Y. Adenovirus vector carrying REIC/DKK-3 gene: neoadjuvant intraprostatic injection for high-risk localized prostate cancer undergoing radical prostatectomy. Cancer Gene Ther. 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene 2006, 25, 7469–7481. [Google Scholar] [CrossRef] [PubMed]
- Romero, D.; Al-Shareef, Z.; Gorroño-Etxebarria, I.; Atkins, S.; Turrell, F.; Chhetri, J.; Bengoa-Vergniory, N.; Zenzmaier, C.; Berger, P.; Waxman, J.; et al. Dickkopf-3 regulates prostate epithelial cell acinar morphogenesis and prostate cancer cell invasion by limiting TGF-β-dependent activation of matrix metalloproteases. Carcinogenesis 2016, 37, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Zenzmaier, C.; Sampson, N.; Plas, E.; Berger, P. Dickkopf-related protein 3 promotes pathogenic stromal remodeling in benign prostatic hyperplasia and prostate cancer. Prostate 2013, 73, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; De Gramont, A. Targeting the TGF?? pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.-W.; Cooper, C.R.; Farach-Carson, M.C.; Ogunnaike, B.A. A control engineering approach to understanding the TGF- paradox in cancer. J. R. Soc. Interface 2012, 9, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Y.; Xiao, H.; Li, Y.; Kan, X.; Wang, X.; Wang, Z.; Yang, Q.; Chen, X.; Weng, X.; et al. Chamaejasmenin B, a novel candidate, inhibits breast tumor metastasis by rebalancing TGF-β paradox. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Tian, M.; Schiemann, W.P. Deconstructing the mechanisms and consequences of TGF-β-induced EMT during cancer progression. Cell Tissue Res. 2012, 347, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, N.; Lee, C. Vicious cycle of TGF-β signaling in tumor progression and metastasis. Am. J. Clin. Exp. Urol. 2014, 2, 149–155. [Google Scholar] [PubMed]
- Zhang, Q.; Chen, L.; Helfand, B.T.; Jang, T.L.; Sharma, V.; Kozlowski, J.; Kuzel, T.M.; Zhu, L.J.; Yang, X.J.; Javonovic, B.; et al. TGF-β regulates DNA methyltransferase expression in prostate cancer, correlates with aggressive capabilities, and predicts disease recurrence. PLoS ONE 2011, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Zhang, Q.; Zi, X.; Dash, A.; Soares, M.B.; Rahmatpanah, F.; Jia, Z.; McClelland, M.; Mercola, D. TGF-beta mediated DNA methylation in prostate cancer. Transl. Androl. Urol. 2012, 1, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.-P.J.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, S.J.; Haendler, B. Exploiting Epigenetic Alterations in Prostate Cancer. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Marques-Magalhães, Â.; Graça, I.; Henrique, R.; Jerónimo, C. Targeting DNA methyltranferases in urological tumors. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Bikard, D.; Jiang, W.; Samai, P.; Hochschild, A.; Zhang, F.; Marraffini, L.A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013, 41, 7429–7437. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Asano, H.; Toyooka, S.; Tsukuda, K.; Soh, J.; Shien, T.; Taira, N.; Maki, Y.; Tanaka, N.; Doihara, H.; et al. DNA methylation status of REIC/Dkk-3 gene in human malignancies. J. Cancer Res. Clin. Oncol. 2012, 138, 799–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, H.; Liang, Y.; Peng, P.; Ma, X.; Zhang, X. DKK3 regulates cell proliferation, apoptosis and collagen synthesis in keloid fibroblasts via TGF-β1/Smad signaling pathway. Biomed. Pharmacother. 2017, 91, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Wang, H.; Krebs, T.L.; Kim, S.-J.; Danielpour, D. Androgenic control of transforming growth factor-beta signaling in prostate epithelial cells through transcriptional suppression of transforming growth factor-β receptor II. Cancer Res. 2008, 68, 8173–8182. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA Methylation in the Mammalian Genome. Cell 2016, 167, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Al-Shareef, Z.; Kardooni, H.; Murillo-Garzon, V.; Domenici, G.; Stylianakis, E.; Steel, J.; Rabano, M.; Gorrono-Etxebarria, I.; Zabalza, I.; Vivanco, M.; et al. Protective effect of stromal Dickkopf-3 in prostate cancer: opposing roles for TGFBI and ECM-1. Oncogene 2018, in press. [Google Scholar]
- Virginia, M.-G.; Irantzu, G.-E.; Malin, Å.; Mikael, C.P.; Lea, S.; Matthias, N.; James, C.; Jonathan, W.; Robert, M.K. Frizzled-8 integrates Wnt-11 and transforming growth factor-β signaling in prostate cancer. Nat. Commun. 2018, 9, 1747. [Google Scholar]
- Taylor, B.; Schultz, N.; Hieronymus, H. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, R.J.; Gorroño-Etxebarria, I.; Rabano, M.; Vivanco, M.D.; Kypta, R. Dickkopf-3 alters the morphological response to retinoic acid during neuronal differentiation of human embryonal carcinoma cells. Dev. Neurobiol. 2014, 74, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.L.; Leonard, D.M.; Wolfe, S.A.; Liu, J.; Rivera, J.; Yang, M.; Leonard, R.T.; Johnson, J.P.S.; Kumar, P.; Liebmann, K.L.; et al. The Dkk3 gene encodes a vital intracellular regulator of cell proliferation. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.K.; Hatanaka, F.; Araoka, T.; Reddy, P.; Wu, M.Z.; Sui, Y.; Yamauchi, T.; Sakurai, M.; O’Keefe, D.D.; Núñez-Delicado, E.; et al. In Vivo Target Gene Activation via CRISPR/Cas9-Mediated Trans-epigenetic Modulation. Cell 2017, 171, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.; Millena, A.C.; Strong, N.; Khan, S.A. Expression of TGFβ3 and its effects on migratory and invasive behaviour of prostate cancer cells: involvement of PI3-kinase/AKT signaling pathway. Clin. Exp. Metastasis 2013, 30, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Mori, A.; Watanabe, M.; Sadahira, T.; Kobayashi, Y.; Ariyoshi, Y.; Ueki, H.; Wada, K.; Ochiai, K.; Li, S.A.; Nasu, Y. The downregulation of the expression of CD147 by tumor suppressor REIC/Dkk-3, and its implication in human prostate cancer cell growth inhibition. Acta Med. Okayama 2017, 71, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Kardooni, H.; Imperial College London, London, UK; Kypta, R.; Imperial College London, London, UK. Unpublished results. 2018.
