Ovarian Cancer Immunotherapy: Preclinical Models and Emerging Therapeutics

 , , and
, , and
Abstract
:1. Introduction
2. Adoptive Cell Therapy
Chimeric Antigen Receptor T Cells (CAR-T)
3. Strategies Targeting Immunosuppression in the TME
4. Increasing Tumor Immunogenicity
4.1. Chemotherapy
4.2. Oncolytic Viruses
5. Preclinical Models for Ovarian Cancer Immunotherapy
5.1. Syngeneic Murine Models
5.1.1. Spontaneously Transformed Syngeneic Models
5.1.2. Genetically Engineered Mouse Models (GEMM)
6. Human-Derived and Autologous Cultures
7. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Odunsi, K. Immunotherapy in ovarian cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, viii1–viii7. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.C.; Bean, S.M.; Whitaker, R.S.; Kondoh, E.; Baba, T.; Fujii, S.; Marks, J.R.; Dressman, H.K.; Murphy, S.K.; Berchuck, A. Ovarian cancer tumor infiltrating T-regulatory (T(reg)) cells are associated with a metastatic phenotype. Gynecol. Oncol. 2010, 116, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Abiko, K.; Matsumura, N.; Baba, T.; Yoshioka, Y.; Kosaka, K.; Konishi, I. The comprehensive assessment of local immune status of ovarian cancer by the clustering of multiple immune factors. Clin. Immunol. 2011, 141, 338–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermans, C.; Anz, D.; Engel, J.; Kirchner, T.; Endres, S.; Mayr, D. Analysis of FoxP3+ T-regulatory cells and CD8+ T-cells in ovarian carcinoma: Location and tumor infiltration patterns are key prognostic markers. PLoS ONE 2014, 9, e111757. [Google Scholar] [CrossRef] [PubMed]
- Leffers, N.; Gooden, M.J.M.; de Jong, R.A.; Hoogeboom, B.-N.; ten Hoor, K.A.; Hollema, H.; Boezen, H.M.; van der Zee, A.G.J.; Daemen, T.; Nijman, H.W. Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol. Immunother. 2009, 58, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.S.; Sahota, R.A.; Milne, K.; Kost, S.E.; Nesslinger, N.J.; Watson, P.H.; Nelson, B.H. CD20+ tumor-infiltrating lymphocytes have an atypical CD27− memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin. Cancer Res. 2012, 18, 3281–3292. [Google Scholar] [CrossRef] [PubMed]
- Raspollini, M.R.; Castiglione, F.; Rossi Degl’innocenti, D.; Amunni, G.; Villanucci, A.; Garbini, F.; Baroni, G.; Taddei, G.L. Tumour-infiltrating gamma/delta T-lymphocytes are correlated with a brief disease-free interval in advanced ovarian serous carcinoma. Ann. Oncol. 2005, 16, 590–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stumpf, M.; Hasenburg, A.; Riener, M.-O.; Jütting, U.; Wang, C.; Shen, Y.; Orlowska-Volk, M.; Fisch, P.; Wang, Z.; Gitsch, G.; et al. Intraepithelial CD8-positive T lymphocytes predict survival for patients with serous stage III ovarian carcinomas: Relevance of clonal selection of T lymphocytes. Br. J. Cancer 2009, 101, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Tomsová, M.; Melichar, B.; Sedláková, I.; Steiner, I. Prognostic significance of CD3+ tumor-infiltrating lymphocytes in ovarian carcinoma. Gynecol. Oncol. 2008, 108, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, X.; Han, W.; Zhang, Y. Tisagenlecleucel, an approved anti-CD19 chimeric antigen receptor T-cell therapy for the treatment of leukemia. Drugs Today 2017, 53, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.M.; Galpin, K.J.C.; McCloskey, C.W.; Vanderhyden, B.C. The tumour microenvironment of epithelial ovarian cancer and its influence on response to immunotherapy. Cancers 2018, 10, 230. [Google Scholar] [CrossRef]
- Yigit, R.; Massuger, L.F.A.G.; Figdor, C.G.; Torensma, R. Ovarian cancer creates a suppressive microenvironment to escape immune elimination. Gynecol. Oncol. 2010, 117, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Besser, M.J.; Shapira-Frommer, R.; Treves, A.J.; Zippel, D.; Itzhaki, O.; Hershkovitz, L.; Levy, D.; Kubi, A.; Hovav, E.; Chermoshniuk, N.; et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2010, 16, 2646–2655. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Dudley, M.E. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr. Opin. Immunol. 2009, 21, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, K.; Ikarashi, H.; Takakuwa, K.; Kodama, S.; Tokunaga, A.; Takahashi, T.; Tanaka, K. Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin. Cancer Res. 1995, 1, 501–507. [Google Scholar]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Takakuwa, K.; Kodama, S.; Tanaka, K.; Takahashi, M.; Tokunaga, A.; Takahashi, T. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res. 1991, 51, 1934–1939. [Google Scholar] [PubMed]
- Freedman, R.S.; Edwards, C.L.; Kavanagh, J.J.; Kudelka, A.P.; Katz, R.L.; Carrasco, C.H.; Atkinson, E.N.; Scott, W.; Tomasovic, B.; Templin, S. Intraperitoneal adoptive immunotherapy of ovarian carcinoma with tumor-infiltrating lymphocytes and low-dose recombinant interleukin-2: A pilot trial. J. Immunother. Emphas. Tumor Immunol. 1994, 16, 198–210. [Google Scholar] [CrossRef]
- Santoiemma, P.P.; Powell, D.J. Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol. Ther. 2015, 16, 807–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.C.; Rosenberg, S.A. Adoptive T-Cell therapy for cancer. Adv. Immunol. 2016, 130, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, C.S.; Rosenberg, S.A. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol. Rev. 2014, 257, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Rivière, I. The promise and potential pitfalls of chimeric antigen receptors. Curr. Opin. Immunol. 2009, 21, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.E.; Powell, D.J.; Coukos, G. A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. J. Transl. Med. 2012, 10, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanitis, E.; Dangaj, D.; Hagemann, I.S.; Song, D.-G.; Best, A.; Sandaltzopoulos, R.; Coukos, G.; Powell, D.J. Primary human ovarian epithelial cancer cells broadly express HER2 at immunologically-detectable levels. PLoS ONE 2012, 7, e49829. [Google Scholar] [CrossRef] [PubMed]
