Evolving Treatment Strategies for Elderly Leukemia Patients with IDH Mutations

Abstract
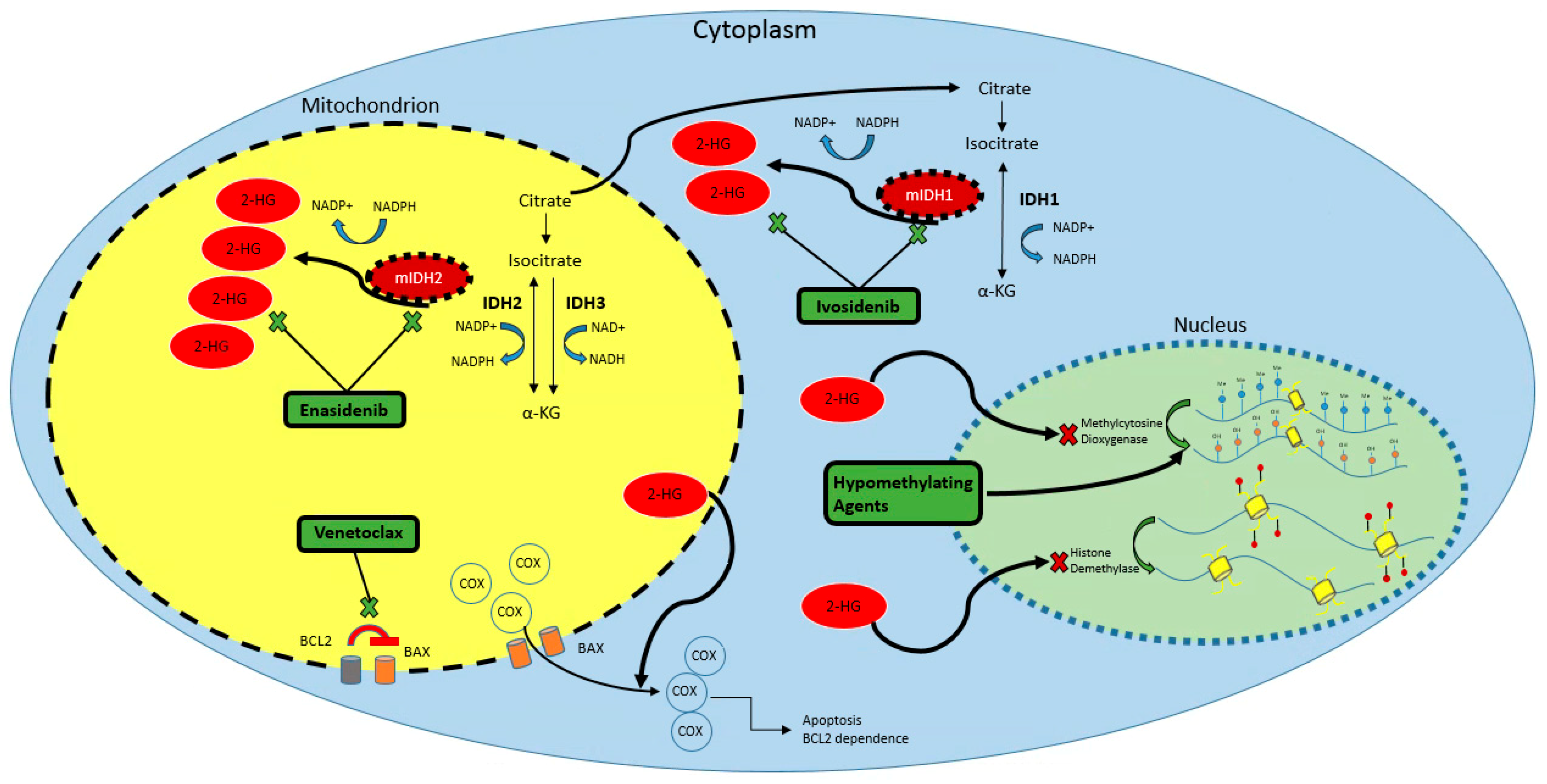
:1. Introduction
2. IDH, R-2-hydroxyglutarate, and Leukemogenesis
2.1. IDH in Normal Cellular Processes
2.2. Mutant IDH Function
3. Targeted Therapies for IDH-Mutant AML
3.1. Rationale for Combination with Hypomethylating Agents
3.2. Enasidenib
3.2.1. Mechanism
3.2.2. Clinical Activity
3.2.3. Safety and Tolerability—General
3.2.4. Differentiation Syndrome
3.3. Ivosidenib
3.3.1. Mechanism
3.3.2. Clinical Activity
3.4. Venetoclax
3.4.1. Mechanism and Clinical Activity
3.4.2. Safety and Tolerability
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Bishop, K.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; et al. Seer Cancer Statistics Review, 1975–2014; National Cancer Institute: Bethesda, MD, USA, 2018. [Google Scholar]
- Almeida, A.M.; Ramos, F. Acute myeloid leukemia in the older adults. Leuk. Res. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xie, H.; Wood, B.L.; Walter, R.B.; Pagel, J.M.; Becker, P.S.; Sandhu, V.K.; Abkowitz, J.L.; Appelbaum, F.R.; Estey, E.H. Relation of clinical response and minimal residual disease and their prognostic impact on outcome in acute myeloid leukemia. J. Clin. Oncol. 2015, 33, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F. Is complete remission key in elderly patients with AML? Lancet Haematol. 2016, 3, e212–e213. [Google Scholar] [CrossRef]
- Walter, R.B.; Kantarjian, H.M.; Huang, X.; Pierce, S.A.; Sun, Z.; Gundacker, H.M.; Ravandi, F.; Faderl, S.H.; Tallman, M.S.; Appelbaum, F.R.; et al. Effect of complete remission and responses less than complete remission on survival in acute myeloid leukemia: A combined eastern cooperative oncology group, southwest oncology group, and m. D. Anderson cancer center study. J. Clin. Oncol. 2010, 28, 1766–1771. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Cao, Y.; Glimelius, I.; Bottai, M.; Eloranta, S.; Smedby, K.E. The impact of comorbid disease history on all-cause and cancer-specific mortality in myeloid leukemia and myeloma–A swedish population-based study. BMC Cancer 2015, 15, 850. [Google Scholar] [CrossRef]
