The Role of Signal Transducer and Activator of Transcription 3 (STAT3) and Its Targeted Inhibition in Hematological Malignancies


 ,
, 
Abstract
:1. Introduction
2. Domain Structure and Signaling Cascade of STAT3
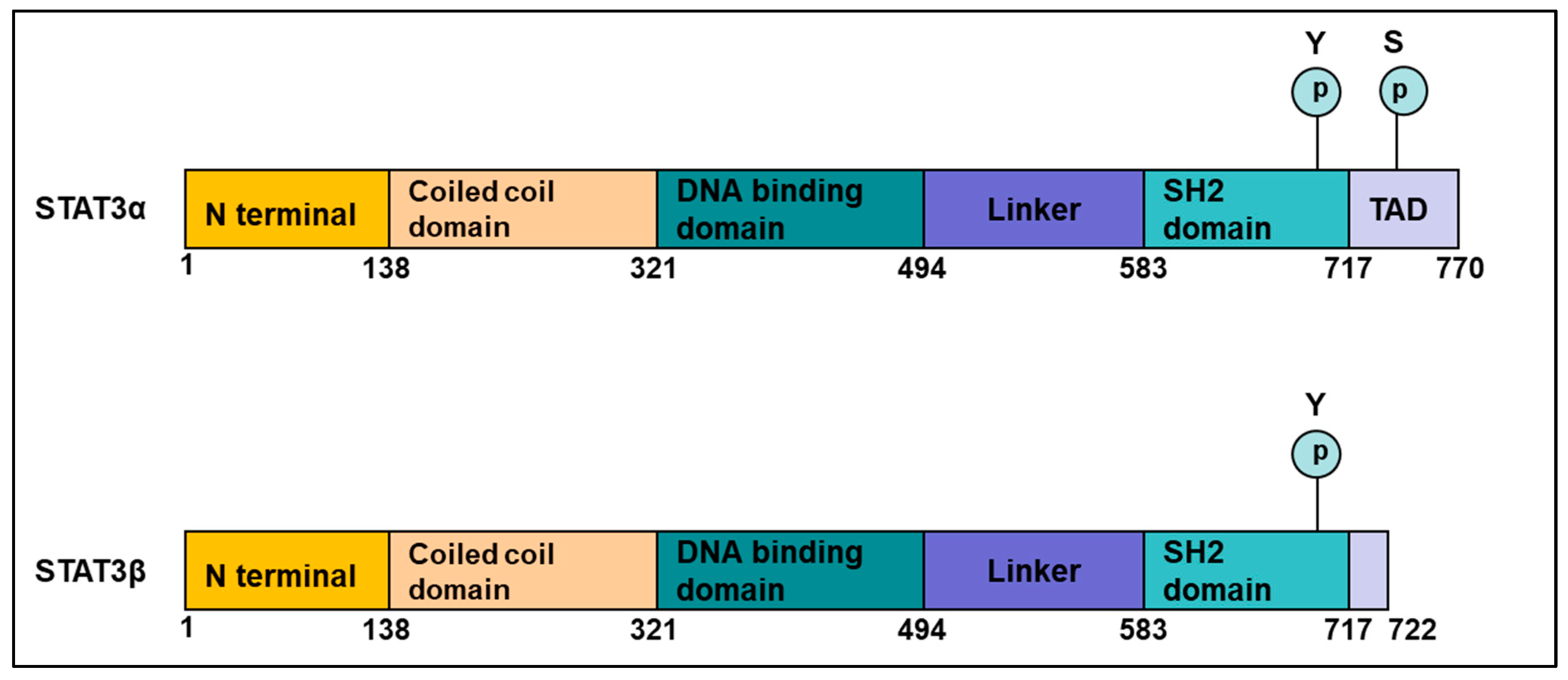
2.1. Structure of STAT3
- The N-terminal domain—composed of an oligomerization and a coiled-coil domain.
- The DNA-binding domain—that can recognize a specific molecular motif in the DNA.
- A linker domain.
- The Src homology 2 (SH2) domain—important for the formation of dimers; phospho-tyrosine 705 is important for this dimerization.
- The C-terminal transactivation domain—This domain differs between the α and β isoforms, with the β form having a unique truncated C-terminal sequence. STAT3 protein also has a Ser 727 phosphorylation site, which though less studied than Tyr 705, is important for regulating STAT3 activation by serine/threonine kinases such as the MAPKs (mitogen-activated protein kinases) [15].