- Chekmasova, A.A.; Rao, T.D.; Nikhamin, Y.; Park, K.J.; Levine, D.A.; Spriggs, D.R.; Brentjens, R.J. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin. Cancer Res. 2010, 16, 3594–3606. [Google Scholar] [CrossRef] [PubMed]
- Felder, M.; Kapur, A.; Gonzalez-Bosquet, J.; Horibata, S.; Heintz, J.; Albrecht, R.; Fass, L.; Kaur, J.; Hu, K.; Shojaei, H.; et al. MUC16 (CA125): Tumor biomarker to cancer therapy, a work in progress. Mol. Cancer 2014, 13, 129. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Pastan, I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc. Natl. Acad. Sci. USA 1996, 93, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Song, D.-G.; Ye, Q.; Santoro, S.; Fang, C.; Best, A.; Powell, D.J. Chimeric NKG2D CAR-expressing T cell-mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Hum. Gene Ther. 2013, 24, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Patriarca, C.; Macchi, R.M.; Marschner, A.K.; Mellstedt, H. Epithelial cell adhesion molecule expression (CD326) in cancer: A short review. Cancer Treat. Rev. 2012, 38, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.L.; Sheard, V.E.; Kalaitsidou, M.; Blount, D.; Lad, Y.; Cheadle, E.J.; Edmondson, R.J.; Kooner, G.; Gilham, D.E.; Harrop, R. Preclinical assessment of Car T-cell therapy targeting the tumor antigen 5t4 in ovarian cancer. J. Immunother. 2018, 41, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Genta, S.; Ghisoni, E.; Giannone, G.; Mittica, G.; Valabrega, G. Reprogramming T-cells for adoptive immunotherapy of ovarian cancer. Expert Opin. Biol. Ther. 2018, 18, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X. Impact of IL-12 in Cancer. Curr. Cancer Drug Targets 2017, 17, 682–697. [Google Scholar] [CrossRef] [PubMed]
- Koneru, M.; O’Cearbhaill, R.; Pendharkar, S.; Spriggs, D.R.; Brentjens, R.J. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J. Transl. Med. 2015, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Tanyi, J.L.; Stashwick, C.; Plesa, G.; Morgan, M.A.; Porter, D.; Maus, M.V.; June, C.H. Possible compartmental cytokine release syndrome in a patient with recurrent ovarian cancer after Treatment with Mesothelin-targeted CAR-T Cells. J. Immunother. 2017, 40, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Ang, W.X.; Li, Z.; Chi, Z.; Du, S.-H.; Chen, C.; Tay, J.C.K.; Toh, H.C.; Connolly, J.E.; Xu, X.H.; Wang, S. Intraperitoneal immunotherapy with T cells stably and transiently expressing anti-EpCAM CAR in xenograft models of peritoneal carcinomatosis. Oncotarget 2017, 8, 13545–13559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewari, D.; Java, J.J.; Salani, R.; Armstrong, D.K.; Markman, M.; Herzog, T.; Monk, B.J.; Chan, J.K. Long-term survival advantage and prognostic factors associated with intraperitoneal chemotherapy treatment in advanced ovarian cancer: A gynecologic oncology group study. J. Clin. Oncol. 2015, 33, 1460–1466. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-J.; Song, D.-G.; Poussin, M.; Ye, Q.; Sharma, P.; Rodríguez-García, A.; Tang, Y.-M.; Powell, D.J. Multiparameter comparative analysis reveals differential impacts of various cytokines on CART cell phenotype and function ex vivo and in vivo. Oncotarget 2016, 7, 82354–82368. [Google Scholar] [CrossRef] [PubMed]
- Jindal, V.; Arora, E.; Gupta, S.; Lal, A.; Masab, M.; Potdar, R. Prospects of chimeric antigen receptor T cell therapy in ovarian cancer. Med. Oncol. 2018, 35, 70. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qiu, J.; Sun, Y. The basics of CAR T design and challenges in immunotherapy of solid tumors—Ovarian cancer as a model. Hum. Vaccines Immunother. 2017, 13, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): Combination or built-in CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.-K.; Peterson, E.; Sun, J.; Goudie, C.; Drapkin, R.I.; Liu, J.F.; Matulonis, U.; Zhu, Q.; Marasco, W.A. Anti-CCR4 monoclonal antibody enhances antitumor immunity by modulating tumor-infiltrating Tregs in an ovarian cancer xenograft humanized mouse model. Oncoimmunology 2015, 5, e1090075. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Idorn, M.; Olsen, M.; Halldórsdóttir, H.R.; Skadborg, S.K.; Pedersen, M.; Høgdall, C.; Høgdall, E.; Met, Ö.; Thor Straten, P. Improved migration of tumor ascites lymphocytes to ovarian cancer microenvironment by CXCR2 transduction. Oncoimmunology 2018, 7, e1412029. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.H.; Kolomeyevskaya, N.; Singel, K.L.; Grimm, M.J.; Moysich, K.B.; Daudi, S.; Grzankowski, K.S.; Lele, S.; Ylagan, L.; Webster, G.A.; et al. Targeting myeloid cells in the tumor microenvironment enhances vaccine efficacy in murine epithelial ovarian cancer. Oncotarget 2015, 6, 11310–11326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okła, K.; Wertel, I.; Polak, G.; Surówka, J.; Wawruszak, A.; Kotarski, J. Tumor-associated macrophages and myeloid-derived suppressor cells as immunosuppressive mechanism in ovarian cancer patients: Progress and challenges. Int. Rev. Immunol. 2016, 35, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Massagué, J. TGF-β inhibition and immunotherapy: Checkmate. Immunity 2018, 48, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Ros, X.R.; Vermeulen, L. Turning cold rumors hot by blocking TGF-β. Trends Cancer 2018, 4, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Lieber, S.; Reinartz, S.; Raifer, H.; Finkernagel, F.; Dreyer, T.; Bronger, H.; Jansen, J.M.; Wagner, U.; Worzfeld, T.; Müller, R.; et al. Prognosis of ovarian cancer is associated with effector memory CD8+ T cell accumulation in ascites, CXCL9 levels and activation-triggered signal transduction in T cells. Oncoimmunology 2018, 7, e1424672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronger, H.; Singer, J.; Windmüller, C.; Reuning, U.; Zech, D.; Delbridge, C.; Dorn, J.; Kiechle, M.; Schmalfeldt, B.; Schmitt, M.; et al. CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br. J. Cancer 2016, 115, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 2015, 6, 27359–27377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latha, K.; Yan, J.; Yang, Y.; Gressot, L.V.; Kong, L.