Current Molecular-Targeted Therapies in NSCLC and Their Mechanism of Resistance

 , ,
, ,
Abstract
:1. Introduction
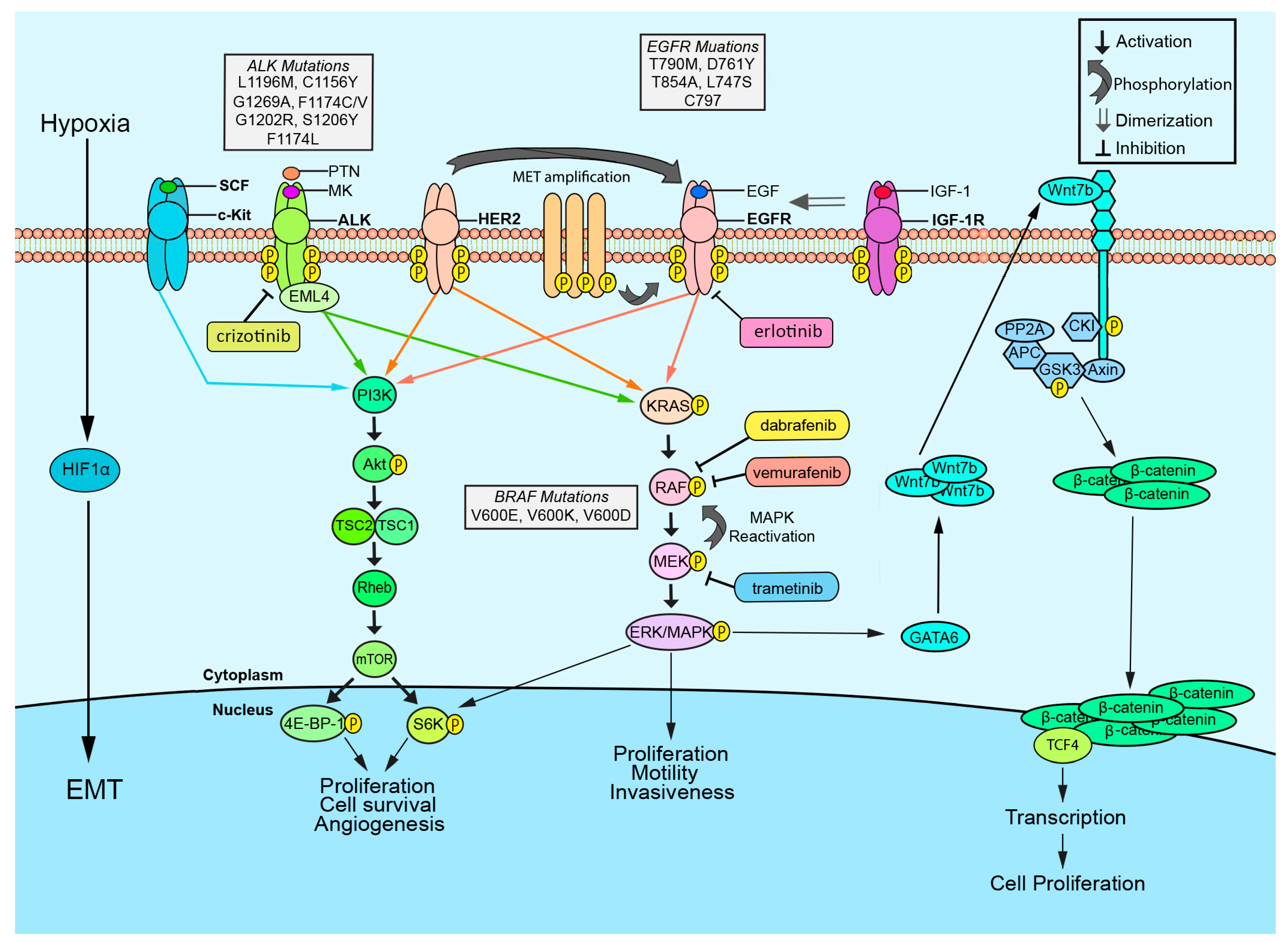
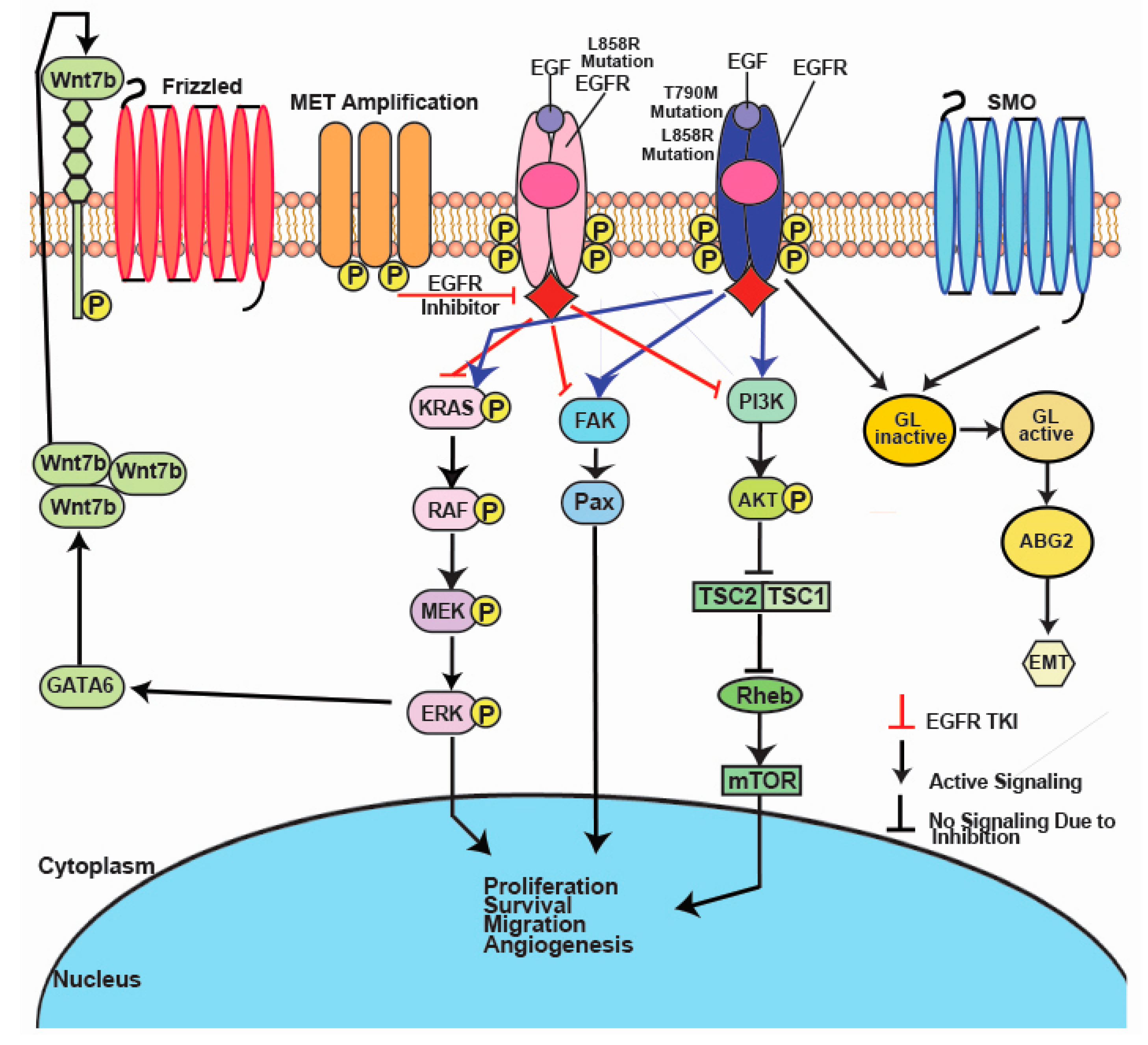
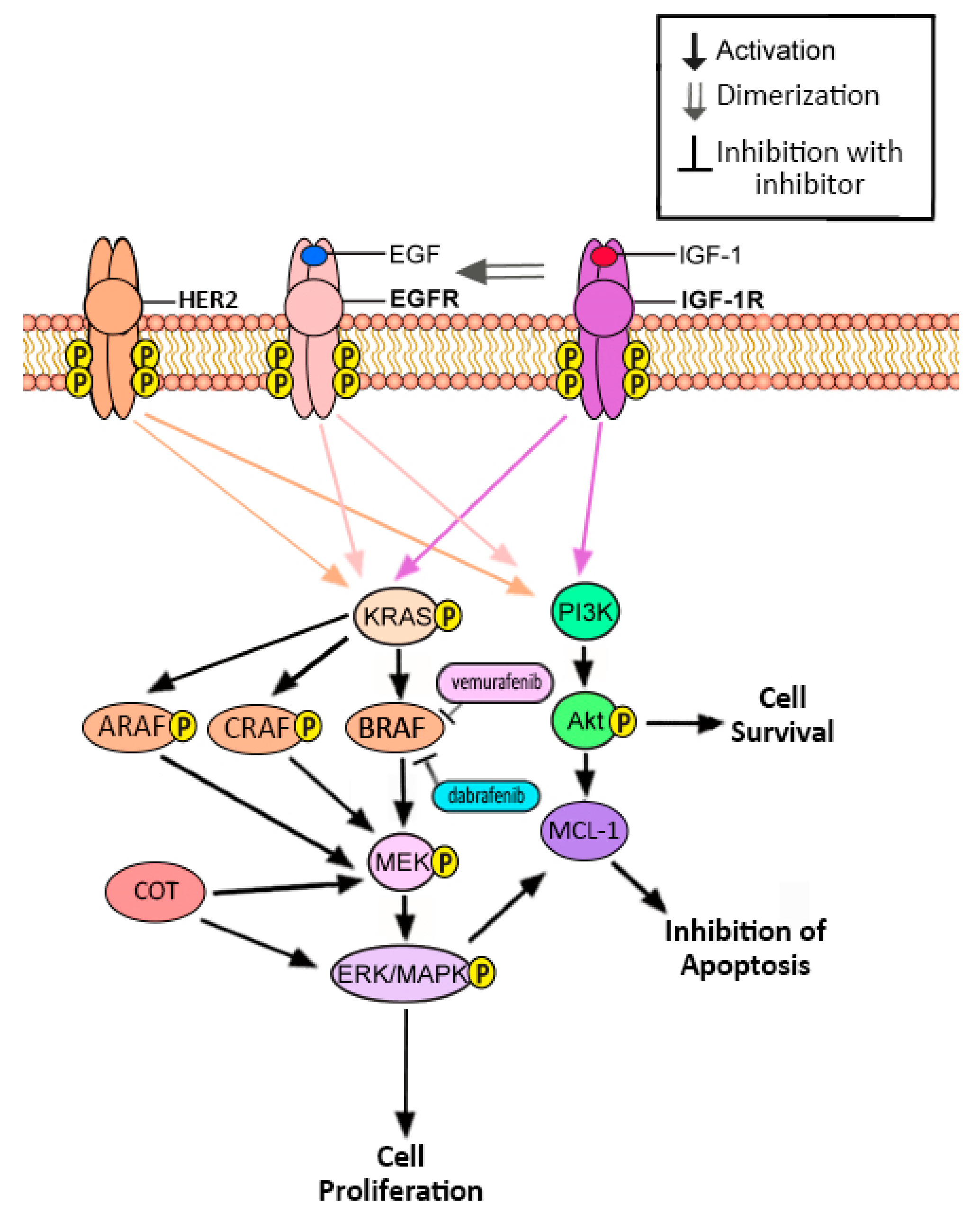
2. EGFR
3. ALK
4. BRAF
5. Conclusions
Funding
Conflicts of Interest
References
- Kratzke, R.; Franklin, M.J. Lung cancer epidemiology. In Encyclopedia of Cancer; Schwab, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–8. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Gadgeel, S.M.; Ramalingam, S.S.; Kalekerian, G.P. Treatment of lung cancer. Radiol. Clin. N. Am. 2012, 50, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Daga, A.; Ansari, A.; Patel, S.; Mirza, S.; Rawal, R.; Umrania, V. Current drugs and drug targets in non-small cell lung cancer: Limitations and opportunities. APJCP 2015, 16, 4147–4156. [Google Scholar] [CrossRef] [PubMed]
- Domvri, K.; Zarogoulidis, P.; Darwiche, K.; Browning, R.F.; Li, Q.; Turner, J.F.; Kioumis, I.; Spyratos, D.; Porpodis, K.; Papaiwannou, A.; et al. Molecular targeted drugs and biomarkers in NSCLC, the evolving role of individualized therapy. J. Cancer 2013, 4, 736–754. [Google Scholar] [CrossRef] [PubMed]