- Tao, L.; Huang, G.; Chen, Y.; Chen, L. DNA methylation of DKK3 modulates docetaxel chemoresistance in human nonsmall cell lung cancer cell. Cancer Biother. Radiopharm. 2015, 30, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Montanari, M.; Rossetti, S.; Cavaliere, C.; D’Aniello, C.; Malzone, M.G.; Vanacore, D.; Di Franco, R.; La Mantia, E.; Iovane, G.; Piscitelli, R.; et al. Epithelial-mesenchymal transition in prostate cancer: An overview. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chen, Y.; Chen, Q.; Xu, H.; Wang, Y.; Yu, J.; Zhou, J.; Wang, Z.; Xu, B. DNMT1 regulates IL-6- and TGF-β1-induced epithelial mesenchymal transition in prostate epithelial cells. Eur. J. Histochem. 2017, 61, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Kirschenbaum, A.; Liu, X.H.; Yao, S.; Levine, A.C. The role of cyclooxygenase-2 in prostate cancer. Urology 2001, 58, 127–131. [Google Scholar] [CrossRef]
- Bastian, P.J.; Ellinger, J.; Heukamp, L.C.; Kahl, P.; Müller, S.C.; von Rücker, A. Prognostic Value of CpG Island Hypermethylation at PTGS2, RAR-β, EDNRB, and Other Gene Loci in Patients Undergoing Radical Prostatectomy. Eur. Urol. 2007, 51, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Mason, M.D.; Clarke, N.W.; James, N.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Attard, G.; Cross, W.; Jones, R.J.; Parker, C.C.; et al. Adding celecoxib with or without zoledronic acid for hormone-naïve prostate cancer: Long-term survival results from an adaptive, multiarm, multistage, platform, randomized controlled trial. J. Clin. Oncol. 2017, 35, 1530–1541. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Tsai, Y.C.; Yeh, H.L.; Suau, F.; Jiang, K.C.; Shao, A.N.; Huang, J.L.Y. Loss of SPDEF and gain of TGFBI activity after androgen deprivation therapy promote EMT and bone metastasis of prostate cancer. Sci. Signal. 2017, 10, 492. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, Y.; Shan, B.; Han, J.; Zhu, H.; Lv, Y.; Fan, X.; Sang, M.; Liu, X.D.; Liu, W. The downregulation of miR-200c/141 promotes ZEB1/2 expression and gastric cancer progression. Med. Oncol. 2015, 32, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, Y. Decitabine reverses TGF- β 1-induced epithelial—Mesenchymal transition in non-small-cell lung cancer by regulating miR-200/ZEB axis. Drug Des. Devel. Ther. 2017, 11, 969–983. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Dickson, J.; Din, S.; Macleod, K.; Jodrell, D.; Ramsahoye, B. Targeting of 5-aza-2′-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res. 2010, 38, 4313–4324. [Google Scholar] [CrossRef] [PubMed]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Investig. Urol. 1979, 17, 16–23. [Google Scholar]
- Thalmann, G.N.; Anezinis, P.E.; Chang, S.M.; Zhau, H.E.; Kim, E.E.; Hopwood, V.L.; Pathak, S.; von Eschenbach, A.C.; Chung, L.K.W. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994, 54, 2577–2581. [Google Scholar] [CrossRef] [PubMed]
- Rowther, F.B.; Kardooni, H.; Warr, T. TOUCH-UP gradient amplification method. J. Biomol. Tech. 2012, 23, 1–3. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kardooni, H.; Gonzalez-Gualda, E.; Stylianakis, E.; Saffaran, S.; Waxman, J.; Kypta, R.M. CRISPR-Mediated Reactivation of DKK3 Expression Attenuates TGF-β Signaling in Prostate Cancer. Cancers 2018, 10, 165. https://doi.org/10.3390/cancers10060165
Kardooni H, Gonzalez-Gualda E, Stylianakis E, Saffaran S, Waxman J, Kypta RM. CRISPR-Mediated Reactivation of DKK3 Expression Attenuates TGF-β Signaling in Prostate Cancer. Cancers. 2018; 10(6):165. https://doi.org/10.3390/cancers10060165
Chicago/Turabian StyleKardooni, Hoda, Estela Gonzalez-Gualda, Emmanouil Stylianakis, Sina Saffaran, Jonathan Waxman, and Robert M. Kypta. 2018. "CRISPR-Mediated Reactivation of DKK3 Expression Attenuates TGF-β Signaling in Prostate Cancer" Cancers 10, no. 6: 165. https://doi.org/10.3390/cancers10060165
APA StyleKardooni, H., Gonzalez-Gualda, E., Stylianakis, E., Saffaran, S., Waxman, J., & Kypta, R. M. (2018). CRISPR-Mediated Reactivation of DKK3 Expression Attenuates TGF-β Signaling in Prostate Cancer. Cancers, 10(6), 165. https://doi.org/10.3390/cancers10060165