-Y.; Manyam, G.; Ezhilarasan, R.; Wang, Q.; Sulman, E.P.; Eric Davis, R.; et al. The Role of Fibrinogen-Like Protein 2 on Immunosuppression and Malignant Progression in Glioma. J. Natl. Cancer Inst. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Kong, L.-Y.; Hu, J.; Gabrusiewicz, K.; Dibra, D.; Xia, X.; Heimberger, A.B.; Li, S. FGL2 as a multimodality regulator of tumor-mediated immune suppression and therapeutic target in gliomas. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, L.; Zha, H.; Yang, F.; Hu, C.; Chen, L.; Guo, B.; Zhu, B. Stroma-derived fibrinogen-like protein 2 activates cancer-associated fibroblasts to promote tumor growth in lung cancer. Int. J. Biol. Sci. 2017, 13, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3—Potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wei, S.; Hurt, E.M.; Green, M.D.; Zhao, L.; Vatan, L.; Szeliga, W.; Herbst, R.; Harms, P.W.; Fecher, L.A.; et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Investig. 2018, 128, 1708. [Google Scholar] [CrossRef] [PubMed]
- Davoodzadeh Gholami, M.; Kardar, G.A.; Saeedi, Y.; Heydari, S.; Garssen, J.; Falak, R. Exhaustion of T lymphocytes in the tumor microenvironment: Significance and effective mechanisms. Cell. Immunol. 2017, 322, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De novo epigenetic programs inhibit PD-1 blockade-Mediated T cell rejuvenation. Cell 2017, 170, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chiappinelli, K.B.; Guzzetta, A.A.; Easwaran, H.; Yen, R.-W.C.; Vatapalli, R.; Topper, M.J.; Luo, J.; Connolly, R.M.; Azad, N.S.; et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014, 5, 587–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; Vancriekinge, W.; Demeyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.L.; Chiappinelli, K.B.; Li, H.; Murphy, L.M.; Travers, M.E.; Topper, M.J.; Mathios, D.; Lim, M.; Shih, I.-M.; Wang, T.-L.; et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc. Natl. Acad. Sci. USA 2017, 114, E10981–E10990. [Google Scholar] [CrossRef] [PubMed]
- Bezu, L.; Gomes-da-Silva, L.C.; Dewitte, H.; Breckpot, K.; Fucikova, J.; Spisek, R.; Galluzzi, L.; Kepp, O.; Kroemer, G. Combinatorial strategies for the induction of immunogenic cell death. Front. Immunol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebremeskel, S.; Johnston, B. Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: Impact on clinical studies and considerations for combined therapies. Oncotarget 2015, 6, 41600–41619. [Google Scholar] [CrossRef] [PubMed]
- Paroli, M.; Bellati, F.; Videtta, M.; Focaccetti, C.; Mancone, C.; Donato, T.; Antonilli, M.; Perniola, G.; Accapezzato, D.; Napoletano, C.; et al. Discovery of chemotherapy-associated ovarian cancer antigens by interrogating memory T cells. Int. J. Cancer 2014, 134, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Hato, S.V.; Khong, A.; de Vries, I.J.M.; Lesterhuis, W.J. Molecular pathways: The immunogenic effects of platinum-based chemotherapeutics. Clin. Cancer Res. 2014, 20, 2831–2837. [Google Scholar] [CrossRef] [PubMed]
- Lesterhuis, W.J.; Punt, C.J.A.; Hato, S.V.; Eleveld-Trancikova, D.; Jansen, B.J.H.; Nierkens, S.; Schreibelt, G.; de Boer, A.; Van Herpen, C.M.L.; Kaanders, J.H.; et al. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J. Clin. Investig. 2011, 121, 3100–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, S.; Clayton, A.; Mason, M.D.; Jasani, B.; Adams, M.; Tabi, Z. Recovery of CD8+ T-cell function during systemic chemotherapy in advanced ovarian cancer. Cancer Res. 2005, 65, 7000–7006. [Google Scholar] [CrossRef] [PubMed]
- Szajnik, M.; Szczepanski, M.J.; Czystowska, M.; Elishaev, E.; Mandapathil, M.; Nowak-Markwitz, E.; Spaczynski, M.; Whiteside, T.L. TLR4 signaling induced by lipopolysaccharide or paclitaxel regulates tumor survival and chemoresistance in ovarian cancer. Oncogene 2009, 28, 4353–4363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.C.; Su, Q.B.; Wu, F.X.; Zhang, X.L.; Liu, P.S. Role of TLR4 for paclitaxel chemotherapy in human epithelial ovarian cancer cells. Eur. J. Clin. Investig. 2009, 39, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.-C.; Ma, Y.-B.; Wu, F.-X.; Ma, Z.-F.; Liu, N.-F.; Gao, R.; Gao, Y.-S.; Sheng, X.-G. TLR4 induces tumor growth and inhibits paclitaxel activity in MyD88-positive human ovarian carcinoma in vitro. Oncol. Lett. 2014, 7, 871–877. [Google Scholar] [CrossRef] [PubMed]
- D’Adhemar, C.J.; Spillane, C.D.; Gallagher, M.F.; Bates, M.; Costello, K.M.; Barry-O’Crowley, J.; Haley, K.; Kernan, N.; Murphy, C.; Smyth, P.C.; et al. The MyD88+ phenotype is an adverse prognostic factor in epithelial ovarian cancer. PLoS ONE 2014, 9, e100816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.-M.; Zhang, G.-N.; Shi, Y.; Zha, X.; Zhu, Y.; Wang, M.-M.; Lin, Q.; Wang, W.; Lu, H.-Y.; Ma, S.-Q.; et al. Atractylenolide-I sensitizes human ovarian cancer cells to paclitaxel by blocking activation of TLR4/MyD88-dependent pathway. Sci. Rep. 2014, 4, 3840. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, G.; Huang, J.; Ma, S.; Mi, K.; Cheng, J.; Zhu, Y.; Zha, X.; Huang, W. Atractylenolide I modulates ovarian cancer cell-mediated immunosuppression by blocking MD-2/TLR4 complex-mediated MyD88/NF-κB signaling in vitro. J. Transl. Med. 2016, 14, 104. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K.; et al. Chemotherapy induces Programmed Cell Death-Ligand 1 overexpression via the Nuclear Factor-κB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. 2015, 75, 5034–5045. [Google Scholar] [CrossRef] [PubMed]
- Mkrtichyan, M.; Najjar, Y.G.; Raulfs, E.C.; Abdalla, M.Y.; Samara, R.; Rotem-Yehudar, R.; Cook, L.; Khleif, S.N. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur. J. Immunol. 2011, 41, 2977–2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesterhuis, W.J.; Salmons, J.; Nowak, A.K.; Rozali, E.N.; Khong, A.; Dick, I.M.; Harken, J.A.; Robinson, B.W.; Lake, R.A. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS ONE 2013, 8, e61895. [Google Scholar] [CrossRef] [PubMed]