- Cardarella, S.; Ogino, A.; Nishino, M.; Butaney, M.; Shen, J.; Lydon, C.; Yeap, B.Y.; Sholl, L.M.; Johnson, B.E.; Janne, P.A. Clinical, pathologic, and biologic features associated with braf mutations in non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 4532–4540. [Google Scholar] [CrossRef] [PubMed]
- Ciuffreda, L.; Incani, U.C.; Steelman, L.S.; Abrams, S.L.; Falcone, I.; Curatolo, A.D.; Chappell, W.H.; Franklin, R.A.; Vari, S.; Cognetti, F.; et al. Signaling intermediates (MAPK and PI3K) as therapeutic targets in nsclc. Curr. Pharm. Des. 2014, 20, 3944–3957. [Google Scholar] [CrossRef] [PubMed]
- Soucheray, M.; Capelletti, M.; Pulido, I.; Kuang, Y.; Paweletz, C.P.; Becker, J.H.; Kikuchi, E.; Xu, C.; Patel, T.B.; Al-Shahrour, F.; et al. Intratumoral heterogeneity in EGFR-mutant NSCLC results in divergent resistance mechanisms in response to egfr tyrosine kinase inhibition. Cancer Res. 2015, 75, 4372–4383. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Sima, C.S.; Huang, J.; Solomon, S.B.; Rimner, A.; Paik, P.; Pietanza, M.C.; Azzoli, C.G.; Rizvi, N.A.; Krug, L.M.; et al. Local therapy with continued egfr tyrosine kinase inhibitor therapy as a treatment strategy in egfr-mutant advanced lung cancers that have developed acquired resistance to egfr tyrosine kinase inhibitors. J. Thorac. Oncol. 2013, 8, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Godin-Heymann, N.; Ulkus, L.; Brannigan, B.W.; McDermott, U.; Lamb, J.; Maheswaran, S.; Settleman, J.; Haber, D.A. The t790m “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible egfr inhibitor. Mol. Cancer Ther. 2008, 7, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Ray, M.; Salgia, R.; Vokes, E.E. The role of egfr inhibition in the treatment of non-small cell lung cancer. Oncologist 2009, 14, 1116–1130. [Google Scholar] [CrossRef] [PubMed]
- Engle, J.A.; Kolesar, J.M. Afatinib: A first-line treatment for selected patients with metastatic non-small-cell lung cancer. AJHP 2014, 71, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target binding properties and cellular activity of afatinib (bibw 2992), an irreversible erbb family blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. Azd9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- FDA. Fda Approves Osimertinib for First-Line Treatment of Metastatic Nsclc with Most Common EGFR Mutations. Available online: http://www.fda.gov (accessed on 12 June 2018).
- First-line osimertinib beneficial in advanced nsclc. Cancer Dis. 2018, 8, OF1.
