The Role of Immune Checkpoint Inhibitors in Classical Hodgkin Lymphoma



Abstract
:1. Overview of Hodgkin Lymphoma
2. Biology and Diagnosis of Classical Hodgkin Lymphoma
3. Management of Classical Hodgkin Lymphoma
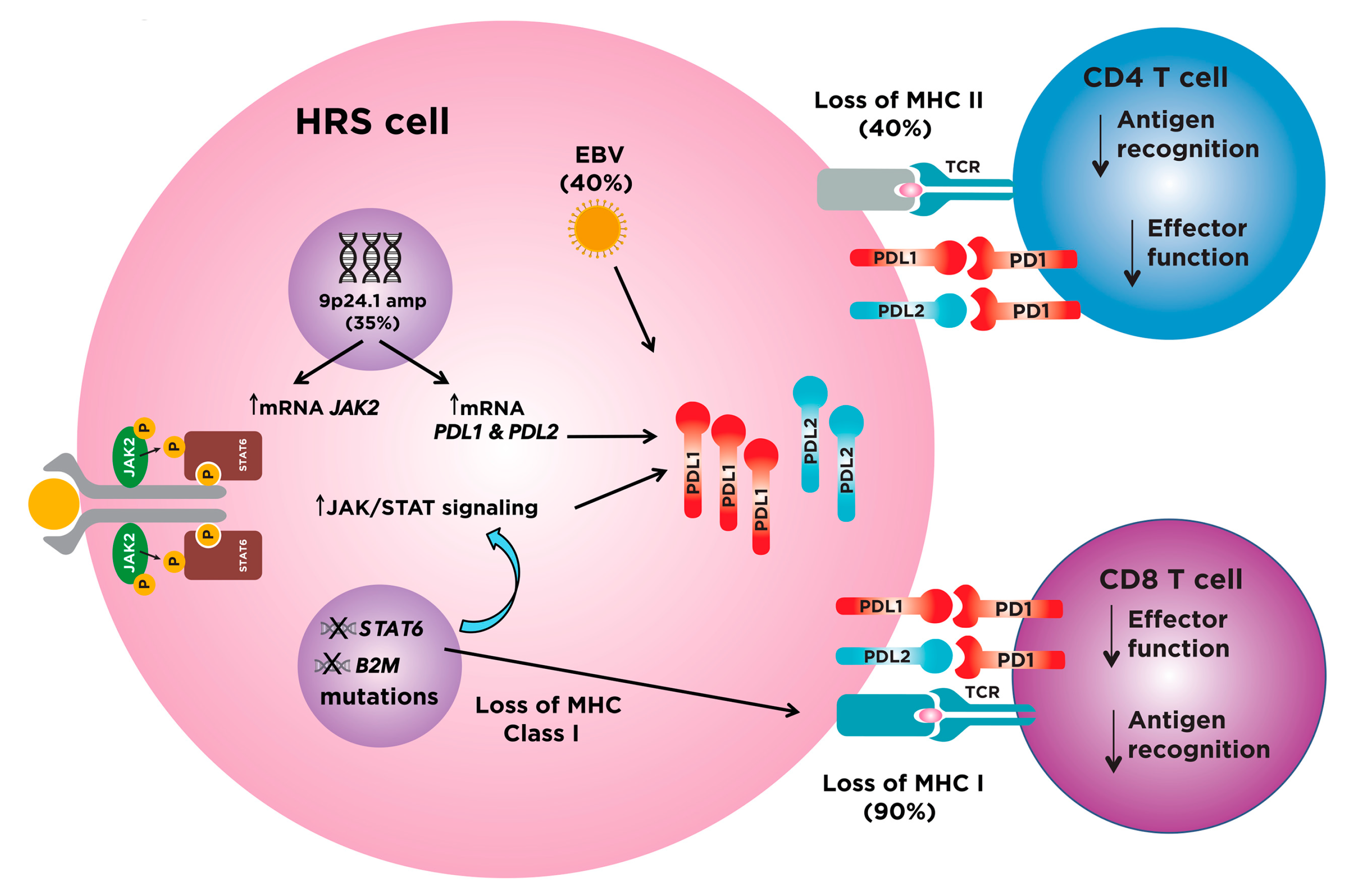
4. Immune Checkpoint Inhibitors in Classical Hodgkin Lymphoma
5. Evaluating Response to Immune Checkpoint Inhibitors in Classical Hodgkin Lymphoma
6. Biomarkers of Response to Immune Checkpoint Blockade in Classical Hodgkin Lymphoma
7. Future Directions in the Management of Classical Hodgkin Lymphoma
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thomas, R.K.; Re, D.; Zander, T.; Wolf, J.; Diehl, V. Epidemiology and etiology of Hodgkin’s lymphoma. Ann. Oncol. 2002, 13 (Suppl. S4), 147–152. [Google Scholar] [CrossRef] [PubMed]
- Morton, L.M.; Wang, S.S.; Devesa, S.S.; Hartge, P.; Weisenburger, D.D.; Linet, M.S. Lymphoma incidence patterns by who subtype in the united states, 1992–2001. Blood 2006, 107, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, R.A.; Watkins, G. Epidemiology of Hodgkin’s disease: A review. Hematol. Oncol. 2004, 22, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Mack, T.M.; Cozen, W.; Shibata, D.K.; Weiss, L.M.; Nathwani, B.N.; Hernandez, A.M.; Taylor, C.R.; Hamilton, A.S.; Deapen, D.M.; Rappaport, E.B. Concordance for Hodgkin’s disease in identical twins suggesting genetic susceptibility to the young-adult form of the disease. N. Engl. J. Med. 1995, 332, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Tirelli, U.; Errante, D.; Dolcetti, R.; Gloghini, A.; Serraino, D.; Vaccher, E.; Franceschi, S.; Boiocchi, M.; Carbone, A. Hodgkin’s disease and human immunodeficiency virus infection: Clinicopathologic and virologic features of 114 patients from the Italian cooperative group on aids and tumors. J. Clin. Oncol. 1995, 13, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Alexander, F.E.; Jarrett, R.F.; Lawrence, D.; Armstrong, A.A.; Freeland, J.; Gokhale, D.A.; Kane, E.; Taylor, G.M.; Wright, D.H.; Cartwright, R.A. Risk factors for Hodgkin’s disease by epstein-barr virus (EBV) status: Prior infection by EBV and other agents. Br. J. Cancer 2000, 82, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.L.; Jaffe, E.S.; Stein, H.; Banks, P.M.; Chan, J.K.; Cleary, M.L.; Delsol, G.; De Wolf-Peeters, C.; Falini, B.; Gatter, K.C.; et al. A revised european-american classification of lymphoid neoplasms: A proposal from the international lymphoma study group. Blood 1994, 84, 1361–1392. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the world health organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Arber, D.A.; Hasserjian, R.P.; Le Beau, M.M.; et al. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2017. [Google Scholar]
- Kanzler, H.; Kuppers, R.; Hansmann, M.L.; Rajewsky, K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J. Exp. Med. 1996, 184, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Yamashita, Y.; Nakayama, A.; Hasegawa, Y.; Kojima, H.; Nagasawa, T.; Mori, N. Varied B-cell immunophenotypes of Hodgkin/Reed-Sternberg cells in classic Hodgkin’s disease. Histopathology 2000, 36, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Schwering, I.; Brauninger, A.; Klein, U.; Jungnickel, B.; Tinguely, M.; Diehl, V.; Hansmann, M.L.; Dalla-Favera, R.; Rajewsky, K.; Kuppers, R. Loss of the b-lineage-specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 2003, 101, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Stein, H.; Gerdes, J.; Kirchner, H.; Schaadt, M.; Diehl, V. Hodgkin and sternberg-reed cell antigen(s) detected by an antiserum to a cell line (l428) derived from Hodgkin’s disease. Int. J. Cancer 1981, 28, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Stein, H.; Uchanska-Ziegler, B.; Gerdes, J.; Ziegler, A.; Wernet, P. Hodgkin and sternberg-reed cells contain antigens specific to late cells of granulopoiesis. Int. J. Cancer 1982, 29, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.M.; Jaffe, E.S. Leu M1 and peanut agglutinin stain the neoplastic cells of Hodgkin’s disease. Am. J. Clin. Pathol. 