- Siew, Y.-Y.; Neo, S.-Y.; Yew, H.-C.; Lim, S.-W.; Ng, Y.-C.; Lew, S.-M.; Seetoh, W.-G.; Seow, S.-V.; Koh, H.-L. Oxaliplatin regulates expression of stress ligands in ovarian cancer cells and modulates their susceptibility to natural killer cell-mediated cytotoxicity. Int. Immunol. 2015, 27, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.S.; Sanii, S.; Kroeger, D.R.; Milne, K.; Talhouk, A.; Chiu, D.S.; Rahimi, K.; Shaw, P.A.; Clarke, B.A.; Nelson, B.H. Neoadjuvant chemotherapy of ovarian cancer results in three patterns of tumor-Infiltrating lymphocyte response with distinct implications for immunotherapy. Clin. Cancer Res. 2017, 23, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Mariya, T.; Hirohashi, Y.; Torigoe, T.; Asano, T.; Kuroda, T.; Yasuda, K.; Mizuuchi, M.; Sonoda, T.; Saito, T.; Sato, N. Prognostic impact of human leukocyte antigen class I expression and association of platinum resistance with immunologic profiles in epithelial ovarian cancer. Cancer Immunol. Res. 2014, 2, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.J.; Barton, S.; Thurgar, E.; Trevor, N. Topotecan, pegylated liposomal doxorubicin hydrochloride, paclitaxel, trabectedin and gemcitabine for advanced recurrent or refractory ovarian cancer: A systematic review and economic evaluation. Health Technol. Assess. 2015, 19, 1–480. [Google Scholar] [CrossRef] [PubMed]
- Salman, L.; Ben-Haroush, A.; Raban, O.; Yeoshoua, E.; Sabah, G.; Jakobson-Setton, A.; Tsoref, D.; Eitan, R. Neoadjuvant chemotherapy treatment modifications in ovarian carcinoma: The impact on surgical outcome and progression-free survival. Am. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kipps, E.; Tan, D.S.P.; Kaye, S.B. Meeting the challenge of ascites in ovarian cancer: New avenues for therapy and research. Nat. Rev. Cancer 2013, 13, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Gwak, H.; Kim, H.S.; Kim, B.; Dhanasekaran, D.N.; Song, Y.S. Malignant ascites enhances migratory and invasive properties of ovarian cancer cells with membrane bound IL-6R in vitro. Oncotarget 2016, 7, 83148–83159. [Google Scholar] [CrossRef] [PubMed]
- Yigit, R.; Figdor, C.G.; Zusterzeel, P.L.M.; Pots, J.M.; Torensma, R.; Massuger, L.F.A.G. Cytokine analysis as a tool to understand tumour-host interaction in ovarian cancer. Eur. J. Cancer 2011, 47, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, H.; Yin, X.; Long, L.; Chen, X.; Feng, F.; Liu, Y.; Zhao, P.; Xu, Y.; Li, M.; et al. IL-6R/STAT3/miR-204 feedback loop contributes to cisplatin resistance of epithelial ovarian cancer cells. Oncotarget 2017, 8, 39154–39166. [Google Scholar] [CrossRef] [PubMed]
- Nishio, H.; Yaguchi, T.; Sugiyama, J.; Sumimoto, H.; Umezawa, K.; Iwata, T.; Susumu, N.; Fujii, T.; Kawamura, N.; Kobayashi, A.; et al. Immunosuppression through constitutively activated NF-κB signalling in human ovarian cancer and its reversal by an NF-κB inhibitor. Br. J. Cancer 2014, 110, 2965–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidthipong, K.; Ma, J.; Yu, W.L.; Wang, Y.F.; Kobayashi, S.; Kishino, S.; Koide, N.; Yokochi, T.; Kato, K.; Okada, S.; et al. Rational design, synthesis and in vitro evaluation of novel exo-methylene butyrolactone salicyloylamide as NF-κB inhibitor. Bioorg. Med. Chem. Lett. 2017, 27, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.-W. Oncolytic virotherapy: A contest between apples and oranges. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.-W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seymour, L.W.; Fisher, K.D. Oncolytic viruses: Finally delivering. Br. J. Cancer 2016, 114, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Lemay, C.G.; Rintoul, J.L.; Kus, A.; Paterson, J.M.; Garcia, V.; Falls, T.J.; Ferreira, L.; Bridle, B.W.; Conrad, D.P.; Tang, V.A.; et al. Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Forbes, N.E.; Abdelbary, H.; Lupien, M.; Bell, J.C.; Diallo, J.-S. Exploiting tumor epigenetics to improve oncolytic virotherapy. Front. Genet. 2013, 4, 184. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Hoeller, C.; Gruter, I.P.; Michielin, O. Combining talimogene laherparepvec with immunotherapies in melanoma and other solid tumors. Cancer Immunol. Immunother. 2017, 66, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Rehman, H.; Silk, A.W.; Kane, M.P.; Kaufman, H.L. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J. Immunother. Cancer 2016, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.D.; Meza-Perez, S.; Bevis, K.S.; Randall, T.D.; Gillespie, G.Y.; Langford, C.; Alvarez, R.D. IL-12 Expressing oncolytic herpes simplex virus promotes anti-tumor activity and immunologic control of metastatic ovarian cancer in mice. J. Ovarian Res. 2016, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Ilkow, C.S.; Marguerie, M.; Batenchuk, C.; Mayer, J.; Neriah, D.B.; Cousineau, S.; Falls, T.; Jennings, V.A.; Boileau, M.; Bellamy, D.; et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat. Med. 2015, 21, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Arulanandam, R.; Batenchuk, C.; Angarita, F.A.; Ottolino-Perry, K.; Cousineau, S.; Mottashed, A.; Burgess, E.; Falls, T.J.; De Silva, N.; Tsang, J.; et al. VEGF-mediated induction of PRD1-BF1/Blimp1 expression sensitizes tumor vasculature to oncolytic virus infection. Cancer Cell 2015, 28, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Arulanandam, R.; De Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.-H.; et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013, 73, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Achard, C.; Surendran, A.; Wedge, M.-E.; Ungerechts, G.; Bell, J.; Ilkow, C.S. Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy. EBioMedicine 2018, 31, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.; Komorowski, M.P.; Seshadri, M.; Rokita, H.; McGray, A.J.R.; Opyrchal, M.; Odunsi, K.O.; Kozbor, D. CXCL12/CXCR4 blockade by oncolytic virotherapy inhibits ovarian cancer growth by decreasing immunosuppression and targeting cancer initiating cells. J. Immunol. 2014, 193, 5327–5337. [Google Scholar] [CrossRef] [PubMed]