- Floc’h, N.; Martin, M.J.; Riess, J.W.; Orme, J.P.; Staniszewska, A.D.; Menard, L.; Cuomo, M.E.; O’Neill, D.J.; Ward, R.A.; Finlay, M.R.V.; et al. Antitumor activity of osimertinib, an irreversible mutant-selective EGFR tyrosine kinase inhibitor, in nsclc harboring egfr exon 20 insertions. Mol. Cancer Ther. 2018, 17, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Loriot, Y.; Andre, F.; Gobert, A.; Auger, N.; Lacroix, L.; Soria, J.C. Egfr-independent mechanisms of acquired resistance to azd9291 in egfr t790m-positive nsclc patients. Ann. Oncol. 2015, 26, 2073–2078. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Hu, M.; Bai, Y.; Zhu, X.; Lu, X.; Wu, C.; Wang, J.; Liu, L.; Wang, Z.; Ni, J.; et al. EGFR g796d mutation mediates resistance to osimertinib. Oncotarget 2017, 8, 49671–49679. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tsui, S.T.; Liu, C.; Song, Y.; Liu, D. EGFR c797s mutation mediates resistance to third-generation inhibitors in t790m-positive non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Steuer, C.E.; Khuri, F.R.; Ramalingam, S.S. The next generation of epidermal growth factor receptor tyrosine kinase inhibitors in the treatment of lung cancer. Cancer 2015, 121, E1–E6. [Google Scholar] [CrossRef] [PubMed]
- Van Der Steen, N.; Caparello, C.; Rolfo, C.; Pauwels, P.; Peters, G.J.; Giovannetti, E. New developments in the management of non-small-cell lung cancer, focus on rociletinib: What went wrong? OncoTargets Ther. 2016, 9, 6065–6074. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, Y.; Liu, D. Eai045: The fourth-generation egfr inhibitor overcoming t790m and c797s resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Eng. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D.; Blumenthal, G.M.; Chen, H.Y.; He, K.; Patel, M.; Justice, R.; Keegan, P.; Pazdur, R. Fda approval summary: Crizotinib for the treatment of metastatic non-small cell lung cancer with anaplastic lymphoma kinase rearrangements. Oncologist 2014, 19, e5–e11. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med. 2012, 4, 120ra117. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with alk gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Dehghanian, F.; Kay, M.; Vallian, S. F1174v mutation alters the alk active conformation in response to crizotinib in NSCLC: Insight from molecular simulations. J. Mol. Graph. Model. 2017, 75, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J. The potential for crizotinib in non-small cell lung cancer: A perspective review. Ther. Adv. Med. Oncol. 2011, 3, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Shen, S.; Shen, L.; Lu, S. An interaction map of small-molecule kinase inhibitors with anaplastic lymphoma kinase (ALK) mutants in ALK-positive non-small cell lung cancer. Biochimie 2015, 112, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Wilson, F.H.; Johannessen, C.M.; Piccioni, F.; Tamayo, P.; Kim, J.W.; van Allen, E.M.; Corsello, S.M.; Capelletti, M.; Calles, A.; Butaney, M.; et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell 2015, 27, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Heuckmann, J.M.; Holzel, M.; Sos, M.L.; Heynck, S.; Balke-Want, H.; Koker, M.; Peifer, M.; Weiss, J.; Lovly, C.M.; Grutter, C.; et al. Alk mutations conferring differential resistance to structurally diverse alk inhibitors. Clin. Cancer Res. 2011, 17, 7394–7401. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Engelman, J.A. Ceritinib in alk-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Shaw, A.T. Alk inhibitors in non-small cell lung cancer: Crizotinib and beyond. Clin. Adv. Hematol. Oncol. 2014, 12, 429–439. [Google Scholar] [PubMed]
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The alk inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Dis. 2014, 4, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Inamasu, E.; Shimamatsu, S.; Yoshida, T.; Nosaki, K.; Hirai, F.; Yamaguchi, M.; Seto, T.; Takenoyama, M.; Ichinose, Y. Identification of a novel ALK g1123s mutation in a patient with ALK-rearranged non-small-cell lung cancer exhibiting resistance to ceritinib. J. Thorac. Oncol. 2015, 10, e55–e57. [Google Scholar] [CrossRef] [PubMed]
- Zhu, V.W.; Cui, J.J.; Fernandez-Rocha, M.; Schrock, A.B.; Ali, S.M.; Ou, S.I. Identification of a novel t1151k alk mutation in a patient with alk-rearranged nsclc with prior exposure to crizotinib and ceritinib. Lung Cancer 2017, 110, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.I.; Perol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Ignatius Ou, S.H.; Azada, M.; Hsiang, D.J.; Herman, J.M.; Kain, T.S.; Siwak-Tapp, C.; Casey, C.; He, J.; Ali, S.M.; Klempner, S.J.; et al. Next-generation sequencing reveals a novel nsclc alk f1174v mutation and confirms ALK g1202r mutation confers high-level resistance to alectinib (ch5424802/ro5424802) in ALK-rearranged nsclc patients who progressed on crizotinib. J. Thorac. Oncol. 2014, 9, 549–553. [Google Scholar] [PubMed]