1984, 82, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Stein, H.; Mason, D.Y.; Gerdes, J.; O’Connor, N.; Wainscoat, J.; Pallesen, G.; Gatter, K.; Falini, B.; Delsol, G.; Lemke, H.; et al. The expression of the Hodgkin’s disease associated antigen KI-1 in reactive and neoplastic lymphoid tissue: Evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985, 66, 848–858. [Google Scholar] [PubMed]
- Schwarting, R.; Gerdes, J.; Durkop, H.; Falini, B.; Pileri, S.; Stein, H. BER-H2: A new anti-KI-1 (CD30) monoclonal antibody directed at a formol-resistant epitope. Blood 1989, 74, 1678–1689. [Google Scholar] [PubMed]
- Von Wasielewski, R.; Mengel, M.; Fischer, R.; Hansmann, M.L.; Hubner, K.; Franklin, J.; Tesch, H.; Paulus, U.; Werner, M.; Diehl, V.; et al. Classical Hodgkin’s disease. Clinical impact of the immunophenotype. Am. J. Pathol. 1997, 151, 1123–1130. [Google Scholar] [PubMed]
- Pileri, S.A.; Ascani, S.; Leoncini, L.; Sabattini, E.; Zinzani, P.L.; Piccaluga, P.P.; Pileri, A., Jr.; Giunti, M.; Falini, B.; Bolis, G.B.; et al. Hodgkin’s lymphoma: The pathologist’s viewpoint. J. Clin. Pathol. 2002, 55, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Karnik, S.; Srinivasan, B.; Nair, S. Hodgkin’s lymphoma: Immunohistochemical features and its association with EBV LMP-1. Experience from a south Indian hospital. Pathology 2003, 35, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Joos, S.; Kupper, M.; Ohl, S.; von Bonin, F.; Mechtersheimer, G.; Bentz, M.; Marynen, P.; Moller, P.; Pfreundschuh, M.; Trumper, L.; et al. Genomic imbalances including amplification of the tyrosine kinase gene JAK2 in CD30+ Hodgkin cells. Cancer Res. 2000, 60, 549–552. [Google Scholar] [PubMed]
- Joos, S.; Menz, C.K.; Wrobel, G.; Siebert, R.; Gesk, S.; Ohl, S.; Mechtersheimer, G.; Trumper, L.; Moller, P.; Lichter, P.; et al. Classical Hodgkin lymphoma is characterized by recurrent copy number gains of the short arm of chromosome 2. Blood 2002, 99, 1381–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jundt, F.; Acikgoz, O.; Kwon, S.H.; Schwarzer, R.; Anagnostopoulos, I.; Wiesner, B.; Mathas, S.; Hummel, M.; Stein, H.; Reichardt, H.M.; et al. Aberrant expression of notch1 interferes with the B-lymphoid phenotype of neoplastic b cells in classical Hodgkin lymphoma. Leukemia 2008, 22, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Bargou, R.C.; Emmerich, F.; Krappmann, D.; Bommert, K.; Mapara, M.Y.; Arnold, W.; Royer, H.D.; Grinstein, E.; Greiner, A.; Scheidereit, C.; et al. Constitutive nuclear factor-kappab-rela activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J. Clin. Investig. 1997, 100, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Martin-Subero, J.I.; Gesk, S.; Harder, L.; Sonoki, T.; Tucker, P.W.; Schlegelberger, B.; Grote, W.; Novo, F.J.; Calasanz, M.J.; Hansmann, M.L.; et al. Recurrent involvement of the REL and BCL11Aloci in classical Hodgkin lymphoma. Blood 2002, 99, 1474–1477. [Google Scholar] [CrossRef] [PubMed]
- Barth, T.F.; Martin-Subero, J.I.; Joos, S.; Menz, C.K.; Hasel, C.; Mechtersheimer, G.; Parwaresch, R.M.; Lichter, P.; Siebert, R.; Mooller, P. Gains of 2p involving the REL locus correlate with nuclear C-Rel protein accumulation in neoplastic cells of classical Hodgkin lymphoma. Blood 2003, 101, 3681–3686. [Google Scholar] [CrossRef] [PubMed]
- Jost, P.J.; Ruland, J. Aberrant NF-kappab signaling in lymphoma: Mechanisms, consequences, and therapeutic implications. Blood 2007, 109, 2700–2707. [Google Scholar] [PubMed]