- Hartkopf, A.D.; Fehm, T.; Wallwiener, M.; Lauer, U. Oncolytic viruses to treat ovarian cancer patients—A review of results from clinical trials. Geburtshilfe Frauenheilkd. 2012, 72, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Jennings, V.A.; Ilett, E.J.; Scott, K.J.; West, E.J.; Vile, R.; Pandha, H.; Harrington, K.; Young, A.; Hall, G.D.; Coffey, M.; et al. Lymphokine-activated killer and dendritic cell carriage enhances oncolytic reovirus therapy for ovarian cancer by overcoming antibody neutralization in ascites. Int. J. Cancer 2014, 134, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, I.; Bauzon, M.; Green, N.; Seymour, L.; Fisher, K.; Hermiston, T. OvAd1, a novel, Potent, and selective chimeric oncolytic virus developed for ovarian cancer by 3D-directed evolution. Mol. Ther. Oncolytics 2017, 4, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Orzechowska, B.U.; Jędryka, M.; Zwolińska, K.; Matkowski, R. VSV based virotherapy in ovarian cancer: The past, the present and …future? J. Cancer 2017, 8, 2369–2383. [Google Scholar] [CrossRef] [PubMed]
- Uusi-Kerttula, H.; Davies, J.A.; Thompson, J.; Wongthida, P.; Evgin, L.; Shim, K.G.; Bradshaw, A.; Baker, A.T.; Rizkallah, P.J.; Jones, R.; et al. Ad5NULL-A20—A tropism-modified, αvβ6 integrin-selective oncolytic adenovirus for epithelial ovarian cancer therapies. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Power, A.T.; Wang, J.; Falls, T.J.; Paterson, J.M.; Parato, K.A.; Lichty, B.D.; Stojdl, D.F.; Forsyth, P.A.J.; Atkins, H.; Bell, J.C. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Alkayyal, A.A.; Tai, L.-H.; Kennedy, M.A.; de Souza, C.T.; Zhang, J.; Lefebvre, C.; Sahi, S.; Ananth, A.A.; Mahmoud, A.B.; Makrigiannis, A.P.; et al. NK-Cell recruitment is necessary for eradication of peritoneal carcinomatosis with an IL12-expressing Maraba virus cellular vaccine. Cancer Immunol. Res. 2017, 5, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Pastor, F.; Rodriguez, A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Melero, I. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin. Oncol. 2015, 42, 640–655. [Google Scholar] [CrossRef] [PubMed]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10, eaao5931. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. IDO inhibitors move center stage in immuno-oncology. Nat. Biotechnol. 2015, 33, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Kapoor, V.; Jassar, A.S.; Kaiser, L.R.; Albelda, S.M. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rébé, C.; Ghiringhelli, F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Germano, G.; Belgiovine, C.; D’Incalci, M.; Mantovani, A. Trabectedin: A drug from the sea that strikes tumor-associated macrophages. Oncoimmunology 2013, 2, e24614. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhao, L.; Hellstrom, I.; Hellstrom, K.E.; Guo, Y. Dual targeting of CD137 co-stimulatory and PD-1 co-inhibitory molecules for ovarian cancer immunotherapy. Oncoimmunology 2014, 3, e28248. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Cheng, D.; Xia, Z.; Luan, M.; Wu, L.; Wang, G.; Zhang, S. Combined TIM-3 blockade and CD137 activation affords the long-term protection in a murine model of ovarian cancer. J. Transl. Med. 2013, 11, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Zhao, L.; Li, W.; Fan, K.; Qian, W.; Hou, S.; Wang, H.; Dai, M.; Hellstrom, I.; Hellstrom, K.E.; et al. Combinatorial PD-1 Blockade and CD137 activation has therapeutic efficacy in murine cancer models and synergizes with cisplatin. PLoS ONE 2013, 8, e84927. [Google Scholar] [CrossRef] [PubMed]
- Hawkridge, A.M. The chicken model of spontaneous ovarian cancer. Proteom. Clin. Appl. 2014, 8, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Edassery, S.L.; Barua, A.; Abramowicz, J.S.; Bahr, J.M.; Hellstrom, I.; Luborsky, J.L. The hen model of human ovarian cancer develops anti-mesothelin autoantibodies in response to mesothelin expressing tumors. J. Ovarian Res. 2011, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradaric, M.J.; Penumatsa, K.; Barua, A.; Edassery, S.L.; Yu, Y.; Abramowicz, J.S.; Bahr, J.M.; Luborsky, J.L. Immune cells in the normal ovary and spontaneous ovarian tumors in the laying hen (Gallus domesticus) model of human ovarian cancer. PLoS ONE 2013, 8, e74147. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.F.; Bahr, J.M.; Yellapa, A.; Bitterman, P.; Abramowicz, J.S.; Edassery, S.L.; Basu, S.; Rotmensch, J.; Barua, A. Expression of leukocyte inhibitory immunoglobulinlike transcript 3 receptors by ovarian tumors in laying hen model of spontaneous ovarian cancer. Transl. Oncol. 2012, 5, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Barua, A.; Yellapa, A.; Bahr, J.M.; Abramowicz, J.S.; Edassery, S.L.; Basu, S.; Rotmensch, J.; Bitterman, P. Expression of death receptor 6 by ovarian rumors in laying hens, a preclinical model of spontaneous ovarian cancer. Transl. Oncol. 2012, 5, 260–268. [Google Scholar] [CrossRef] [PubMed]
- McNeal, S.; Bitterman, P.; Bahr, J.M.; Edassery, S.L.; Abramowicz, J.S.; Basu, S.; Barua, A. Association of Immunosuppression with DR6 Expression during the Development and Progression of Spontaneous Ovarian Cancer in Laying Hen Model. Available online: https://www.hindawi.com/journals/jir/2016/6729379/ (accessed on 29 May 2018).
- Corner, S.; Parys, M.; Agnew, D.; Moresco, A.; Yuzbasiyan-Gurkan, V. Ovarian Adenocarcinoma in Captive North American Jaguars (Panthera Onca): Tumor Characterization and Investigation of Brca1 Mutations. In Proceedings of the 47th Annual Conference of the American Association of Zoo Veterinarians, Portland, OR, USA, 26 September–2 October 2016. [Google Scholar]