- Alibhai, S.M.; Leach, M.; Minden, M.D.; Brandwein, J. Outcomes and quality of care in acute myeloid leukemia over 40 years. Cancer 2009, 115, 2903–2911. [Google Scholar] [PubMed] [Green Version]
- Williams, G.R.; Mackenzie, A.; Magnuson, A.; Olin, R.; Chapman, A.; Mohile, S.; Allore, H.; Somerfield, M.R.; Targia, V.; Extermann, M.; et al. Comorbidity in older adults with cancer. J. Geriatr. Oncol. 2016, 7, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jonge, H.J.; de Bont, E.S.; Valk, P.J.; Schuringa, J.J.; Kies, M.; Woolthuis, C.M.; Delwel, R.; Veeger, N.J.; Vellenga, E.; Lowenberg, B.; et al. AML at older age: Age-related gene expression profiles reveal a paradoxical down-regulation of p16ink4a mRNA with prognostic significance. Blood 2009, 114, 2869–2877. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.V.; Valk, P.J.; Metzeler, K.H.; Acharya, C.R.; Tuchman, S.A.; Stevenson, M.M.; Rizzieri, D.A.; Delwel, R.; Buske, C.; Bohlander, S.K.; et al. Age-specific differences in oncogenic pathway dysregulation and anthracycline sensitivity in patients with acute myeloid leukemia. J. Clin. Oncol. 2009, 27, 5580–5586. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Skrabanek, L.; Li, Y.; Jiemjit, A.; Fandy, T.E.; Paietta, E.; Fernandez, H.; Tallman, M.S.; Greally, J.M.; Carraway, H.; et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 2009, 114, 3448–3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ali, H.K.; Jaekel, N.; Niederwieser, D. The role of hypomethylating agents in the treatment of elderly patients with AML. J. Geriatr. Oncol. 2014, 5, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Nccn Clinical Practice Guidelines in Oncology, Acute myeloid leukemia version 1.2018; National Comprehensive Cancer Network: Jenkintown, PA, USA, 2014.