- Mottok, A.; Steidl, C. Biology of classical Hodgkin lymphoma: Implications for prognosis and novel therapies. Blood 2018, 131, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Kube, D.; Holtick, U.; Vockerodt, M.; Ahmadi, T.; Haier, B.; Behrmann, I.; Heinrich, P.C.; Diehl, V.; Tesch, H. Stat3 is constitutively activated in Hodgkin cell lines. Blood 2001, 98, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, B.F.; Elia, A.J.; Gascoyne, R.D.; Patterson, B.; Trumper, L.; Kapp, U.; Mak, T.W. Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 2002, 99, 618–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheeren, F.A.; Diehl, S.A.; Smit, L.A.; Beaumont, T.; Naspetti, M.; Bende, R.J.; Blom, B.; Karube, K.; Ohshima, K.; van Noesel, C.J.; et al. Il-21 is expressed in Hodgkin lymphoma and activates stat5: Evidence that activated stat5 is required for Hodgkin lymphomagenesis. Blood 2008, 111, 4706–4715. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Ladewig, E.; Schiavoni, G.; Penson, A.; Fortini, E.; Pettirossi, V.; Wang, Y.; Rosseto, A.; Venanzi, A.; Vlasevska, S.; et al. Pervasive mutations of jak-stat pathway genes in classical Hodgkin lymphoma. Blood 2018, 131, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Guffei, A.; Sarkar, R.; Klewes, L.; Righolt, C.; Knecht, H.; Mai, S. Dynamic chromosomal rearrangements in Hodgkin’s lymphoma are due to ongoing three-dimensional nuclear remodeling and breakage-bridge-fusion cycles. Haematologica 2010, 95, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Johnson, N.A.; Haliotis, T.; Lichtensztejn, D.; Mai, S. Disruption of direct 3D telomere-TRF2 interaction through two molecularly disparate mechanisms is a hallmark of primary Hodgkin and Reed-Sternberg cells. Lab. Investig. 2017, 97, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Re, D.; Kuppers, R.; Diehl, V. Molecular pathogenesis of Hodgkin’s lymphoma. J. Clin. Oncol. 2005, 23, 6379–6386. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.K.; Lambley, E.; Duraiswamy, J.; Dua, U.; Smith, C.; Elliott, S.; Gill, D.; Marlton, P.; Seymour, J.; Khanna, R. Expression of LAG-3 by tumor-infiltrating lymphocytes is coincident with the suppression of latent membrane antigen-specific CD8+ T-cell function in Hodgkin lymphoma patients. Blood 2006, 108, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Vardhana, S.; Younes, A. The immune microenvironment in Hodgkin lymphoma: T cells, B cells, and immune checkpoints. Haematologica 2016, 101, 794–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Berg, A.; Visser, L.; Poppema, S. High expression of the cc chemokine TARC in Reed-Sternberg cells. A possible explanation for the characteristic t-cell infiltratein Hodgkin’s lymphoma. Am. J. Pathol. 1999, 154, 1685–1691. [Google Scholar] [CrossRef]
- Morris, C.S.; Stuart, A.E. Reed-Sternberg/lymphocyte rosette: Lymphocyte subpopulations as defined by monoclonal antibodies. J. Clin. Pathol. 1984, 37, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Gloghini, A.; Gattei, V.; Aldinucci, D.; Degan, M.; De Paoli, P.; Zagonel, V.; Pinto, A. Expression of functional CD40 antigen on Reed-Sternberg cells and Hodgkin’s disease cell lines. Blood 1995, 85, 780–789. [Google Scholar] [PubMed]
- Annunziata, C.M.; Safiran, Y.J.; Irving, S.G.; Kasid, U.N.; Cossman, J. Hodgkin disease: Pharmacologic intervention of the CD40-NF kappa b pathway by a protease inhibitor. Blood 2000, 96, 2841–2848. [Google Scholar] [PubMed]