- Ou, S.H.; Klempner, S.J.; Greenbowe, J.R.; Azada, M.; Schrock, A.B.; Ali, S.M.; Ross, J.S.; Stephens, P.J.; Miller, V.A. Identification of a novel hip1-ALK fusion variant in non-small-cell lung cancer (NSCLC) and discovery of alk i1171 (i1171n/s) mutations in two alk-rearranged nsclc patients with resistance to alectinib. J. Thorac. Oncol. 2014, 9, 1821–1825. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Hirai, F.; Inamasu, E.; Yoshida, T.; Nosaki, K.; Takenaka, T.; Yamaguchi, M.; Seto, T.; Takenoyama, M.; Ichinose, Y. Secondary mutations at i1171 in the alk gene confer resistance to both crizotinib and alectinib. J. Thorac. Oncol. 2014, 9, e86–e87. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two novel ALK mutations mediate acquired resistance to the next-generation alk inhibitor alectinib. Clin. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef] [PubMed]
- Sabari, J.K.; Santini, F.C.; Schram, A.M.; Bergagnini, I.; Chen, R.; Mrad, C.; Lai, W.V.; Arbour, K.C.; Drilon, A. The activity, safety, and evolving role of brigatinib in patients with ALK-rearranged non-small cell lung cancers. OncoTargets Ther. 2017, 10, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Felip, E.; Bauer, T.M.; Besse, B.; Navarro, A.; Postel-Vinay, S.; Gainor, J.F.; Johnson, M.; Dietrich, J.; James, L.P.; et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: An international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017, 18, 1590–1599. [Google Scholar] [CrossRef]
- Shaw, A.T.; Friboulet, L.; Leshchiner, I.; Gainor, J.F.; Bergqvist, S.; Brooun, A.; Burke, B.J.; Deng, Y.L.; Liu, W.; Dardaei, L.; et al. Resensitization to crizotinib by the lorlatinib alk resistance mutation l1198f. N. Engl. J. Med. 2016, 374, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Farago, A.F.; Le, L.P.; Zheng, Z.; Muzikansky, A.; Drilon, A.; Patel, M.; Bauer, T.M.; Liu, S.V.; Ou, S.H.; Jackman, D.; et al. Durable clinical response to entrectinib in NTRK1-rearranged non-small cell lung cancer. J. Thorac. Oncol. 2015, 10, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Infante, J.R.; Reckamp, K.L.; Blumenschein, G.R.; Leal, T.A.; Waqar, S.N.; Gitlitz, B.J.; Sanborn, R.E.; Whisenant, J.G.; Du, L.; et al. Ensartinib (x-396) in ALK-positive non-small cell lung cancer: Results from a first-in-human phase I/II, multicenter study. Clin. Cancer Res. 2018, 24, 2771–2779. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Kim, T.M.; Mazieres, J.; Quoix, E.; Riely, G.J.; Barlesi, F.; Souquet, P.; Smit, E.F.; Groen, H.J.M.; Kelly, R.J.; et al. Dabrafenib in patients with braf v600e-mutant advanced non-small cell lung cancer (NSCLC): A multicenter, open-label, phase II trial (BRF113928). Ann. Oncol. 2014, 25. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hong, K.; Streit, M. Molecular characterization of acquired resistance to the BRAF inhibitor dabrafenib in a patient with braf-mutant non-small-cell lung cancer. J. Thorac. Oncol. 2013, 8, e41–e42. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Vultur, A.; Herlyn, M. Resistance to braf inhibitors: Unraveling mechanisms and future treatment options. Cancer Res. 2011, 71, 7137–7140. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT drives resistance to Raf inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Rice, S.J.; Liu, X.; Miller, B.; Belani, C.P. Trametinib with or without vemurafenib in braf mutated non-small cell lung cancer. PLoS ONE 2015, 10, e0118210. [Google Scholar] [CrossRef] [PubMed]
- Noeparast, A.; Teugels, E.; Giron, P.; Verschelden, G.; de Brakeleer, S.; Decoster, L.; de Greve, J. Non-v600 braf mutations recurrently found in lung cancer predict sensitivity to the combination of trametinib and dabrafenib. Oncotarget 2017, 8, 60094–60108. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Baselga, J. Epidermal growth factor receptor targeting in cancer. Semin. Oncol. 2006, 33, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Stiegler, A.L.; Boggon, T.J.; Kobayashi, S.; Halmos, B. EGFR-mutated lung cancer: A paradigm of molecular oncology. Oncotarget 2010, 1, 497–514. [Google Scholar] [PubMed]
- Noronha, V.; Choughule, A.; Patil, V.M.; Joshi, A.; Kumar, R.; Susan Joy Philip, D.; Banavali, S.; Dutt, A.; Prabhash, K. Epidermal growth factor receptor exon 20 mutation in lung cancer: Types, incidence, clinical features and impact on treatment. OncoTargets Ther. 2017, 10, 2903–2908. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F. Activating and resistance mutations of egfr in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28, S24–S31. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wei, S.; Song, Y. T790m and acquired resistance of EGFR TKI: A literature review of clinical reports. J. Thorac. Dis. 2011, 3, 10–18. [Google Scholar] [PubMed]