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; Jin, S.; Su, S.M. IDH mutations in glioma and acute myeloid leukemia. Trends Mol. Med. 2010, 16, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Mondesir, J.; Willekens, C.; Touat, M.; de Botton, S. IDH1 and IDH2 mutations as novel therapeutic targets: Current perspectives. J. Blood Med. 2016, 7, 171–180. [Google Scholar] [PubMed]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; van Noorden, C.J.F. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Madala, H.R.; Punganuru, S.R.; Arutla, V.; Misra, S.; Thomas, T.J.; Srivenugopal, K.S. Beyond brooding on oncometabolic havoc in IDH-mutant gliomas and AML: Current and future therapeutic strategies. Cancers 2018, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; Su, S.M. Isocitrate dehydrogenase mutation and (r)-2-hydroxyglutarate: From basic discovery to therapeutics development. Annu. Rev. Biochem. 2017, 86, 305–331. [Google Scholar] [CrossRef] [PubMed]
- Ragon, B.K.; DiNardo, C.D. Targeting IDH1 and IDH2 mutations in acute myeloid leukemia. Curr. Hematol. Malig. Rep. 2017, 12, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Al-Khallaf, H. Isocitrate dehydrogenases in physiology and cancer: Biochemical and molecular insight. Cell Biosci. 2017, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Nassereddine, S.; Lap, C.J.; Haroun, F.; Tabbara, I. The role of mutant IDH1 and IDH2 inhibitors in the treatment of acute myeloid leukemia. Ann. Hematol. 2017, 96, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M. Enasidenib, a targeted inhibitor of mutant IDH2 proteins for treatment of relapsed or refractory acute myeloid leukemia. Future Oncol. 2018, 14, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Kipp, B.R.; Voss, J.S.; Kerr, S.E.; Barr Fritcher, E.G.; Graham, R.P.; Zhang, L.; Highsmith, W.E.; Zhang, J.; Roberts, L.R.; Gores, G.J.; et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum. Pathol. 2012, 43, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Kosmider, O.; Gelsi-Boyer, V.; Slama, L.; Dreyfus, F.; Beyne-Rauzy, O.; Quesnel, B.; Hunault-Berger, M.; Slama, B.; Vey, N.; Lacombe, C.; et al. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia 2010, 24, 1094–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.C.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Chan, S.M.; Swords, R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017, 31, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.P.; Ravandi, F.; Ma, D.; Paladugu, A.; Barkoh, B.A.; Medeiros, L.J.; Luthra, R. Acute myeloid leukemia with IDH1 or IDH2 mutation: Frequency and clinicopathologic features. Am. J. Clin. Pathol. 2011, 135, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Maharry, K.; Wu, Y.Z.; Radmacher, M.D.; Mrozek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A cancer and leukemia group b study. J. Clin. Oncol. 2010, 28, 2348–2355. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Kronke, J.; Bullinger, L.; Spath, D.; Kayser, S.; Zucknick, M.; Gotze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with npm1 mutation without flt3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt tet2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Platt, M.Y.; Fathi, A.T.; Borger, D.R.; Brunner, A.M.; Hasserjian, R.P.; Balaj, L.; Lum, A.; Yip, S.; Dias-Santagata, D.; Zheng, Z.; et al. Detection of dual IDH1 and IDH2 mutations by targeted next-generation sequencing in acute myeloid leukemia and myelodysplastic syndromes. J. Mol. Diagn. 2015, 17, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Damm, F.; Gohring, G.; Gorlich, K.; Heuser, M.; Schafer, I.; Ottmann, O.; Lubbert, M.; Heit, W.; Kanz, L.; et al. Impact of IDH1 r132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J. Clin. Oncol. 2010, 28, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Willander, K.; Falk, I.J.; Chaireti, R.; Paul, E.; Hermansson, M.; Green, H.; Lotfi, K.; Soderkvist, P. Mutations in the isocitrate dehydrogenase 2 gene and IDH1 SNP 105c > t have a prognostic value in acute myeloid leukemia. Biomark. Res. 2014, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 2001, 1, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Reitman, Z.J.; Duncan, C.G.; Spasojevic, I.; Gooden, D.M.; Rasheed, B.A.; Yang, R.; Lopez, G.Y.; He, Y.; McLendon, R.E.; et al. Disruption of wild-type IDH1 suppresses d-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013, 73, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Rzem, R.; Vincent, M.F.; Van Schaftingen, E.; Veiga-da-Cunha, M. l-2-hydroxyglutaric aciduria, a defect of metabolite repair. J. Inherit. Metab. Dis. 2007, 30, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Van Schaftingen, E.; Rzem, R.; Marbaix, A.; Collard, F.; Veiga-da-Cunha, M.; Linster, C.L. Metabolite proofreading, a neglected aspect of intermediary metabolism. J. Inherit. Metab. Dis. 2013, 36, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Radivoyevitch, T.; Maciejewski, J.P.; van Noorden, C.J.; Bleeker, F.E. The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation. Biochim. Biophys. Acta 2014, 1846, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Grassian, A.R.; Parker, S.J.; Davidson, S.M.; Divakaruni, A.S.; Green, C.R.; Zhang, X.; Slocum, K.L.; Pu, M.; Lin, F.; Vickers, C.; et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014, 74, 3317–3331. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.J.; Metallo, C.M. Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 2015, 152, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, D.; Guan, K.L.; Xiong, Y. Metabolism, activity, and targeting of d- and l-2-hydroxyglutarates. Trends Cancer 2018, 4, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (r)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Venneti, S.; Akalin, A.; Fang, F.; Ward, P.S.; Dematteo, R.G.; Intlekofer, A.M.; Chen, C.; Ye, J.; Hameed, M.; et al. Induction of sarcomas by mutant IDH2. Genes Dev. 2013, 27, 1986–1998. [Google Scholar] [PubMed] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase a silencing in IDH mutant gliomas. Neuro Oncol. 2014, 16, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.C.; Lei, W.C.; Ko, B.S.; Hou, H.A.; Chen, C.Y.; Tang, J.L.; Yao, M.; Tsay, W.; Wu, S.J.; Huang, S.Y.; et al. The prognostic impact and stability of isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia 2011, 25, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.G.; Ribeiro, C.A.; Leipnitz, G.; Dutra-Filho, C.S.; Wyse, A.A.; Wannmacher, C.M.; Sarkis, J.J.; Jakobs, C.; Wajner, M. Inhibition of cytochrome c oxidase activity in rat cerebral cortex and human skeletal muscle by d-2-hydroxyglutaric acid in vitro. Biochim. Biophys. Acta 2002, 1586, 81–91. [Google Scholar] [CrossRef]
- Latini, A.; da Silva, C.G.; Ferreira, G.C.; Schuck, P.F.; Scussiato, K.; Sarkis, J.J.; Dutra Filho, C.S.; Wyse, A.T.; Wannmacher, C.M.; Wajner, M. Mitochondrial energy metabolism is markedly impaired by d-2-hydroxyglutaric acid in rat tissues. Mol. Genet. Metab. 2005, 86, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Lomonosova, E.; Chinnadurai, G. Bh3-only proteins in apoptosis and beyond: An overview. Oncogene 2008, 27 (Suppl. 1), S2–S19. [Google Scholar] [CrossRef] [PubMed]
- Shroff, E.H.; Snyder, C.; Chandel, N.S. Role of Bcl-2 family members in anoxia induced cell death. Cell Cycle 2007, 6, 807–809. [Google Scholar] [CrossRef] [PubMed]
- McClintock, D.S.; Santore, M.T.; Lee, V.Y.; Brunelle, J.; Budinger, G.R.; Zong, W.X.; Thompson, C.B.; Hay, N.; Chandel, N.S. Bcl-2 family members and functional electron transport chain regulate oxygen deprivation-induced cell death. Mol. Cell Biol. 2002, 22, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Saikumar, P.; Dong, Z.; Patel, Y.; Hall, K.; Hopfer, U.; Weinberg, J.M.; Venkatachalam, M.A. Role of hypoxia-induced BAX translocation and cytochrome c release in reoxygenation injury. Oncogene 1998, 17, 3401–3415. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce Bcl-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Eguchi, Y.; Kosaka, H.; Kamiike, W.; Matsuda, H.; Tsujimoto, Y. Prevention of hypoxia-induced cell death by Bcl-2 and Bcl-xl. Nature 1995, 374, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Bian, K.; Tang, Q.; Fedeles, B.I.; Singh, V.; Humulock, Z.T.; Essigmann, J.M.; Li, D. Oncometabolites d- and l-2-hydroxyglutarate inhibit the AlkB family DNA repair enzymes under physiological conditions. Chem. Res. Toxicol. 2017, 30, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.L.; Ye, D.; et al. Oncometabolite d-2-hydroxyglutarate inhibits AlkBH DNA repair enzymes and sensitizes IDH mutant cells to alkylating agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef] [PubMed]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress stat1 and CD8+ t cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Tyrakis, P.A.; Palazon, A.; Macias, D.; Lee, K.L.; Phan, A.T.; Velica, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; et al. S-2-hydroxyglutarate regulates CD8(+) t-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Sun, B.; Shi, W.; Zuo, H.; Cui, D.; Ni, L.; Chen, J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour. Biol. 2015, 36, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.H.; Jiang, Y.; Meydan, C.; Shank, K.; Pandey, S.; Barreyro, L.; Antony-Debre, I.; Viale, A.; Socci, N.; Sun, Y.; et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 2015, 27, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Kats, L.M.; Reschke, M.; Taulli, R.; Pozdnyakova, O.; Burgess, K.; Bhargava, P.; Straley, K.; Karnik, R.; Meissner, A.; Small, D.; et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell 2014, 14, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.H.; Meydan, C.; Shank, K.; Garrett-Bakelman, F.E.; Ward, P.S.; Intlekofer, A.M.; Nazir, A.; Stein, E.M.; Knapp, K.; Glass, J.; et al. Combination targeted therapy to disrupt aberrant oncogenic signaling and reverse epigenetic dysfunction in IDH2- and tet2-mutant acute myeloid leukemia. Cancer Discov. 2017, 7, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, J.; Sun, X.; Wang, Z.; Cheng, X.; Lu, W.; Cai, X.; Hu, C.; Shen, X.; Cao, P. Allosteric inhibitor remotely modulates the conformation of the orthestric pockets in mutant IDH2/r140q. Sci. Rep. 2017, 7, 16458. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research. Application Number 209606orig1s000 Multi-Discipline Review. 28 July 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209606Orig1s000MultidisciplineR.pdf (accessed on 30 March 2018).