- Aldinucci, D.; Gloghini, A.; Pinto, A.; Colombatti, A.; Carbone, A. The role of CD40/CD40L and interferon regulatory factor 4 in Hodgkin lymphoma microenvironment. Leuk. Lymphoma 2012, 53, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Carey, C.D.; Connelly, C.; Gjini, E.; Roemer, M.G.; Stack, E.; Hodi, S.; Shipp, M.A.; Rodig, S.J. Quantitative assessment of PD-L1 expression in classical Hodgkin lymphoma suggests a critical role for tumor associated macrophages in suppressing anti-tumor immunity. Blood 2015, 126, 1440. [Google Scholar]
- Gatalica, Z.; Bilalovic, N.; Vranic, S.; Arguello, D.; Reddy, S.; Ghosh, N. PD-L1 and PD1 expression in lymphomas. Blood 2015, 126, 3899. [Google Scholar]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large b-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Chapuy, B.; Ouyang, J.; Sun, H.H.; Roemer, M.G.; Xu, M.L.; Yu, H.; Fletcher, C.D.; Freeman, G.J.; Shipp, M.A.; et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin. Cancer Res. 2013, 19, 3462–3473. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Gravelle, P.; Burroni, B.; Pericart, S.; Rossi, C.; Bezombes, C.; Tosolini, M.; Damotte, D.; Brousset, P.; Fournie, J.J.; Laurent, C. Mechanisms of PD-1/PD-L1 expression and prognostic relevance in non-Hodgkin lymphoma: A summary of immunohistochemical studies. Oncotarget 2017, 8, 44960–44975. [Google Scholar] [CrossRef] [PubMed]
- Oudejans, J.J.; Jiwa, N.M.; Kummer, J.A.; Horstman, A.; Vos, W.; Baak, J.P.; Kluin, P.M.; van der Valk, P.; Walboomers, J.M.; Meijer, C.J. Analysis of major histocompatibility complex class I expression on Reed-Sternberg cells in relation to the cytotoxic T-cell response in epstein-barr virus-positive and -negative Hodgkin’s disease. Blood 1996, 87, 3844–3851. [Google Scholar] [PubMed]
- Reichel, J.; Chadburn, A.; Rubinstein, P.G.; Giulino-Roth, L.; Tam, W.; Liu, Y.; Gaiolla, R.; Eng, K.; Brody, J.; Inghirami, G.; et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood 2015, 125, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.; Advani, R.H.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin lymphoma with reduced beta2M/MHC class I expression is associated with inferior outcome independent of 9p24.1 status. Cancer Immunol. Res. 2016, 4, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Diepstra, A.; van Imhoff, G.W.; Karim-Kos, H.E.; van den Berg, A.; te Meerman, G.J.; Niens, M.; Nolte, I.M.; Bastiaannet, E.; Schaapveld, M.; Vellenga, E.; et al. Hla class ii expression by Hodgkin Reed-Sternberg cells is an independent prognostic factor in classical Hodgkin’s lymphoma. J. Clin. Oncol. 2007, 25, 3101–3108. [Google Scholar] [CrossRef] [PubMed]
- Barrington, S.F.; Mikhaeel, N.G.; Kostakoglu, L.; Meignan, M.; Hutchings, M.; Mueller, S.P.; Schwartz, L.H.; Zucca, E.; Fisher, R.I.; Trotman, J.; et al. Role of imaging in the staging and response assessment of lymphoma: Consensus of the international conference on malignant lymphomas imaging working group. J. Clin. Oncol. 2014, 32, 3048–3058. [Google Scholar] [CrossRef] [PubMed]
- Skoetz, N.; Will, A.; Monsef, I.; Brillant, C.; Engert, A.; von Tresckow, B. Comparison of first-line chemotherapy including escalated BEACOPP versus chemotherapy including ABVD for people with early unfavourable or advanced stage Hodgkin lymphoma. Cochrane Database Syst. Rev. 2017, 5, CD007941. [Google Scholar] [CrossRef] [PubMed]
- Brockelmann, P.J.; Sasse, S.; Engert, A. Balancing risk and benefit in early-stage classical Hodgkin lymphoma. Blood 2018, 131, 1666–1678. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Johnson, P.W.M. Optimizing therapy in advanced stage Hodgkin lymphoma. Blood 2018, 131, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Engert, A.; Plutschow, A.; Eich, H.T.; Lohri, A.; Dorken, B.; Borchmann, P.; Berger, B.; Greil, R.; Willborn, K.C.; Wilhelm, M.; et al. Reduced treatment intensity in patients with early-stage Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 363, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Von Tresckow, B.; Plutschow, A.; Fuchs, M.; Klimm, B.; Markova, J.; Lohri, A.; Kral, Z.; Greil, R.; Topp, M.S.; Meissner, J.; et al. Dose-intensification in early unfavorable Hodgkin’s lymphoma: Final analysis of the german Hodgkin study group hd14 trial. J. Clin. Oncol. 