- McLean, K.; Mehta, G. Tumor microenvironment and models of ovarian cancer: The 11th Biennial Rivkin Center Ovarian Cancer Research Symposium. Int. J. Gynecol. Cancer 2017, 27, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, C.W.; Goldberg, R.L.; Carter, L.E.; Gamwell, L.F.; Al-Hujaily, E.M.; Collins, O.; Macdonald, E.A.; Garson, K.; Daneshmand, M.; Carmona, E.; et al. A new spontaneously transformed syngeneic model of high-grade serous ovarian cancer with a tumor-initiating cell population. Front. Oncol. 2014, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Roby, K.F.; Taylor, C.C.; Sweetwood, J.P.; Cheng, Y.; Pace, J.L.; Tawfik, O.; Persons, D.L.; Smith, P.G.; Terranova, P.F. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 2000, 21, 585–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leinster, D.A.; Kulbe, H.; Everitt, G.; Thompson, R.; Perretti, M.; Gavins, F.N.E.; Cooper, D.; Gould, D.; Ennis, D.P.; Lockley, M.; et al. The peritoneal tumour microenvironment of high-grade serous ovarian cancer. J. Pathol. 2012, 227, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.-L.; Kandalaft, L.E.; Tanyi, J.; Hagemann, A.R.; Motz, G.T.; Svoronos, N.; Montone, K.; Mantia-Smaldone, G.M.; Smith, L.; Nisenbaum, H.L.; et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: From bench to bedside. Clin. Cancer Res. 2013, 19, 4801–4815. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Wang, X.; Cheng, D.; Xia, Z.; Luan, M.; Zhang, S. PD-1 Blockade and OX40 Triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PLoS ONE 2014, 9, e89350. [Google Scholar] [CrossRef] [PubMed]
- Nounamo, B.; Liem, J.; Cannon, M.; Liu, J. Myxoma Virus Optimizes Cisplatin for the treatment of ovarian cancer in vitro and in a syngeneic murine dissemination model. Mol. Ther. Oncolytics 2017, 6, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.B.; Meza-Perez, S.; Londoño, A.; Katre, A.; Peabody, J.E.; Smith, H.J.; Forero, A.; Norian, L.A.; Straughn, J.M.; Buchsbaum, D.J.; et al. Epigenetic modifiers upregulate MHC II and impede ovarian cancer tumor growth. Oncotarget 2017, 8, 44159–44170. [Google Scholar] [CrossRef] [PubMed]
- Benencia, F.; Courrèges, M.C.; Fraser, N.W.; Coukos, G. Herpes virus oncolytic therapy reverses tumor immune dysfunction and facilitates tumor antigen presentation. Cancer Biol. Ther. 2008, 7, 1194–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Ravindranathan, R.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017, 8, 14754. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.D.; Brown, S.D.; Wick, D.A.; Nielsen, J.S.; Kroeger, D.R.; Twumasi-Boateng, K.; Holt, R.A.; Nelson, B.H. Low mutation burden in ovarian cancer may limit the utility of neoantigen-targeted vaccines. PLoS ONE 2016, 11, e0155189. [Google Scholar] [CrossRef] [PubMed]
- Conejo-Garcia, J.R.; Benencia, F.; Courreges, M.-C.; Kang, E.; Mohamed-Hadley, A.; Buckanovich, R.J.; Holtz, D.O.; Jenkins, A.; Na, H.; Zhang, L.; et al. Tumor-infiltrating dendritic cell precursors recruited by a β-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat. Med. 2004, 10, 950. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.R.; Barnden, M.; Kurts, C.; Carbone, F.R.; Miller, J.F.; Heath, W.R. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol. Cell Biol. 2000, 78, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.-A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, J.; Blagih, J.; Ennis, D.; Leung, E.; Dowson, S.; Farquharson, M.; Tookman, L.A.; Orange, C.; Athineos, D.; Mason, S.; et al. CRISPR/Cas9-mediated Trp53 and Brca2 knockout to generate improved murine models of ovarian high-grade serous carcinoma. Cancer Res. 2016, 76, 6118–6129. [Google Scholar] [CrossRef] [PubMed]
- Walton, J.B.; Farquharson, M.; Mason, S.; Port, J.; Kruspig, B.; Dowson, S.; Stevenson, D.; Murphy, D.; Matzuk, M.; Kim, J.; et al. CRISPR/Cas9-derived models of ovarian high grade serous carcinoma targeting Brca1, Pten and Nf1, and correlation with platinum sensitivity. Sci. Rep. 2017, 7, 16827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroeger, D.R.; Milne, K.; Nelson, B.H. Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic T-Cell responses, and superior prognosis in ovarian cancer. Clin. Cancer Res. 2016, 22, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, P.C.; Mottillo, E.P.; Baxa, A.C.; Heng, H.H.Q.; Doyon-Reale, N.; Gregoire, L.; Lancaster, W.D.; Rabah, R.; Schmelz, E.M. Sequential molecular and cellular events during neoplastic progression: A mouse syngeneic ovarian cancer model. Neoplasia 2005, 7, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Endsley, M.P.; Moyle-Heyrman, G.; Karthikeyan, S.; Lantvit, D.D.; Davis, D.A.; Wei, J.-J.; Burdette, J.E. Spontaneous transformation of murine oviductal epithelial cells: A model system to investigate the onset of fallopian-derived tumors. Front. Oncol. 2015, 5, 154. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Czarnecki, A.A.; Dean, M.; Modi, D.A.; Lantvit, D.D.; Hardy, L.; Baligod, S.; Davis, D.A.; Wei, J.-J.; Burdette, J.E. PTEN loss in the fallopian tube induces hyperplasia and ovarian tumor formation. Oncogene 2018, 37, 1976–1990. [Google Scholar] [CrossRef] [PubMed]
- Garson, K.; Gamwell, L.F.; Pitre, E.M.; Vanderhyden, B.C. Technical challenges and limitations of current mouse models of ovarian cancer. J. Ovarian Res. 2012, 5, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, P.J.; Weeraratna, A.T. Genetically-defined ovarian cancer mouse models. J. Pathol. 2016, 238, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Connolly, D.C.; Bao, R.; Nikitin, A.Y.; Stephens, K.C.; Poole, T.W.; Hua, X.; Harris, S.S.; Vanderhyden, B.C.; Hamilton, T.C. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res. 2003, 63, 1389–1397. [Google Scholar] [PubMed]
- Laviolette, L.A.; Garson, K.; Macdonald, E.A.; Senterman, M.K.; Courville, K.; Crane, C.A.; Vanderhyden, B.C. 17β-Estradiol accelerates tumor onset and decreases survival in a transgenic mouse model of ovarian cancer. Endocrinology 2010, 151, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Baust, C.A.; Kuhn, E.; Valle, B.L.; Shih, I.-M.; Kurman, R.J.; Wang, T.-L.; Amano, T.; Ko, M.S.H.; Miyoshi, I.; Araki, Y.; et al. A genetically engineered ovarian cancer mouse model based on fallopian tube transformation mimics human high-grade serous carcinoma development. J. Pathol. 2014, 233, 228–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schietinger, A.; Philip, M.; Krisnawan, V.E.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hämmerling, G.J.; et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, B.; Goode, E.L.; Kalli, K.R.; Knutson, K.L.; DeRycke, M.S. The immune system in the pathogenesis of ovarian cancer. Crit. Rev. Immunol. 2013, 33, 137–164. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flesken-Nikitin, A.; Choi, K.-C.; Eng, J.P.; Shmidt, E.N.; Nikitin, A.Y. Induction of Carcinogenesis by Concurrent Inactivation of p53 and Rb1 in the Mouse Ovarian Surface Epithelium. Cancer Res. 2003, 63, 3459–3463. [Google Scholar] [PubMed]