- Rastogi, I.; Rajanna, S.; Webb, A.; Chhabra, G.; Foster, B.; Webb, B.; Puri, N. Mechanism of c-MET and EGFR tyrosine kinase inhibitor resistance through epithelial mesenchymal transition in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2016, 477, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Fenoglio, S.; Gao, D.C.; Camiolo, M.; Stiles, B.; Lindsted, T.; Schlederer, M.; Johns, C.; Altorki, N.; Mittal, V.; et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 15535–15540. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Han, R.; Xiao, H.; Lin, C.; Wang, Y.; Liu, H.; Li, K.; Chen, H.; Sun, F.; Yang, Z.; et al. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and emt reversal. Clin. Cancer Res. 2014, 20, 2714–2726. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Mukohara, T.; Zejnullahu, K.; Lifshits, E.; Borras, A.M.; Gale, C.M.; Naumov, G.N.; Yeap, B.Y.; Jarrell, E.; Sun, J.; et al. Allelic dilution obscures detection of a biologically significant resistance mutation in egfr-amplified lung cancer. J. Clin. Investig. 2006, 116, 2695–2706. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.T.; Jacobs, R.J.; Moravec, D.N.; Uppada, S.B.; Botting, G.M.; Nlend, M.; Puri, N. Alternative signaling pathways as potential therapeutic targets for overcoming egfr and c-met inhibitor resistance in non-small cell lung cancer. PLoS ONE 2013, 8, e78398. [Google Scholar] [CrossRef] [PubMed]
- Botting, G.M.; Rastogi, I.; Chhabra, G.; Nlend, M.; Puri, N. Mechanism of resistance and novel targets mediating resistance to egfr and c-met tyrosine kinase inhibitors in non-small cell lung cancer. PLoS ONE 2015, 10, e0136155. [Google Scholar] [CrossRef] [PubMed]
- Civenni, G.; Holbro, T.; Hynes, N.E. Wnt1 and wnt5a induce cyclin d1 expression through erbb1 transactivation in hc11 mammary epithelial cells. EMBO 2003, 4, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.Y.; Zhang, X.C.; Yang, S.Q.; An, S.J.; Chen, Z.H.; Su, J.; Xie, Z.; Gou, L.Y.; Wu, Y.L. Blockade of hedgehog signaling synergistically increases sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer cell lines. PLoS ONE 2016, 11, e0149370. [Google Scholar] [CrossRef] [PubMed]
- Palma, V.; Ruiz i Altaba, A. Hedgehog-Gli signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development 2004, 131, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Narasanna, A.; Perez-Torres, M.; Xiang, B.; Wu, F.Y.; Yang, S.; Carpenter, G.; Gazdar, A.F.; Muthuswamy, S.K.; Arteaga, C.L. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to egfr tyrosine kinase inhibitors. Cancer Cell 2006, 10, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. MET, metastasis, motility and more. Nat. Rev. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. Met amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Stolz, D.B.; Esplen, J.E.; Dorko, K.; Michalopoulos, G.K.; Strom, S.C. Cross-talk between epidermal growth factor receptor and c-MET signal pathways in transformed cells. J. Biol. Chem. 2000, 275, 8806–8811. [Google Scholar] [CrossRef] [PubMed]
- Puri, N.; Salgia, R. Synergism of EGFR and c-MET pathways, cross-talk and inhibition, in non-small cell lung cancer. J. Carcinog. 2008, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Cascone, T.; Xu, L.; Lin, H.Y.; Liu, W.; Tran, H.T.; Liu, Y.; Howells, K.; Haddad, V.; Hanrahan, E.; Nilsson, M.B.; et al. The HGF/c-MET pathway is a driver and biomarker of VEGFR-inhibitor resistance and vascular remodeling in non-small cell lung cancer. Clin. Cancer Res. 2017, 23, 5489–5501. [Google Scholar] [CrossRef] [PubMed]
- McDermott, U.; Pusapati, R.V.; Christensen, J.G.; Gray, N.S.; Settleman, J. Acquired resistance of non-small cell lung cancer cells to met kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010, 70, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Takeuchi, S.; Yamada, T.; Nanjo, S.; Ishikawa, D.; Sano, T.; Kita, K.; Nakamura, T.; Matsumoto, K.; Suda, K.; et al. Combined therapy with mutant-selective EGFR inhibitor and MET kinase inhibitor for overcoming erlotinib resistance in EGFR-mutant lung cancer. Mol. Cancer Ther. 2012, 11, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Rho, J.K.; Jeon, B.S.; Choi, S.J.; Park, S.C.; Lee, S.S.; Kim, H.R.; Kim, C.H.; Lee, J.C. Combined inhibition of igfr enhances the effects of gefitinib in h1650: A lung cancer cell line with egfr mutation and primary resistance to EGFR-TK inhibitors. Cancer Chemother. Pharmacol. 2010, 66, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, R.; Visser, L.E.; van Herk-Sukel, M.P.; Coebergh, J.W.; Haak, H.R.; Geelhoed-Duijvestijn, P.H.; Straus, S.M.; Herings, R.M.; Stricker, B.H. Lower risk of cancer in patients on metformin in comparison with those on sulfonylurea derivatives: Results from a large population-based follow-up study. Diabetes Care 2012, 35, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Sasso, F.C.; Della Corte, C.M.; Vitagliano, D.; D’Aiuto, E.; Troiani, T.; Martinelli, E.; de Vita, F.; Orditura, M.; de Palma, R.; et al. Synergistic effects of metformin treatment in combination with gefitinib, a selective egfr tyrosine kinase inhibitor, in LKB1 wild-type nsclc cell lines. Clin. Cancer Res. 2013, 19, 3508–3519. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Gu, C.; Gu, H.; Hu, H.; Han, Y.; Li, Q. Metformin induces apoptosis of lung cancer cells through activating jnk/p38 MAPK pathway and gadd153. Neoplasma 2011, 58, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Huang, W.; Li, K.; Zhang, K.; Lin, C.; Han, R.; Lu, C.; Wang, Y.; Chen, H.; Sun, F.; et al. Metformin attenuates gefitinib-induced exacerbation of pulmonary fibrosis by inhibition of TGF-beta signaling pathway. Oncotarget 2015, 6, 43605–43619. [Google Scholar] [CrossRef] [PubMed]