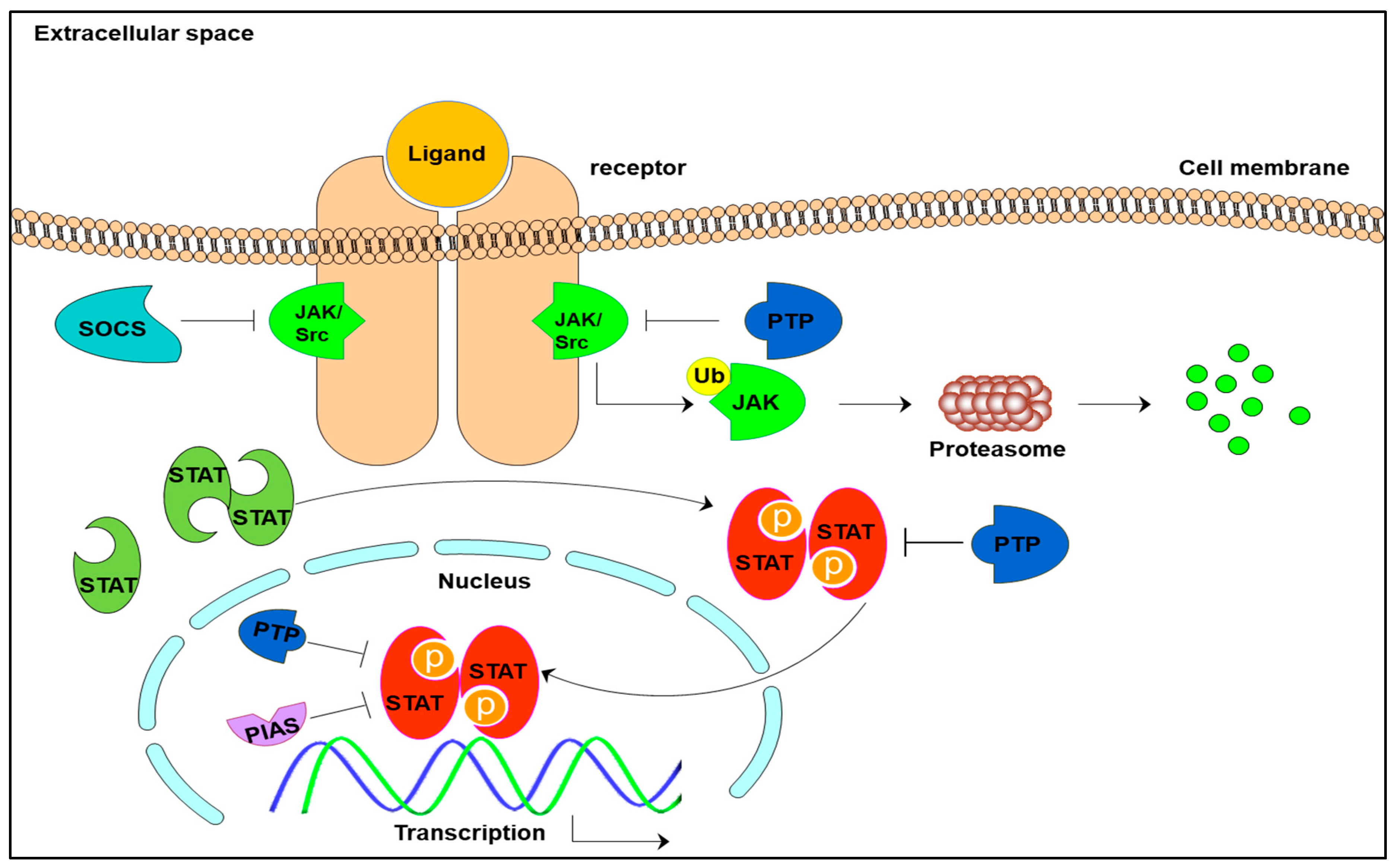
2.2. STAT3 Signaling Cascade
3. Role of STAT3 in Tumorigenesis: Solid and Hematological Tumors
3.1. Role of STAT3 in Leukaemia
3.2. Role of STAT3 in Lymphoma
3.3. Role of STAT3 in Multiple Myeloma (MM)
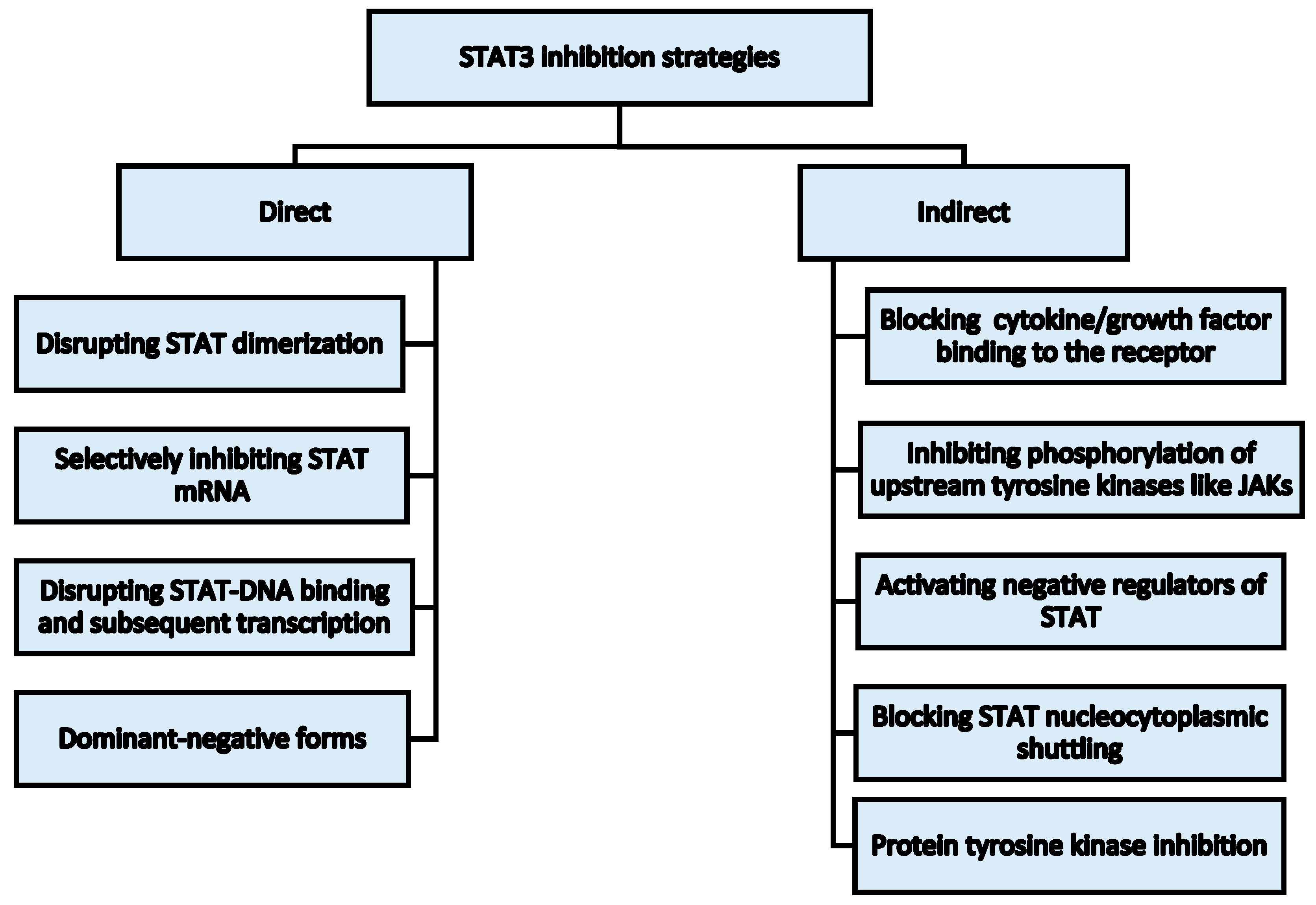
4. STAT3 as an Anti-Cancer Target and Selected Inhibition Strategies in Hematological Malignancies
4.1. Strategies for STAT3 Inhibition
4.1.1. Selected Natural Compounds as Examples of STAT3 Signaling Inhibitors
Bavachin
Butein
Celastrol
Cucurbitacins
Guggulsterone (GS)
Honokiol (HNK)
Withaferin A
4.1.2. Synthetic Inhibitors
Direct Inhibitors of STAT3 Activation
Indirect Inhibitors
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Darnell, J.E., Jr. The JAK-STAT pathway: Summary of initial studies and recent advances. Recent Prog. Horm. Res. 1996, 51, 391–403. [Google Scholar] [PubMed]
- Siveen, K.S.; Nguyen, A.H.; Lee, J.H.; Li, F.; Singh, S.S.; Kumar, A.P.; Low, G.; Jha, S.; Tergaonkar, V.; Ahn, K.S.; et al. Negative regulation of signal transducer and activator of transcription-3 signalling cascade by lupeol inhibits growth and induces apoptosis in hepatocellular carcinoma cells. Br. J. Cancer 2014, 111, 1327–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.H.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Rev. Cancer 2014, 1845, 136–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihle, J.N. The STAT family in cytokine signaling. Curr. Opin. Cell Biol. 2001, 13, 211–217. [Google Scholar] [CrossRef]
- Subramaniam, A.; Shanmugam, M.K.; Ong, T.H.; Li, F.; Perumal, E.; Chen, L.; Vali, S.; Abbasi, T.; Kapoor, S.; Ahn, K.S.; et al. Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3. Br. J. Pharmacol. 2013, 170, 807–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, A.; Yang, Z.; Shen, Y.; Zhou, J.; Shen, Q. Transcription factor STAT3 as a novel molecular target for cancer prevention. Cancers 2014, 6, 926–957. [Google Scholar] [CrossRef] [PubMed]
- Fagard, R.; Metelev, V.; Souissi, I.; Baran-Marszak, F. STAT3 inhibitors for cancer therapy: Have all roads been explored? Jakstat 2013, 2, e22882. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.; Domoszlai, T.; Kleshchanok, D.; Lehmann, S.; Schmitt, A.; Poli, V.; Richtering, W.; Müller-Newen, G. The role of the n-terminal domain in dimerization and nucleocytoplasmic shuttling of latent STAT3. J. Cell Sci. 2011, 124, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, J.; Brutsaert, S.; Olson, R.; Schindler, C. STATs dimerize in the absence of phosphorylation. J. Biol. Chem. 2003, 278, 34133–34140. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Bharathkumar, H.; Bulusu, K.C.; Pandey, V.; Rangappa, S.; Fuchs, J.E.; Shanmugam, M.K.; Dai, X.; Li, F.; Deivasigamani, A.; et al. Development of a novel azaspirane that targets the janus kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 34296–34307. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Ahn Kwang, S.; Kim, C.; Siveen Kodappully, S.; Ong Tina, H.; Shanmugam Muthu, K.; Li, F.; Shi, J.; Kumar Alan, P.; Wang Ling, Z.; et al. Ascochlorin, an isoprenoid antibiotic inhibits growth and invasion of hepatocellular carcinoma by targeting STAT3 signaling cascade through the induction of PIAS3. Mol. Oncol. 2015, 9, 818–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoni, M.; Massari, F.; Del Re, M.; Ciccarese, C.; Piva, F.; Principato, G. Investigational therapies targeting signal transducer and activator of transcription 3 for the treatment of cancer. Expert Opin. Investig. Drugs 2015, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Leeman, R.J.; Lui, V.W.Y.; Grandis, J.R. STAT3 as a therapeutic target in head and neck cancer. Expert Opin. Biol. Ther. 2006, 6, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.A. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007, 251, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Wakahara, R.; Kunimoto, H.; Tanino, K.; Kojima, H.; Inoue, A.; Shintaku, H.; Nakajima, K. Phospho-Ser727 of STAT3 regulates STAT3 activity by enhancing dephosphorylation of phospho-Tyr705 largely through TC45. Genes Cells 2012, 17, 132–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rane, S.G.; Reddy, E.P. JAKs, STATs and Src kinases in hematopoiesis. Oncogene 2002, 21, 3334–3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse STAT3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, A.P.; Diez, D.; Miranda-Saavedra, D. Genomic and computational approaches to dissect the mechanisms of STAT3’s universal and cell type-specific functions. Jakstat 2013, 2, e25097. [Google Scholar] [CrossRef] [PubMed]
- Mali, S.B. Review of STAT3 (signal transducers and activators of transcription) in head and neck cancer. Oral Oncol. 2015, 51, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Hinde, E.; Pandzic, E.; Yang, Z.; Ng, I.H.; Jans, D.A.; Bogoyevitch, M.A.; Gratton, E.; Gaus, K. Quantifying the dynamics of the oligomeric transcription factor STAT3 by pair correlation of molecular brightness. Nat. Commun. 2016, 7, 11047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruserud, Ø.; Nepstad, I.; Hauge, M.; Hatfield, K.J.; Reikvam, H. STAT3 as a possible therapeutic target in human malignancies: Lessons from acute myeloid leukemia. Expert Rev. Hematol. 2015, 8, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; McBride, K.M.; Reich, N.C. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-α3. Proc. Natl. Acad. Sci. USA 2005, 102, 8150–8155. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Cao, X. Regulation of STAT3 nuclear import by importin alpha5 and importin α 7 via two different functional sequence elements. Cell. Signal. 2006, 18, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Reich, N.C.; Liu, L. Tracking STAT nuclear traffic. Nat. Rev. Immunol. 