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef] [PubMed]
- Celgene Corporation. Idhifa Prescribing Information August 2017. Available online: http://media.celgene.com/content/uploads/idhifa-pi.pdf (accessed on 30 March 2018).
- Shih, A.H.; Shank, K.R.; Meydan, C.; Intlekofer, A.M.; Ward, P.; Thompson, C.B.; Melnick, A.M.; Travins, J.; Straley, K.; Gliser, C.; et al. Ag-221, a small molecule mutant idh2 inhibitor, remodels the epigenetic state of idh2-mutant cells and induces alterations in self-renewal/differentiation in IDH2-mutant AML model in vivo. Blood 2014, 124, 437. [Google Scholar]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.D.; Marteyn, B.; Farnoud, N.R.; de Botton, S.; Bernard, O.A.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNardo, C.D.; Ravandi, F.; Agresta, S.; Konopleva, M.; Takahashi, K.; Kadia, T.; Routbort, M.; Patel, K.P.; Mark, B.; Pierce, S.; et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am. J. Hematol. 2015, 90, 732–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinicaltrials.Gov. Phase 1/2 Study of Ag-221 in Subjects with Advanced Hematologic Malignancies with an IDH2 Mutation (nct01915498). Available online: https://clinicaltrials.gov/ct2/show/NCT01915498. (accessed on 2 April 2018).
- Pollyea, D.A.; Tallman, M.S.; De Botton, S.; DiNardo, C.D.; Kantarjian, H.M.; Collins, R.H.; Stein, A.S.; Xu, Q.; Tosolini, A.; Gupta, I.; et al. 638 enasidenib monotherapy is effective and well-tolerated in patients with previously untreated mutant-IDH2 (mIDH2) acute myeloid leukemia (AML). In Proceedings of the Ash Annual Meeting & Exposition, Atlanta, GA, USA, 9–12 December 2017. [Google Scholar]
- Stein, E.M.; DiNardo, C.D.; Mims, A.S.; Savona, M.R.; Pratz, K.; Stein, A.S.; Fathi, A.T.; Stone, R.M.; Pollyea, D.A.; Odenike, O.; et al. Ivosidenib or enasidenib combined with standard induction chemotherapy is well tolerated and active in patients with newly diagnosed AML with an IDH1 or IDH2 mutation: Initial results from a phase 1 trial. Blood 2017, 130, 726. [Google Scholar]
- Clinicaltrials.Gov. Safety Study of Ag-120 or Ag-221 in Combination with Induction and Consolidation Therapy in Patients with Newly Diagnosed Acute Myeloid Leukemia with an IDH1 and/or IDH2 Mutation (nct02632708). Available online: https://clinicaltrials.gov/ct2/show/NCT02632708 (accessed on 2 April 2018).
- DiNardo, C.D.; Stein, A.S.; Fathi, A.T.; Montesinos, P.; Odenike, O.; Kantarjian, H.M.; Stone, R.M.; Koralek, D.O.; Van Oostendorp, J.; Gong, J.; et al. Mutant isocitrate dehydrogenase (mIDH) inhibitors, enasidenib or ivosidenib, in combination with azacitidine (AZA): Preliminary results of a phase 1b/2 study in patients with newly diagnosed acute myeloid leukemia (AML). Blood 2017, 130, 639. [Google Scholar]
- Clinicaltrials.Gov. A Safety and Efficacy Study of Oral Ag-120 plus Subcutaneous Azacitidine and Oral Ag-221 Plus Subcutaneous Azacitidine in Subjects with Newly Diagnosed Acute Myeloid Leukemia (AML) (nct02677922). Available online: https://clinicaltrials.gov/ct2/show/NCT02677922 (accessed on 2 April 2018).