- Flesken-Nikitin, A.; Hwang, C.-I.; Cheng, C.-Y.; Michurina, T.V.; Enikolopov, G.; Nikitin, A.Y. Ovarian surface epithelium at the junction area contains cancer-prone stem cell niche. Nature 2013, 495, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Perets, R.; Wyant, G.A.; Muto, K.W.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.H.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten Models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Szabova, L.; Yin, C.; Bupp, S.; Guerin, T.M.; Schlomer, J.J.; Householder, D.B.; Baran, M.L.; Yi, M.; Song, Y.; Sun, W.; et al. Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Res. 2012, 72, 4141–4153. [Google Scholar] [CrossRef] [PubMed]
- Wieser, V.; Gaugg, I.; Fleischer, M.; Shivalingaiah, G.; Wenzel, S.; Sprung, S.; Lax, S.F.; Zeimet, A.G.; Fiegl, H.; Marth, C. BRCA1/2 and TP53 mutation status associates with PD-1 and PD-L1 expression in ovarian cancer. Oncotarget 2018, 9, 17501–17511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, D.-S.; Kabir, S.M.; Dong, Y.-L.; Lee, E.; Adunyah, S.E. Inhibitory effect of tumor suppressor p53 on proinflammatory chemokine expression in ovarian cancer cells by Reducing Proteasomal Degradation of IκB. PLoS ONE 2012, 7, e51116. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Wu, R.; Kuick, R.; Sessine, M.S.; Schulman, S.; Green, M.; Fearon, E.R.; Cho, K.R. High-grade serous carcinomas arise in the mouse oviduct via defects linked to the human disease. J. Pathol. 2017, 243, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Budiu, R.A.; Elishaev, E.; Brozick, J.; Lee, M.; Edwards, R.P.; Kalinski, P.; Vlad, A.M. Immunobiology of human mucin 1 in a preclinical ovarian tumor model. Oncogene 2013, 32, 3664. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, L.; Chen, H.; Li, L.; Ma, Y.; Ni, J.; Li, Y. The role of tumour-associated MUC1 in epithelial ovarian cancer metastasis and progression. Cancer Metastasis Rev. 2013, 32, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Snijdewint, F.G.; von Mensdorff-Pouilly, S.; Karuntu-Wanamarta, A.H.; Verstraeten, A.A.; van Zanten-Przybysz, I.; Hummel, P.; Nijman, H.W.; Kenemans, P.; Hilgers, J. Cellular and humoral immune responses to MUC1 mucin and tandem-repeat peptides in ovarian cancer patients and controls. Cancer Immunol. Immunother. 1999, 48, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Vlad, A.M.; Diaconu, I.; Gantt, K.R. MUC1 in endometriosis and ovarian cancer. Immunol. Res. 2006, 36, 229–236. [Google Scholar] [CrossRef]
- Barua, A.; Bitterman, P.; Abramowicz, J.S.; Dirks, A.L.; Bahr, J.M.; Hales, D.B.; Bradaric, M.J.; Edassery, S.L.; Rotmensch, J.; Luborsky, J.L. Histopathology of ovarian tumors in laying hens, a preclinical model of human ovarian cancer. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2009, 19, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Engle, X.; Scarlett, U.K.; Martinez, D.; Barber, A.; Elgueta, R.; Wang, L.; Nesbeth, Y.; Durant, Y.; Gewirtz, A.T.; et al. Polyethylenimine-based siRNA nanocomplexes reprogram tumor-associated dendritic cells via TLR5 to elicit therapeutic antitumor immunity. J. Clin. Investig. 2009, 119, 2231–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubillos-Ruiz, J.R.; Baird, J.R.; Tesone, A.J.; Rutkowski, M.R.; Scarlett, U.K.; Camposeco-Jacobs, A.L.; Anadon-Arnillas, J.; Harwood, N.M.; Korc, M.; Fiering, S.N.; et al. Reprogramming tumor-associated dendritic cells in vivo using microrna mimetics triggers protective immunity against ovarian cancer. Cancer Res. 2012, 72, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Baert, T.; Verschuere, T.; Van Hoylandt, A.; Gijsbers, R.; Vergote, I.; Coosemans, A. The dark side of ID8-Luc2: Pitfalls for luciferase tagged murine models for ovarian cancer. J. Immunother. Cancer 2015, 3, 57. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Barham, W.; Saskowski, J.; Tikhomirov, O.; Chen, L.; Lee, H.-J.; Yull, F.; Khabele, D. Tracking NF-κB activity in tumor cells during ovarian cancer progression in a syngeneic mouse model. J. Ovarian Res. 2013, 6, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, I.; Takahashi, K.; Kon, Y.; Okamura, T.; Mototani, Y.; Araki, Y.; Kasai, N. Mouse transgenic for murine oviduct-specific glycoprotein promoter-driven simian virus 40 large T-antigen: Tumor formation and its hormonal regulation. Mol. Reprod. Dev. 2002, 63, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Tirodkar, T.S.; Budiu, R.A.; Elishaev, E.; Zhang, L.; Mony, J.T.; Brozick, J.; Edwards, R.P.; Vlad, A.M. MUC1 positive, Kras and Pten driven mouse gynecologic tumors replicate human tumors and vary in survival and nuclear grade based on anatomical location. PLoS ONE 2014, 9, e102409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Fleury, H.; Communal, L.; Carmona, E.; Portelance, L.; Arcand, S.L.; Rahimi, K.; Tonin, P.N.; Provencher, D.; Mes-Masson, A.-M. Novel high-grade serous epithelial ovarian cancer cell lines that reflect the molecular diversity of both the sporadic and hereditary disease. Genes Cancer 2015, 6, 378–398. [Google Scholar] [PubMed]