2006, 6, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Ng, I.H.; Ng, D.C.; Jans, D.A.; Bogoyevitch, M.A. Selective STAT3-α or -β expression reveals spliceform-specific phosphorylation kinetics, nuclear retention and distinct gene expression outcomes. Biochem. J. 2012, 447, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croker, B.A.; Kiu, H.; Nicholson, S.E. Socs regulation of the JAK/STAT signalling pathway. Semin. Cell Dev. Biol. 2008, 19, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Junicho, A.; Matsuda, T.; Yamamoto, T.; Kishi, H.; Korkmaz, K.; Saatcioglu, F.; Fuse, H.; Muraguchi, A. Protein inhibitor of activated STAT3 regulates androgen receptor signaling in prostate carcinoma cells. Biochem. Biophys. Res. Commun. 2000, 278, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Beaulieu, D.R.; Paudel, H.; Barankov, R.; Bifano, T.G.; Mertz, J. Conjugate adaptive optics in widefield microscopy with an extended-source wavefront sensor. Optica 2015, 2, 682–688. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.L.; Meyer, D.J.; Campbell, G.S.; Larner, A.C.; Carter-Su, C.; Schwartz, J.; Jove, R. Enhanced DNA-binding activity of a STAT3-related protein in cells transformed by the Src oncoprotein. Science 1995, 269, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Al Zaid Siddiquee, K.; Turkson, J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008, 18, 254–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redell, M.S.; Ruiz, M.J.; Alonzo, T.A.; Gerbing, R.B.; Tweardy, D.J. STAT3 signaling in acute myeloid leukemia: Ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule STAT3 inhibitor. Blood 2011, 117, 5701–5709. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Turkson, J. Targeting STAT3 in cancer: How successful are we? Expert Opin. Investig. Drugs 2009, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Chai, E.Z.; Shanmugam, M.K.; Arfuso, F.; Dharmarajan, A.; Wang, C.; Kumar, A.P.; Samy, R.P.; Lim, L.H.; Wang, L.; Goh, B.C.; et al. Targeting transcription factor STAT3 for cancer prevention and therapy. Pharmacol. Ther. 2016, 162, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Jiang, T.; Zhu, L.; Liu, J.; Cao, J.; Huang, K.J.; Qiu, Z.J. STAT3-targeting RNA interference inhibits pancreatic cancer angiogenesis in vitro and in vivo. Int. J. Oncol. 2011, 38, 1637–1644. [Google Scholar] [PubMed] [Green Version]
- Du, W.; Hong, J.; Wang, Y.C.; Zhang, Y.J.; Wang, P.; Su, W.Y.; Lin, Y.W.; Lu, R.; Zou, W.P.; Xiong, H.; et al. Inhibition of JAK2/STAT3 signalling induces colorectal cancer cell apoptosis via mitochondrial pathway. J. Cell. Mol. Med. 2012, 16, 1878–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, B.; Zhou, L.; Vogler, T.O.; Ben-Neriah, S.; Schinke, C.; Tamari, R.; Yu, Y.; Bhagat, T.D.; Bhattacharyya, S.; Barreyro, L.; et al. Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood 2012, 120, 2076–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benekli, M.; Baumann, H.; Wetzler, M. Targeting signal transducer and activator of transcription signaling pathway in leukemias. J. Clin. Oncol. 2009, 27, 4422–4432. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, D.W.; Gilliland, D.G. The role of signal transducer and activator of transcription factors in leukemogenesis. J. Clin. Oncol. 2004, 22, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.; Dhillon, N.; Janku, F.; Watowich, S.S.; Hong, D.S. STAT3 inhibitors: Finding a home in lymphoma and leukemia. Oncologist 2014, 19, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Chen, S.J.; Tweardy, D.J. Cross-talk between retinoic acid and STAT3 signaling pathways in acute promyelocytic leukemia. Leuk. Lymphoma 2003, 44, 2023–2029. [Google Scholar] [CrossRef] [PubMed]
- Gouilleux-Gruart, V.; Gouilleux, F.; Desaint, C.; Claisse, J.F.; Capiod, J.C.; Delobel, J.; Weber-Nordt, R.; Dusanter-Fourt, I.; Dreyfus, F.; Groner, B.; et al. STAT-related transcription factors are constitutively activated in peripheral blood cells from acute leukemia patients. Blood 1996, 87, 1692–1697. [Google Scholar] [PubMed]
- Frank, D.A.; Mahajan, S.; Ritz, J. B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues. J. Clin. Investig. 1997, 100, 3140–3148. [Google Scholar] [CrossRef] [PubMed]
- Shastri, A.; Teixeira, M.; Bhattacharyya, S.; Ramachandra, N.; Lopez, R.; Ravipati, G.; Feld, J.; Dhar, Y.; Bhagat, T.D.; Choudhary, G.; et al. Targeting MDS and AML stem cells with AZD-9150 mediated inhibition of STAT3. Blood 2016, 128, 4314. [Google Scholar]
- Koskela, H.L.M.; Eldfors, S.; Ellonen, P.; Van Adrichem, A.J.; Kuusanmäki, H.; Andersson, E.I.; Lagström, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Shahmarvand, N.; Nagy, A.; Shahryari, J.; Ohgami, R.S. Mutations in the signal transducer and activator of transcription family of genes in cancer. Cancer Sci. 2018, 109, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Shastri, A.; Schinke, C.; Varshavsky Yanovsky, A.; Bhagat, T.D.; Giricz, O.; Barreyro, L.; Boultwood, J.; Pellagati, A.; Yu, Y.; Brown, J.R.; et al. Targeting of MDS and AML stem cells via inhibition of STAT3 by pyrimethamine. Blood 2014, 124, 3602. [Google Scholar]
- Shankland, K.R.; Armitage, J.O.; Hancock, B.W. Non-Hodgkin lymphoma. Lancet 2012, 380, 848–857. [Google Scholar] [CrossRef]
- Scott, L.M.; Gandhi, M.K. Deregulated JAK/STAT signalling in lymphomagenesis, and its implications for the development of new targeted therapies. Blood Rev. 2015, 29, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanzler, H.; Kuppers, R.; Hansmann, M.L.; Rajewsky, K. Hodgkin and reed-sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J. Exp. Med. 1996, 184, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.F.; Camacho, F.I.; Morente, M.; Fraga, M.; Montalban, C.; Alvaro, T.; Bellas, C.; Castano, A.; Diez, A.; Flores, T.; et