2012, 30, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.M.; Gospodarowicz, M.K.; Connors, J.M.; Pearcey, R.G.; Wells, W.A.; Winter, J.N.; Horning, S.J.; Dar, A.R.; Shustik, C.; Stewart, D.A.; et al. ABVD alone versus radiation-based therapy in limited-stage Hodgkin’s lymphoma. N. Engl. J. Med. 2012, 366, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Radford, J.; Illidge, T.; Counsell, N.; Hancock, B.; Pettengell, R.; Johnson, P.; Wimperis, J.; Culligan, D.; Popova, B.; Smith, P.; et al. Results of a trial of pet-directed therapy for early-stage Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Andre, M.P.E.; Girinsky, T.; Federico, M.; Reman, O.; Fortpied, C.; Gotti, M.; Casasnovas, O.; Brice, P.; van der Maazen, R.; Re, A.; et al. Early positron emission tomography response-adapted treatment in stage I and II Hodgkin lymphoma: Final results of the randomized EORTC/LYSA/FIL H10 trial. J. Clin. Oncol. 2017, 35, 1786–1794. [Google Scholar] [CrossRef] [PubMed]
- Viviani, S.; Zinzani, P.L.; Rambaldi, A.; Brusamolino, E.; Levis, A.; Bonfante, V.; Vitolo, U.; Pulsoni, A.; Liberati, A.M.; Specchia, G.; et al. ABVD versus beacopp for Hodgkin’s lymphoma when high-dose salvage is planned. N. Engl. J. Med. 2011, 365, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.; Federico, M.; Kirkwood, A.; Fossa, A.; Berkahn, L.; Carella, A.; d’Amore, F.; Enblad, G.; Franceschetto, A.; Fulham, M.; et al. Adapted treatment guided by interim PET-CT scan in advanced Hodgkin’s lymphoma. N. Engl. J. Med. 2016, 374, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illes, A.; Picardi, M.; et al. Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin’s lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, N.; Pfistner, B.; Sextro, M.; Sieber, M.; Carella, A.M.; Haenel, M.; Boissevain, F.; Zschaber, R.; Muller, P.; Kirchner, H.; et al. Aggressive conventional chemotherapy compared with high-dose chemotherapy with autologous haemopoietic stem-cell transplantation for relapsed chemosensitive Hodgkin’s disease: A randomised trial. Lancet 2002, 359, 2065–2071. [Google Scholar] [CrossRef]
- Nikolaenko, L.; Chen, R.; Herrera, A.F. Current strategies for salvage treatment for relapsed classical Hodgkin lymphoma. Ther. Adv. Hematol. 2017, 8, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reece, D.E.; Barnett, M.J.; Shepherd, J.D.; Hogge, D.E.; Klasa, R.J.; Nantel, S.H.; Sutherland, H.J.; Klingemann, H.G.; Fairey, R.N.; Voss, N.J.; et al. High-dose cyclophosphamide, carmustine (BCNU), and etoposide (VP16-213) with or without cisplatin (CBV +/− p) and autologous transplantation for patients with Hodgkin’s disease who fail to enter a complete remission after combination chemotherapy. Blood 1995, 86, 451–456. [Google Scholar] [PubMed]
- Andre, M.; Henry-Amar, M.; Pico, J.L.; Brice, P.; Blaise, D.; Kuentz, M.; Coiffier, B.; Colombat, P.; Cahn, J.Y.; Attal, M.; et al. Comparison of high-dose therapy and autologous stem-cell transplantation with conventional therapy for Hodgkin’s disease induction failure: A case-control study. Societe francaise de greffe de moelle. J. Clin. Oncol. 1999, 17, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Majhail, N.S.; Weisdorf, D.J.; Defor, T.E.; Miller, J.S.; McGlave, P.B.; Slungaard, A.; Arora, M.; Ramsay, N.K.; Orchard, P.J.; MacMillan, M.L.; et al. Long-term results of autologous stem cell transplantation for primary refractory or relapsed Hodgkin’s lymphoma. Biol. Blood Marrow Transplant. 