- Ince, T.A.; Sousa, A.D.; Jones, M.A.; Harrell, J.C.; Agoston, E.S.; Krohn, M.; Selfors, L.M.; Liu, W.; Chen, K.; Yong, M.; et al. Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nat. Commun. 2015, 6, 7419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloudová, K.; Hromádková, H.; Partlová, S.; Brtnický, T.; Rob, L.; Bartůňková, J.; Hensler, M.; Halaška, M.J.; Špíšek, R.; Fialová, A. Expression of tumor antigens on primary ovarian cancer cells compared to established ovarian cancer cell lines. Oncotarget 2016, 7, 46120–46126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nham, T.; Poznanski, S.M.; Fan, I.Y.; Shenouda, M.M.; Chew, M.V.; Lee, A.J.; Vahedi, F.; Karimi, Y.; Butcher, M.; Lee, D.A.; et al. Ex vivo-expanded NK cells from blood and ascites of ovarian cancer patients are cytotoxic against autologous primary ovarian cancer cells. Cancer Immunol. Immunother. 2018, 67, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.D.; Hagel, J.; Scott, E.M.; Psallidas, I.; Gupta, A.; Spiers, L.; Miller, P.; Kanellakis, N.; Ashfield, R.; Fisher, K.D.; et al. Oncolytic adenovirus expressing bispecific antibody targets T-cell cytotoxicity in cancer biopsies. EMBO Mol. Med. 2017, 9, 1067–1087. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Sánchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell 2017, 170, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. Evolutionary determinants of cancer. Cancer Discov. 2015, 5, 806–820. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Cancer evolution constrained by the immune microenvironment. Cell 2017, 170, 825–827. [Google Scholar] [CrossRef] [PubMed]


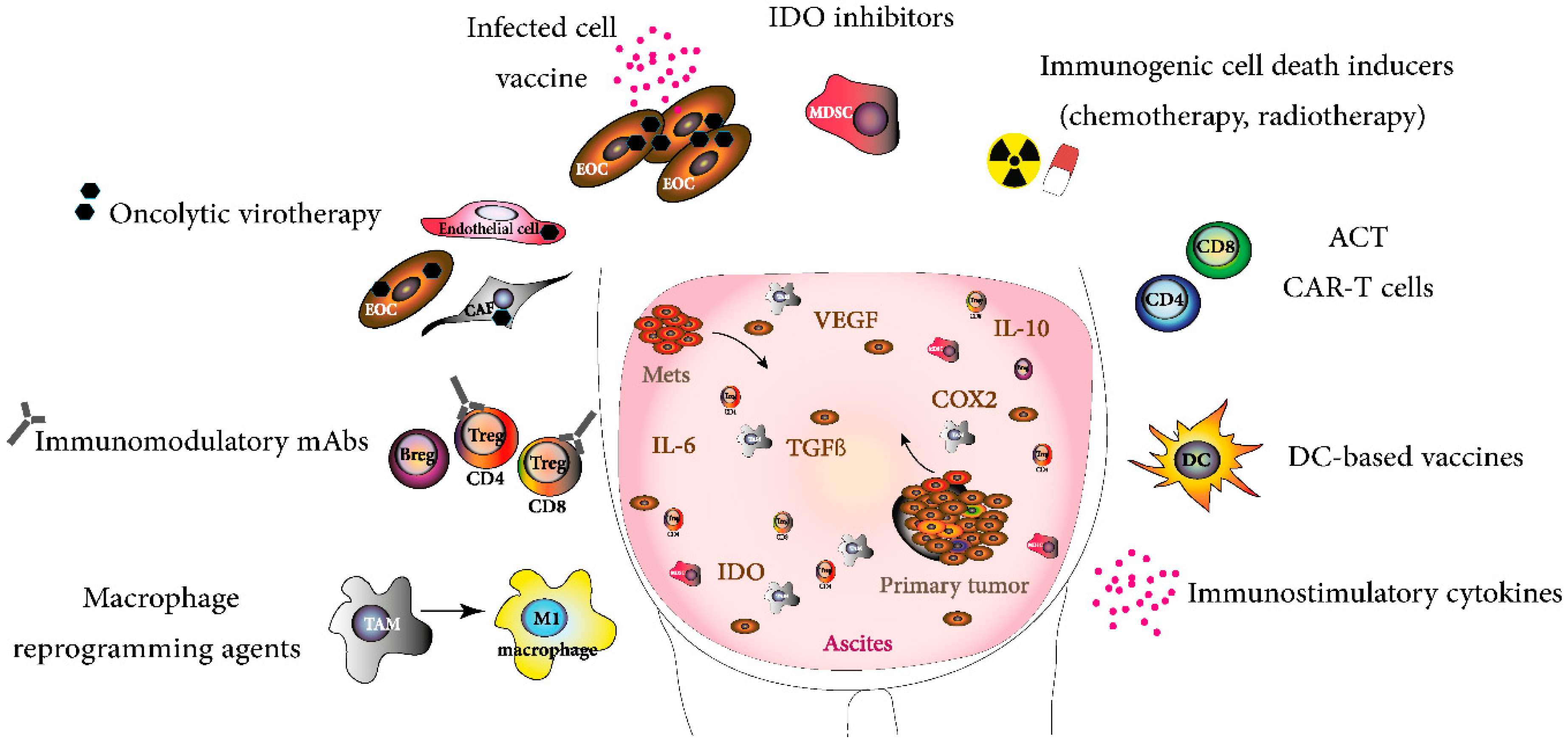{kind=link}
| Immune Cell Type | Antitumoral Function | Tumor-Promoting Function |
|---|---|---|
| CD4+ Th1 cells | Help to CTLs in tumor rejection and production of TNFα, IFNγ, and IL-2 | Production of cytokines |
| CD4+ Th2 cells | Education of macrophages, production of cytokines, B cell activation | |
| CD4+ Treg Cells | Suppression of inflammation (cytokines and other suppressive mechanisms) | Immunosuppression: causes IL-2 and other cytokine deprivation, production of TGFβ, IL-10, impaired activation of CTLs |
| CD8+ T Cells | Direct lysis of cancer cells and production of pro-inflammatory cytokines TNFα, IFNγ, and IL-2 | FOXP3+ CTLA-4+ CD25+, convert effector CD8+ T cells into suppressor cells, suppressive function through TGF-β1 |
| B Cells | Production of tumor specific antibodies, IFNγ, TAA presentation, Th1 polarization, promotes T cell expansion | Production of IL-6, IL-10, IL-35, TGFβ, CCL22, immunosuppression, T cell conversion to Tregs, promotes Th2 inhibitory responses |
| Macrophages, DCs | TAA sampling and presentation; T-cell priming; and production of IL-12 and type I IFN, lympho-attracting chemokines CXCL9, CXCL10, CXCL11 | Promotes metastasis and invasion. Produces CSF-1, arginase, IL-6, IL-10, and CCL22. B7-H4+ TAMs suppress antitumoral responses. |
| MDSCs | Immunosuppression, induces Tregs differentiation, M2 TAM, cancer stemness, sphere formation, and metastasis. Defective TAA presentation. Production of arginase-1, nitric oxide, reactive oxygen and nitrogen species, prostaglandin E2, CXCL12. Deplete cysteine, induce Tregs, inhibit T-cell activation and proliferation, and attenuate the cytolytic ability of NK cells. | |
| NK Cells | Direct cytotoxicity toward cancer cells and production of pro-inflammatory cytokines GM-CSF, TNFα, IFNγ, IL-2 and chemokine CCL5 |
| Model | Genetic Engineering | Key Features of Tumor Immune Landscape | Mutation/Neoantigen Burden | Advantages | Limitations | References |
|---|---|---|---|---|---|---|
| Laying Hen | None |
| Unknown |
|
| [131,132,133,134,178] |
| Jaguar | None |
| Familial BRCA mutations |
|
| [136,137] |
| ID8-(original) | None |
| Low |
|
| [139,147] |
| ID8-Defb29/Vegf-A | Stable Defb29 and Vegf-A expression |
| Unknown |
|
| [148,149,179,180] |
| ID8-OVA | Stable ovalbumin expression |
| OVA |
|
| [141] |
| ID8-Trp53−/− Brca2−/− | Trp53 and Brca2 deletion |
| Unknown |
|
| [152,153] |
| ID8-NGL | NF-kappaB-dependent GFP/luciferase expression |
| Unknown |
|
| [181,182] |
| STOSE | None |
| Unknown |
|
| [138] |
| Model | Genetic Engineering | Key Features of Tumor Immune Landscape | Mutation/Neoantigen Burden | Advantages | Disadvantages | References |
|---|---|---|---|---|---|---|
| TgMISIIRTAg | SV40TAg driven from reproductive tract-specific MISIIR (Amhr2) promoter during development | Epigenetic modifiers enhance MHCII expression on cancer cells | Unknown |
|
| [144,161] |
| TgCAG-LS-TAg | SV40TAg with lox-stop cassette driven from ubiquitous CAG promoter * | Unknown | Unknown |
|
| [162] |
| mogp-TAg | SV40TAg driven from oviduct-specific Ovgp1 promoter | Unknown | Unknown |
|
| [163,183] |
| TgK18-GT121-Brca-Trp53 | Inducible SV40TAg and either Trp53−/− or Trp53mut and Brca1 or 2 deletions driven from epithelial specific cytokeratin 18 expression * | Unknown | Unknown |
|
| [170] |
| Trp53loxP/loxP-Rb1loxP/loxP | Inducible deletion of Trp53 and Rb1 * | Unknown | Unknown |
|
| [167,168] |
| Pax8-Cre-Brca1(2) −/−; Trp53mut(−/−);Pten −/− * | Doxycyline inducible Cre-mediated deletion of Brca, Pten, and Trp53, driven from oviduct-specific Pax8 promoter. | Unknown | Copy number alterations similar to HGSC, Neoantigen and mutation burden unknown |
|
| [169] |
| Ovgp1-iCre-ERT2 + tumor suppressor genes | Conditional deletion of Brca1, Pten, Rb1, and Nf1 (BPRN mice) or Brca1, Pten, and p53 (BPP mice), driven from the oviduct-specific Ovgp1 promoter | Unknown | Unknown |
|
| [173] |
| MUC1KrasPTEN | Constitutive expression of human MUC1 and inducible oncogenic KRASG12D and Pten deletion. * | Robust Tregs among TILs and dysfunctional DCs | unknown |
|
| [174,184] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCloskey, C.W.; Rodriguez, G.M.; Galpin, K.J.C.; Vanderhyden, B.C. Ovarian Cancer Immunotherapy: Preclinical Models and Emerging Therapeutics. Cancers 2018, 10, 244. https://doi.org/10.3390/cancers10080244
McCloskey CW, Rodriguez GM, Galpin KJC, Vanderhyden BC. Ovarian Cancer Immunotherapy: Preclinical Models and Emerging Therapeutics. Cancers. 2018; 10(8):244. https://doi.org/10.3390/cancers10080244
Chicago/Turabian StyleMcCloskey, Curtis W., Galaxia M. Rodriguez, Kristianne J. C. Galpin, and Barbara C. Vanderhyden. 2018. "Ovarian Cancer Immunotherapy: Preclinical Models and Emerging Therapeutics" Cancers 10, no. 8: 244. https://doi.org/10.3390/cancers10080244
APA StyleMcCloskey, C. W., Rodriguez, G. M., Galpin, K. J. C., & Vanderhyden, B. C. (2018). Ovarian Cancer Immunotherapy: Preclinical Models and Emerging Therapeutics. Cancers, 10(8), 244. https://doi.org/10.3390/cancers10080244