- Clinicaltrials.Gov. An Efficacy and Safety Study of Ag-221 (cc-90007) versus Conventional Care Regimens in Older Subjects with Late Stage Acute Myeloid Leukemia Harboring an Isocitrate Dehydrogenase 2 Mutation (Idhentify) (nct02577406). Available online: https://clinicaltrials.gov/ct2/show/NCT02577406. (accessed on 30 March 2018).
- Clinicaltrials.Gov. Study of Orally Administered Ag-120 in Subjects with Advanced Hematologic Malignancies with an Idh1 Mutation (nct02074839). Available online: https://clinicaltrials.gov/ct2/show/NCT02074839 (accessed on 30 March 2018).
- DiNardo, C.D.; de Botton, S.; Stein, E.M.; Roboz, G.J.; Mims, A.S.; Pollyea, D.A.; Swords, R.; Altman, J.K.; Collins, R.H.; Mannis, G.N.; et al. 725 ivosidenib (AG-120) in mutant IDH1 AML and advanced hematologic malignancies: Results of a phase 1 dose escalation and expansion study clinically relevant abstract. In Proceedings of the Ash Annual Meeting & Exposition, Atlanta, GA, USA, 9–12 December 2017. [Google Scholar]
- Clinicaltrials.Gov. Study of Ag-120 (Ivosidenib) vs. Placebo in Combination with Azacitidine in Patients with Previously Untreated Acute Myeloid Leukemia with an IDH1 Mutation (Agile) (nct03173248). Available online: https://clinicaltrials.gov/ct2/show/NCT03173248 (accessed on 30 March 2018).
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and biological correlates of response in a phase ii study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.A.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- Clinicaltrials.Gov. A Study of Abt-199 in Combination with Azacitidine or Decitabine (Chemo Combo) in Subjects with Acute Myelogenous Leukemia (AML) (nct02203773). Available online: https://clinicaltrials.gov/ct2/show/NCT02203773 (accessed on 2 April 2018).
- Stein, E.M.; Stone, R.M.; Pollyea, D.A.; Roboz, G.J.; Altman, J.K.; DiNardo, C.D.; de Botton, S.; Tu, N.; Swern, A.S.; Tosolini, A.; et al. Continuing enasidenib treatment for patients with mutant-IDH2 (m IDH2) relapsed or refractory acute myeloid leukemia (r/r AML) with stable disease may result in improved survival and responses over time. Blood 2017, 130, 1299. [Google Scholar]
- Patatanian, E.; Thompson, D.F. Retinoic acid syndrome: A review. J. Clin. Pharm. Ther. 2008, 33, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Nasr, R.; Lallemand-Breitenbach, V.; Zhu, J.; Guillemin, M.C.; de The, H. Therapy-induced PML/RARA proteolysis and acute promyelocytic leukemia cure. Clin. Cancer Res. 2009, 15, 6321–6326. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.E.; Yang, D. Differentiation syndrome in patients with acute promyelocytic leukemia. J. Oncol. Pharm. Pract. 2012, 18, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, P.; Sanz, M.A. The differentiation syndrome in patients with acute promyelocytic leukemia: Experience of the pethema group and review of the literature. Mediterr. J. Hematol. Infect. Dis. 2011, 3, e2011059. [Google Scholar] [CrossRef] [PubMed]
- Camacho, L.H.; Soignet, S.L.; Chanel, S.; Ho, R.; Heller, G.; Scheinberg, D.A.; Ellison, R.; Warrell, R.P., Jr. Leukocytosis and the retinoic acid syndrome in patients with acute promyelocytic leukemia treated with arsenic trioxide. J. Clin. Oncol. 2000, 18, 2620–2625. [Google Scholar] [CrossRef] [PubMed]
- Tallman, M.S.; Andersen, J.W.; Schiffer, C.A.; Appelbaum, F.R.; Feusner, J.H.; Ogden, A.; Shepherd, L.; Rowe, J.M.; Francois, C.; Larson, R.S.; et al. Clinical description of 44 patients with acute promyelocytic leukemia who developed the retinoic acid syndrome. Blood 2000, 95, 90–95. [Google Scholar] [PubMed]