2006, 12, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Forero-Torres, A.; Leonard, J.P.; Younes, A.; Rosenblatt, J.D.; Brice, P.; Bartlett, N.L.; Bosly, A.; Pinter-Brown, L.; Kennedy, D.; Sievers, E.L.; et al. A phase ii study of SGN-30 (anti-CD30 mAb) in Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Br. J. Haematol. 2009, 146, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Connors, J.M.; Engert, A.; Larsen, E.K.; Huebner, D.; et al. Five-year survival and durability results of brentuximab vedotin in patients with relapsed or refractory Hodgkin lymphoma. Blood 2016, 128, 1562–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, O.A.; Lue, J.K.; Sawas, A.; Amengual, J.E.; Deng, C.; Kalac, M.; Falchi, L.; Marchi, E.; Turenne, I.; Lichtenstein, R.; et al. Brentuximab vedotin plus bendamustine in relapsed or refractory Hodgkin’s lymphoma: An international, multicentre, single-arm, phase 1–2 trial. Lancet Oncol. 2018, 19, 257–266. [Google Scholar] [CrossRef]
- LaCasce, A.S.; Bociek, R.G.; Sawas, A.; Caimi, P.; Agura, E.; Matous, J.; Ansell, S.M.; Crosswell, H.E.; Islas-Ohlmayer, M.; Behler, C.; et al. Brentuximab vedotin plus bendamustine: A highly active first salvage regimen for relapsed or refractory Hodgkin lymphoma. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.H.; Chen, A.I.; Stiff, P.; Gianni, A.M.; Carella, A.; et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Odorizzi, P.M.; Wherry, E.J. Inhibitory receptors on lymphocytes: Insights from infections. J. Immunol. 2012, 188, 2957–2965. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Nishikori, M.; Kitawaki, T.; Sakai, T.; Hishizawa, M.; Tashima, M.; Kondo, T.; Ohmori, K.; Kurata, M.; Hayashi, T.; et al. PD-1-PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood 2008, 111, 3220–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ t cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Shipp, M.A.; Ribrag, V.; Michot, J.M.; Zinzani, P.L.; Kuruvilla, J.; Snyder, E.S.; Ricart, A.D.; Balakumaran, A.; Rose, S.; et al. Programmed death-1 blockade with pembrolizumab in patients with classical Hodgkin lymphoma after brentuximab vedotin failure. J. Clin. Oncol. 2016, 34, 3733–3739. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, C.H.; Chen, R.W.; Armand, P.; Zinzani, P.L.; Vassilakopoulos, T.P.; Goldmacher, G.V.; Lin, J.; Nahar, A.; Balakumaran, A.; Salles, G. Pembrolizumab antitumor activity in relapsed/refractory classical Hodgkin lymphoma in keynote-087: Revised response criteria for malignant lymphoma 2007 criteria versus lugano 2014 classification. Blood 2017, 130, 4085. [Google Scholar]
- Armand, P.; Engert, A.; Younes, A.; Fanale, M.; Santoro, A.; Zinzani, P.L.; Timmerman, J.M.; Collins, G.P.; Ramchandren, R.; Cohen, J.B.; et al. Nivolumab for relapsed/refractory classic Hodgkin lymphoma after failure of autologous hematopoietic cell transplantation: Extended follow-up of the multicohort single-arm phase II checkmate 205 trial. J. Clin. Oncol. 2018, 36, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Gibb, A.L.; Collins, G.P.; Popat, R.; El-Sharkawi, D.; Burton, C.; Lewis, D.; Miall, F.M.; Forgie, A.; Compagnoni, A.; et al. Blockade of the PD-1 checkpoint with anti-PD-L1 antibody avelumab is sufficienct for clinical activity in relapsed/refractory classical Hodgkin lymphoma (CHL). Hematol. Oncol. 2017, 35, 67. [Google Scholar] [CrossRef]
- Younes, A.; Santoro, A.; Shipp, M.; Zinzani, P.L.; Timmerman, J.M.; Ansell, S.; Armand, P.; Fanale, M.; Ratanatharathorn, V.; Kuruvilla, J.; et al. Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: A multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol. 2016, 17, 1283–1294. [Google Scholar] [CrossRef]