al. Hodgkin and reed-sternberg cells harbor alterations in the major tumor suppressor pathways and cell-cycle checkpoints: Analyses using tissue microarrays. Blood 2003, 101, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Kube, D.; Holtick, U.; Vockerodt, M.; Ahmadi, T.; Haier, B.; Behrmann, I.; Heinrich, P.C.; Diehl, V.; Tesch, H. STAT3 is constitutively activated in Hodgkin cell lines. Blood 2001, 98, 762–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, G.G.; Queisser, N.; Wolfson, M.L.; Fraga, C.G.; Adamo, A.M.; Oteiza, P.I. Curcumin induces cell-arrest and apoptosis in association with the inhibition of constitutively active NF-κB and STAT3 pathways in Hodgkin’s lymphoma cells. Int. J. Cancer 2008, 123, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Zhang, M.; Wilson, K.M.; Petrus, M.N.; Bamford, R.N.; Zhang, X.; Guha, R.; Ferrer, M.; Thomas, C.J.; Waldmann, T.A. Augmented efficacy of brentuximab vedotin combined with ruxolitinib and/or navitoclax in a murine model of human Hodgkin’s lymphoma. Proc. Natl. Acad. Sci. USA 2016, 113, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom-Smedby, K. Epidemiology and etiology of Non-Hodgkin lymphoma—A review. Acta Oncol. 2006, 45, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, Z.; Jia, Z.; Liu, C.; Guo, C.; Lu, H.; Chen, P.; Ma, K.; Wang, W.; Zhou, C. ISL-1 is overexpressed in Non-Hodgkin lymphoma and promotes lymphoma cell proliferation by forming a p-STAT3/pc-JUN/ISL-1 complex. Mol. Cancer 2014, 13, 181. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, F. Non-Hodgkin’s lymphoma: The old and the new. Clin. Lymphoma Myeloma Leuk. 2011, 11, S87–S90. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Z.; Moreira, D.; Su, Y.-L.; Won, H.; Adamus, T.; Dong, Z.; Liang, Y.; Yin, H.H.; Swiderski, P.; et al. B cell lymphoma immunotherapy using TLR9-targeted oligonucleotide STAT3 inhibitors. Mol. Ther. 2018, 26, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.J.; John, S. Signal transducer and activator of transcription (STAT) signalling and T-cell lymphomas. Immunology 2005, 114, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roodman, G.D. Pathogenesis of myeloma bone disease. Leukemia 2008, 23, 435–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Hu, W.X. Targeting signaling pathways in multiple myeloma: Pathogenesis and implication for treatments. Cancer Lett. 2018, 414, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Chauhan, D.; Anderson, K.C. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia 2008, 23, 10–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, V.; Kimlinger, T.; Haug, J.; Timm, M.; Wellik, L.; Halling, T.; Pardanani, A.; Tefferi, A.; Vincent Rajkumar, S.; Kumar, S. TG101209, a novel JAK2 inhibitor, has significant in vitro activity in multiple myeloma and displays preferential cytotoxicity for CD45+ myeloma cells. Am. J. Hematol. 