- Luesink, M.; Pennings, J.L.; Wissink, W.M.; Linssen, P.C.; Muus, P.; Pfundt, R.; de Witte, T.J.; van der Reijden, B.A.; Jansen, J.H. Chemokine induction by all-trans retinoic acid and arsenic trioxide in acute promyelocytic leukemia: Triggering the differentiation syndrome. Blood 2009, 114, 5512–5521. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T.; DiNardo, C.D.; Kline, I.; Kenvin, L.; Gupta, I.; Attar, E.C.; Stein, E.M.; de Botton, S.; Investigators, A.C.S. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: Analysis of a phase 1/2 study. JAMA Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hansen, E.; Quivoron, C.; Straley, K.; Lemieux, R.M.; Popovici-Muller, J.; Sadrzadeh, H.; Fathi, A.T.; Gliser, C.; David, M.; Saada, V.; et al. AG-120, an oral, selective, first-in-class, potent inhibitor of mutant IDH1, reduces intracellular 2HG and induces cellular differentiation in tf-1 r132h cells and primary human IDH1 mutant AML patient samples treated ex vivo. Blood 2014, 124, 3734. [Google Scholar]
- Fan, B.; Le, K.; Manyak, E.; Liu, H.; Prahl, M.; Bowden, C.J.; Biller, S.; Agresta, S.; Yang, H. Longitudinal pharmacokinetic/pharmacodynamic profile of ag-120, a potent inhibitor of the IDH1 mutant protein, in a phase 1 study of IDH1-mutant advanced hematologic malignancies. Blood 2015, 126, 1310. [Google Scholar]
- Stone, R.M.; Choe, S.; Zhang, V.; Marteyn, B.; Penard-Lacronique, V.; Wang, H.; DiNardo, C.D.; Stein, E.M.; Fathi, A.T.; Tallman, M.S.; et al. Genetic profiling and deep IDH1 mutation clearance to ≤0.04% in ivosidenib (ag-120)-treated patients with mutant idh1 relapsed or refractory and untreated AML. Blood 2017, 130, 2684. [Google Scholar]
- Birendra, K.C.; DiNardo, C.D. Evidence for clinical differentiation and differentiation syndrome in patients with acute myeloid leukemia and IDH1 mutations treated with the targeted mutant idh1 inhibitor, AG-120. Clin. Lymphoma Myeloma Leuk. 2016, 16, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective Bcl-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the bh3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef] [PubMed]
- King, A.C.; Peterson, T.J.; Horvat, T.Z.; Rodriguez, M.; Tang, L.A. Venetoclax: A first-in-class oral Bcl-2 inhibitor for the management of lymphoid malignancies. Ann. Pharmacother. 2017, 51, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Tiong, I.S. Midostaurin, enasidenib, cpx-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood 2017, 130, 2469–2474. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Rausch, C.R.; Benton, C.; Kadia, T.; Jain, N.; Pemmaraju, N.; Daver, N.; Covert, W.; Marx, K.R.; Mace, M.; et al. Clinical experience with the Bcl2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am. J. Hematol. 2018, 93, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.C.; Jones, D.P.; Pui, C.H. The tumor lysis syndrome. N. Engl. J. Med. 2011, 364, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, J.; Cairo, M.S. Tumor lysis syndrome: Current perspective. Haematologica 2008, 93, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting Bcl2 with venetoclax in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Cheson, B.D.; Heitner Enschede, S.; Cerri, E.; Desai, M.; Potluri, J.; Lamanna, N.; Tam, C. Tumor lysis syndrome in chronic lymphocytic leukemia with novel targeted agents. Oncologist 2017, 22, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Davids, M.S.; Roberts, A.W.; Hallek, M.; Wierda, W.G.; Hillmen, P.; Gerecitano, J.F.; Cerri, E.; Potluri, J.; Kim, S.Y.; et al. Safety profile of venetoclax monotherapy in patients with chronic lymphocytic leukemia. Blood 2016, 128, 4395. [Google Scholar]
- Rausch, C.R.; DiNardo, C.D.; Kadia, T.; Takahashi, K.; Jain, N.; Benton, C.B.; Thompson, P.A.; Ohanian, M.; Covert, W.; Mace, M.; et al. 1356 results of off-label venetoclax use in combination with low-intensity chemotherapy in patients with relapsed and refractory myeloid malignancies. In Proceedings of the Ash Annual Meeting & Exposition, Atlanta, GA, USA, 9–12 December 2017. [Google Scholar]
- Center for Drug Evaluation and Research. Application Number 2085373orig1s000 Clinical Pharmacology and Biopharmaceutics Review(s). 14 March 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208573Orig1s000ClinPharmR.pdf (accessed on 30 March 2018).