- Ansell, S.; Gutierrez, M.E.; Shipp, M.A.; Gladstone, D.; Moskowitz, A.; Borello, I.; Popa-Mckiver, M.; Farsaci, B.; Zhu, L.; Lesokhin, A.M.; et al. A phase 1 study of nivolumab in combination with ipilimumab for relapsed or refractory hematologic malignancies (checkmate 039). Blood 2016, 128, 183. [Google Scholar]
- Herrera, A.F.; Moskowitz, A.J.; Bartlett, N.L.; Vose, J.M.; Ramchandren, R.; Feldman, T.A.; LaCasce, A.S.; Ansell, S.M.; Moskowitz, C.H.; Fenton, K.; et al. Interim results of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2018, 131, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, C.S.; Hong, F.; Cohen, J.B.; Robertson, M.J.; Ambinder, R.F.; Fenske, T.S.; Advani, R.H.; Kahl, B.S.; Ansell, S. Preliminary safety and efficacy of the combination of brentuximab vedotin and ipilimumab in relapsed/refractory Hodgkin lymphoma: A trial of the ECOG-ACRIN cancer research group (E4412). Blood 2015, 126, 585. [Google Scholar]
- Ramchandren, R. Nivolumab for newly diagnosed advanced-stage classical Hodgkin lymphoma (CHL): Results from the phase 2 checkmate 205 study. In Proceedings of the 59th Annual American Society of Hematology Conference, San Diego, CA, USA, 6 December 2017. [Google Scholar]
- Diefenbach, C.S.; Hong, F.; David, K.; Cohen, J.; Roberston, M.; Advani, R.; Palmisano, N.; Ambinder, R.; Kahl, B.; Ansell, S. Safety and efficacy of combination of brentuximab vedotin and nivolumab in relapsed/refractory Hodgkin lymphoma: A trial of the ecog-acrin cancer research group (e4412). Hematol. Oncol. 2017, 35, 84–85. [Google Scholar] [CrossRef]
- Cheson, B.D.; Pfistner, B.; Juweid, M.E.; Gascoyne, R.D.; Specht, L.; Horning, S.J.; Coiffier, B.; Fisher, R.I.; Hagenbeek, A.; Zucca, E.; et al. Revised response criteria for malignant lymphoma. J. Clin. Oncol. 2007, 25, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Cheson, B.D.; Fisher, R.I.; Barrington, S.F.; Cavalli, F.; Schwartz, L.H.; Zucca, E.; Lister, T.A.; Alliance, A.L.; Lymphoma, G.; Eastern Cooperative Oncology, G.; et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: The lugano classification. J. Clin. Oncol. 2014, 32, 3059–3068. [Google Scholar] [CrossRef] [PubMed]
- Cheson, B.D.; Ansell, S.; Schwartz, L.; Gordon, L.I.; Advani, R.; Jacene, H.A.; Hoos, A.; Barrington, S.F.; Armand, P. Refinement of the lugano classification lymphoma response criteria in the era of immunomodulatory therapy. Blood 2016, 128, 2489–2496. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Zinzani, P.L.; Fanale, M.A.; Chen, R.; Armand, P.; Johnson, N.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Pembrolizumab monotherapy in patients with primary refractory classical Hodgkin lymphoma: Subgroup analysis of the phase 2 keynote-087 study [abstract 126]. In Proceedings of the 14th International Conference on Malignant Lymphoma, Lugano, Switzerland, 14–17 June 2017. [Google Scholar]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to pd-1 inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [PubMed]
- Hude, I.; Sasse, S.; Brockelmann, P.J.; von Tresckow, B.; Momotow, J.; Engert, A.; Borchmann, S. Leucocyte and eosinophil counts predict progression-free survival in relapsed or refractory classical Hodgkin lymphoma patients treated with PD1 inhibition. Br. J. Haematol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.M.; Redd, R.A.; Cader, F.Z.; Pak, C.J.; Abdelrahman, S.; Ouyang, J.; Sasse, S.; Younes, A.; Fanale, M.; Santoro, A.; et al. Major histocompatibility complex class II and programmed death ligand 1 expression predict outcome after programmed death 1 blockade in classic Hodgkin lymphoma. J. Clin. Oncol. 