- Codony-Servat, C.; Codony-Servat, J.; Karachaliou, N.; Molina, M.A.; Chaib, I.; Ramirez, J.L.; de Los Llanos Gil, M.; Solca, F.; Bivona, T.G.; Rosell, R. Activation of signal transducer and activator of transcription 3 (STAT3) signaling in egfr mutant non-small-cell lung cancer (NSCLC). Oncotarget 2017, 8, 47305–47316. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired resistance to the mutant-selective EGFR inhibitor azd9291 is associated with increased dependence on Ras signaling in preclinical models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Marti, A.; Felip, E.; Matito, J.; Mereu, E.; Navarro, A.; Cedres, S.; Pardo, N.; Martinez de Castro, A.; Remon, J.; Miquel, J.M.; et al. Dual MET and ERBB inhibition overcomes intratumor plasticity in osimertinib-resistant-advanced non-small-cell lung cancer (NSCLC). Ann. Oncol. 2017, 28, 2451–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.I.; Agarwal, N.; Ali, S.M. High met amplification level as a resistance mechanism to osimertinib (azd9291) in a patient that symptomatically responded to crizotinib treatment post-osimertinib progression. Lung Cancer 2016, 98, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Wellstein, A. Alk receptor activation, ligands and therapeutic targeting in glioblastoma and in other cancers. Front. Oncol. 2012, 2, 192. [Google Scholar] [CrossRef] [PubMed]
- Souttou, B.; Carvalho, N.B.; Raulais, D.; Vigny, M. Activation of anaplastic lymphoma kinase receptor tyrosine kinase induces neuronal differentiation through the mitogen-activated protein kinase pathway. J. Biol. Chem. 2001, 276, 9526–9531. [Google Scholar] [CrossRef] [PubMed]
- Morales La Madrid, A.; Campbell, N.; Smith, S.; Cohn, S.L.; Salgia, R. Targeting alk: A promising strategy for the treatment of non-small cell lung cancer, non-hodgkin’s lymphoma, and neuroblastoma. Target. Oncol. 2012, 7, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Solomon, B. Targeting anaplastic lymphoma kinase in lung cancer. Clin. Cancer Res. 2011, 17, 2081–2086. [Google Scholar] [CrossRef] [PubMed]
- Wellstein, A.; Toretsky, J.A. Hunting ALK to feed targeted cancer therapy. Nat. Med. 2011, 17, 290–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proietti, A.; Ali, G.; Pelliccioni, S.; Lupi, C.; Sensi, E.; Boldrini, L.; Servadio, A.; Chella, A.; Ribechini, A.; Cappuzzo, F.; et al. Anaplastic lymphoma kinase gene rearrangements in cytological samples of non-small cell lung cancer: Comparison with histological assessment. Cancer Cytopathol. 2014, 122, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogita, A.; Togashi, Y.; Hayashi, H.; Sogabe, S.; Terashima, M.; de Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Takeyama, Y.; et al. Hypoxia induces resistance to alk inhibitors in the h3122 non-small cell lung cancer cell line with an ALK rearrangement via epithelial-mesenchymal transition. Int. J. Oncol. 2014, 45, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; McDonald, N.T.; Chen, H.; Ortiz-Cuaran, S.; Heukamp, L.C.; Yan, Y.; Florin, A.; Ozretic, L.; Lim, D.; Wang, L.; et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat. Med. 2014, 20, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Zhang, L.; Cheng, Y.; Patel, R.; Wu, H.; Zhang, Y.; Wang, M.; Ji, S.; Belani, C.P.; Yang, J.M.; et al. Induction of autophagy contributes to crizotinib resistance in alk-positive lung cancer. Cancer Biol. Ther. 2014, 15, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kim, D.H.; Choi, Y.J.; Kim, S.Y.; Lee, J.E.; Sung, K.J.; Kim, W.S.; Choi, C.M.; Rho, J.K.; Lee, J.C. Multiple receptor tyrosine kinase activation related to ALK inhibitor resistance in lung cancer cells with alk rearrangement. Oncotarget 2017, 8, 58771–58780. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Ou, S.I.; Shaw, A.T.; Barlesi, F.; Dingemans, A.C.; Kim, D.W.; Camidge, D.R.; Hughes, B.G.M.; Yang, J.C.; de Castro, J.; et al. Efficacy of alectinib in central nervous system metastases in crizotinib-resistant alk-positive non-small-cell lung cancer: Comparison of recist 1.1 and RANO-HGG criteria. Eur. J. Cancer 2017, 82, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Gouji, T.; Takashi, S.; Mitsuhiro, T.; Yukito, I. Crizotinib can overcome acquired resistance to ch5424802: Is amplification of the met gene a key factor? J. Thorac. Oncol. 2014, 9, e27–e28. [Google Scholar] [CrossRef] [PubMed]