- Abbvie, Inc. Venclexta Prescribing Information. December 2017. Available online: http://www.rxabbvie.com/pdf/venclexta.pdf (accessed on 30 March 2018).
- Gomes, M.Z.; Mulanovich, V.E.; Jiang, Y.; Lewis, R.E.; Kontoyiannis, D.P. Incidence density of invasive fungal infections during primary antifungal prophylaxis in newly diagnosed acute myeloid leukemia patients in a tertiary cancer center, 2009 to 2011. Antimicrob. Agents Chemother. 2014, 58, 865–873. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Phase b | Treatment Setting c | Intervention | No. of Patients d | Age, y | ORR, % e | Combined CR/CRi/CRp, % | Time to CR, mo | OS, mo (Median, 95% CI) f | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Enasidenib | |||||||||
| 1/2 | R/R | Enasidenib monotherapy | 109 | 67 (19–100) | 38.5 | 26.6 | 3.7 (0.7–11.2) | 9.3 | [91,92] |
| Front-line | 37 | 77 (58–87) | 37.8 | 18.9 | 5.6 (3.4–12.9) | 10.4 (5.7–15.1) | [92,93] | ||
| 1 | Front-line | Enasidenib plus 7 + 3 g | 38 | 63 (32–76) | 81 | 62 | NR h | NR | [94,95] |
| 1/2 | Front-line | Enasidenib plus 5-Aza | 6 | 68 (64–79) | 50 | 33.3 | NR | NR | [96,97] |
| 3 | R/R | Enasidenib monotherapy vs BSC i, 5-Aza, or cytarabine | No results available | [98] | |||||
| Ivosidenib | |||||||||
| 1 | R/R | Ivosidenib monotherapy | 125 | NR | 41.6 | 30.4 | NR | NR | [99,100] |
| 1 | Front-line | Ivosidenib plus 7 + 3 | 27 | 60 (24–76) | 83 | 70 | NR | NR | [94,95] |
| 1/2 | Front-line | Ivosidenib plus 5-Aza | 5 | 81 (72–88) | 60 | 60 | NR | NR | [96,97] |
| 3 | Front-line | Ivosidenib plus 5-Aza vs. 5-Aza plus placebo | No results available | [101] | |||||
| Venetoclax | |||||||||
| 2 j | R/R | Venetoclax monotherapy | 12 | 71 (19–84) k | NR | 33 | NR | NR | [102] |
| 1 | Front-line | Venetoclax plus 5-Aza or DAC | 17 | ≥65 l | 77 | 59 | NR | NR | [103,104] |
| Adverse Event | Any Grade, % | Grade ≥ 3, % |
|---|---|---|
| Hyperbilirubinemia b | 32–83 | 7–15 |
| Differentiation syndrome b | 13–33 | 7–17 |
| Nausea | 28 | 2 |
| Decreased appetite | 19 | 2 |
| Fatigue | 18 | 2 |
| Vomiting | 17 | 1 |
| Diarrhea | 16 | 1 |
| Hepatic injury c | 14 | 3 |
| Rash | 13 | 2 |
| Dysgeusia | 10 | 0 |
| Dyspnea c | 10 | 6 |
| Leukocytosis | 7 | 2 |
| Peripheral neuropathy | 7 | 0 |
| Anemia | 7 | 6 |
| Pyrexia | 7 | 1 |
| Hyperuricemia | 6 | 1 |
| Renal insufficiency | 5 | 1 |
| Weight decrease | 5 | 0 |
| Edema c | 5 | 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buege, M.J.; DiPippo, A.J.; DiNardo, C.D. Evolving Treatment Strategies for Elderly Leukemia Patients with IDH Mutations. Cancers 2018, 10, 187. https://doi.org/10.3390/cancers10060187
Buege MJ, DiPippo AJ, DiNardo CD. Evolving Treatment Strategies for Elderly Leukemia Patients with IDH Mutations. Cancers. 2018; 10(6):187. https://doi.org/10.3390/cancers10060187
Chicago/Turabian StyleBuege, Michael J., Adam J. DiPippo, and Courtney D. DiNardo. 2018. "Evolving Treatment Strategies for Elderly Leukemia Patients with IDH Mutations" Cancers 10, no. 6: 187. https://doi.org/10.3390/cancers10060187
APA StyleBuege, M. J., DiPippo, A. J., & DiNardo, C. D. (2018). Evolving Treatment Strategies for Elderly Leukemia Patients with IDH Mutations. Cancers, 10(6), 187. https://doi.org/10.3390/cancers10060187