2018, 36, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Manson, G.; Herbaux, C.; Brice, P.; Bouabdallah, K.; Stamatoullas, A.; Schiano, J.M.; Ghesquieres, H.; Dercle, L.; Houot, R. Prolonged remissions after anti-PD1 discontinuation in patients with Hodgkin lymphoma. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Evens, A.M.; Helenowski, I.; Ramsdale, E.; Nabhan, C.; Karmali, R.; Hanson, B.; Parsons, B.; Smith, S.; Larsen, A.; McKoy, J.M.; et al. A retrospective multicenter analysis of elderly Hodgkin lymphoma: Outcomes and prognostic factors in the modern era. Blood 2012, 119, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Thyss, A.; Saada, E.; Gastaud, L.; Peyrade, F.; Re, D. Hodgkin’s lymphoma in older patients: An orphan disease? Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014050. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.B.; Gordon, L.I. Frontline therapy for classical Hodgkin lymphoma by stage and prognostic factors. Clin. Med. Insights Oncol. 2017, 11, 1179554917731072. [Google Scholar] [CrossRef] [PubMed]
- Derer, A.; Frey, B.; Fietkau, R.; Gaipl, U.S. Immune-modulating properties of ionizing radiation: Rationale for the treatment of cancer by combination radiotherapy and immune checkpoint inhibitors. Cancer Immunol. Immunother. 2016, 65, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Barker, C.A.; Yamada, Y.; Yuan, J.; Kitano, S.; Mu, Z.; Rasalan, T.; Adamow, M.; Ritter, E.; et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 2012, 366, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockelmann, P.J.; Engert, A. Checkpoint inhibition in Hodgkin lymphoma—A review. Oncol. Res. Treat. 2017, 40, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating tumor DNA reveals genetics, clonal evolution and residual disease in classical Hodgkin lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug | Phase | ORR% | CR% | Ref. |
|---|---|---|---|---|
| Monotherapy | ||||
| Nivolumab | I | 87 | 17 | [86] |
| Nivolumab | II | 68 | 9 | [92] |
| Pembrolizumab | I | 65 | 16 | [87] |
| Pembrolizumab | II | 69 | 22 | [88] |
| Avelumab | I | 54.8 | 6.5 | [91] |
| Combination Therapy | ||||
| Nivolumab + Ipilimumab | I | 74 | 19 | [93] |
| Nivolumab + Brentuximab Vedotin | I/II | 82 | 61 | [94] |
| Ipilimumab + Brentuximab Vedotin | I/II | 72 | 50 | [95] |
| Nivolumab + AVD | II | 84 | 67 | [96] |
| Combination ICI Clinical Trials | Phase | Trial ID |
|---|---|---|
| Nivolumab + Ipilimumab + BV | I | NCT01896999 |
| Nivolumab + Ipilimumab + Daratumumab | I | NCT01592370 |
| Nivolumab + Ipilimumab + Daratumumab + Pomalidomide | I | NCT01592370 |
| Nivolumab + Ibrutinib | II | NCT02940301 |
| Nivolumab + ICE chemotherapy | II | NCT03016871 |
| Pembrolizumab + ISRT | II | NCT03179917 |
| Pembrolizumab + AFM13 | I | NCT02665650 |
| Pembrolizumab + Lenalidomide | I/II | NCT02875067 |
| Pembrolizumab + ICE chemotherapy | II | NCT03077828 |
| Pembrolizumab + BV | III | NCT02684292 |
| Pembrolizumab + Vorinostat | I | NCT03150329 |
| Nivolumab + Bendamustine | I/II | NCT03343652 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meti, N.; Esfahani, K.; Johnson, N.A. The Role of Immune Checkpoint Inhibitors in Classical Hodgkin Lymphoma. Cancers 2018, 10, 204. https://doi.org/10.3390/cancers10060204
Meti N, Esfahani K, Johnson NA. The Role of Immune Checkpoint Inhibitors in Classical Hodgkin Lymphoma. Cancers. 2018; 10(6):204. https://doi.org/10.3390/cancers10060204
Chicago/Turabian StyleMeti, Nicholas, Khashayar Esfahani, and Nathalie A. Johnson. 2018. "The Role of Immune Checkpoint Inhibitors in Classical Hodgkin Lymphoma" Cancers 10, no. 6: 204. https://doi.org/10.3390/cancers10060204
APA StyleMeti, N., Esfahani, K., & Johnson, N. A. (2018). The Role of Immune Checkpoint Inhibitors in Classical Hodgkin Lymphoma. Cancers, 10(6), 204. https://doi.org/10.3390/cancers10060204