- Kogita, A.; Togashi, Y.; Hayashi, H.; Banno, E.; Terashima, M.; de Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Takeyama, Y.; et al. Activated MET acts as a salvage signal after treatment with alectinib, a selective alk inhibitor, in alk-positive non-small cell lung cancer. Int. J. Oncol. 2015, 46, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, A.; Yamada, T.; Nanjo, S.; Takeuchi, S.; Ebi, H.; Kita, K.; Matsumoto, K.; Yano, S. Receptor ligand-triggered resistance to alectinib and its circumvention by HSP90 inhibition in EML4-ALK lung cancer cells. Oncotarget 2014, 5, 4920–4928. [Google Scholar] [CrossRef] [PubMed]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.; et al. Ras-mapk dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Jocolle, G.; Conti, A.; Tiseo, M.; Zito Marino, F.; Donati, G.; Franco, R.; Bono, F.; Barbisan, F.; Facchinetti, F. Detection of ROS1 rearrangement in non-small cell lung cancer: Current and future perspectives. Lung Cancer 2017, 8, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Misale, S.; Wei, G.; Siravegna, G.; Crisafulli, G.; Lazzari, L.; Corti, G.; Rospo, G.; Novara, L.; Mussolin, B.; et al. Acquired resistance to the trk inhibitor entrectinib in colorectal cancer. Cancer Discov. 2016, 6, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Li, G.; Dogan, S.; Gounder, M.; Shen, R.; Arcila, M.; Wang, L.; Hyman, D.M.; Hechtman, J.; Wei, G.; et al. What hides behind the masc: Clinical response and acquired resistance to entrectinib after etv6-ntrk3 identification in a mammary analogue secretory carcinoma (MASC). Ann. Oncol. 2016, 27, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Chmielecki, J.; Gay, L.; Johnson, A.; Chudnovsky, J.; Yelensky, R.; Lipson, D.; Ali, S.M.; Elvin, J.A.; et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int. J. Cancer 2016, 138, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Arcila, M.E.; Fara, M.; Sima, C.S.; Miller, V.A.; Kris, M.G.; Ladanyi, M.; Riely, G.J. Clinical characteristics of patients with lung adenocarcinomas harboring braf mutations. J. Clin. Oncol. 2011, 29, 2046–2051. [Google Scholar] [CrossRef] [PubMed]
- Luk, P.P.; Yu, B.; Ng, C.C.; Mercorella, B.; Selinger, C.; Lum, T.; Kao, S.; O’Toole, S.A.; Cooper, W.A. Braf mutations in non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 142–148. [Google Scholar] [PubMed]
- Zheng, Y.; Zhou, J.; Tong, Y. Gene signatures of drug resistance predict patient survival in colorectal cancer. Pharmacogenomics J. 2015, 15, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Gilligan, D.; Pacey, S. Treatment approaches for egfr-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 2015, 16, e447–e459. [Google Scholar] [CrossRef]
- Mehta, S.; Shelling, A.; Muthukaruppan, A.; Lasham, A.; Blenkiron, C.; Laking, G.; Print, C. Predictive and prognostic molecular markers for cancer medicine. Ther. Adv. Med. Oncol. 2010, 2, 125–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, A.S.; Dinis, J.; Gromicho, M.; Martins, C.; Laires, A.; Rueff, J. Genomics and cancer drug resistance. Curr. Pharm. Biotechnol. 2012, 13, 651–673. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, Z. Understanding the genetic mechanisms of cancer drug resistance using genomic approaches. TIG 2016, 32, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhao, K.; Kirberger, M.; Liao, W.; Yan, Y. Next generation sequencing technologies in cancer diagnostics and therapeutics: A mini review. Cell. Mol. Biol. 2015, 61, 91–102. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Target | Acquired Mutations Conferring Resistance | References |
|---|---|---|---|
| Erlotinib | EGFR | T790M, D761Y, T854A, L747S | [9,10,11] |
| Gefitinib | EGFR | T790M, D761Y, T854A, L747S | [9,10,11] |
| Afatinib | EGFR, HER2 | T790M | [12,13] |
| Osimertinib (AZD9291) | EGFR | C797, G796D | [14,15,16,17,18,19,20] |
| Rociletinib | EGFR | C797 | [21,22] |
| EAI045 | EGFR | Under Investigation | [20,23] |
| Crizotinib | ALK, MET, ROS1 | L1196M, C1156Y, F1174L, F1174V T1151K | [24,25,26,27,28,29,30] |
| TAE684 | ALK | G1123S, G1123SD | [31,32] |
| Ceritinib | ALK | G1202R, F1174C/V, G1123S, T1151K | [33,34,35,36,37] |
| Alectinib | ALK | G1202R, I1171T/N/S, V1180L | [34,38,39,40,41,42] |
| Brigatinib (AP26113) | ALK, ROS1 | Under Investigation | [34,43] |
| Lorlatinib (PF-06463922) | ALK, ROS1 | L1198F | [44,45] |
| Entrectinib (RxDx-101) | ALK, ROS1, NTRK1–3 | NTRK1, NTRK2, NTRK3 | [46] |
| Ensartinib (X-398) | ALK, ROS1, MET, SLK | Under Investigation | [47] |
| Dabrafenib | BRAF | G12D KRAS | [48,49] |
| Vemurafenib | BRAF | Alternate isoforms of RAF proteins | [50,51,52] |
| Trametinib | MEK | Under Investigation | [52,53] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schrank, Z.; Chhabra, G.; Lin, L.; Iderzorig, T.; Osude, C.; Khan, N.; Kuckovic, A.; Singh, S.; Miller, R.J.; Puri, N. Current Molecular-Targeted Therapies in NSCLC and Their Mechanism of Resistance. Cancers 2018, 10, 224. https://doi.org/10.3390/cancers10070224
Schrank Z, Chhabra G, Lin L, Iderzorig T, Osude C, Khan N, Kuckovic A, Singh S, Miller RJ, Puri N. Current Molecular-Targeted Therapies in NSCLC and Their Mechanism of Resistance. Cancers. 2018; 10(7):224. https://doi.org/10.3390/cancers10070224
Chicago/Turabian StyleSchrank, Zachary, Gagan Chhabra, Leo Lin, Tsatsral Iderzorig, Chike Osude, Nabiha Khan, Adijan Kuckovic, Sanjana Singh, Rachel J. Miller, and Neelu Puri. 2018. "Current Molecular-Targeted Therapies in NSCLC and Their Mechanism of Resistance" Cancers 10, no. 7: 224. https://doi.org/10.3390/cancers10070224
APA StyleSchrank, Z., Chhabra, G., Lin, L., Iderzorig, T., Osude, C., Khan, N., Kuckovic, A., Singh, S., Miller, R. J., & Puri, N. (2018). Current Molecular-Targeted Therapies in NSCLC and Their Mechanism of Resistance. Cancers, 10(7), 224. https://doi.org/10.3390/cancers10070224