2010, 85, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansone, P.; Bromberg, J. Targeting the interleukin-6/JAK/STAT pathway in human malignancies. J. Clin. Oncol. 2012, 30, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; De Veirman, K.; Faict, S.; Frassanito Maria, A.; Ribatti, D.; Vacca, A.; Menu, E. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J. Pathol. 2016, 239, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-H.; Ahn, S.-Y.; Choi, H.-W.; Shin, M.-G.; Lee, S.-S.; Yang, D.-H.; Ahn, J.-S.; Kim, Y.-K.; Kim, H.-J.; Lee, J.-J. STAT3 expression is associated with poor survival in non-elderly adult patients with newly diagnosed multiple myeloma. Blood Res. 2017, 52, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannaiyan, R.; Hay, H.S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Vali, S.; Abbasi, T.; Kapoor, S.; Sharma, A.; Kumar, A.P.; et al. Celastrol inhibits proliferation and induces chemosensitization through down-regulation of NF-κB and STAT3 regulated gene products in multiple myeloma cells. Br. J. Pharmacol. 2011, 164, 1506–1521. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Tsubaki, M.; Tomonari, Y.; Kawashima, K.; Itoh, T.; Imano, M.; Satou, T.; Nishida, S. Bavachin induces the apoptosis of multiple myeloma cell lines by inhibiting the activation of nuclear factor kappa B and signal transducer and activator of transcription 3. Biomed. Pharmacother. 2018, 100, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wu, H.; Li, B.; Song, D.; Yang, G.; Chen, G.; Xie, B.; Xu, Z.; Zhang, Y.; Yu, D.; et al. Dihydrocelastrol inhibits multiple myeloma cell proliferation and promotes apoptosis through Erk1/2 and IL-6/STAT3 pathways in vitro and in vivo. Acta Biochim. Biophys. Sin. 2017, 49, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: How intimate is the relationship? Ann. N. Y. Acad. Sci. 2009, 1171, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Jing, N.; Tweardy, D.J. Targeting STAT3 in cancer therapy. Anti-Cancer Drugs 2005, 16, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Frank, D.A. Screening approaches to generating STAT inhibitors: Allowing the hits to identify the targets. Jakstat 2012, 1, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.S.; Mahajan, S.; Frank, D.A. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene 2000, 19, 2496–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Z.; Baer, M.R.; Block, A.W.; Baumann, H.; Wetzler, M. Expression of signal transducers and activators of transcription proteins in acute myeloid leukemia blasts. Cancer Res. 1998, 58, 3173–3180. [Google Scholar] [PubMed]
- Frank, D.A.; Varticovski, L. BCR/Abl leads to the constitutive activation of STAT proteins, and shares an epitope with tyrosine phosphorylated STATs. Leukemia 1996, 10, 1724–1730. [Google Scholar] [PubMed]
- Lacronique, V.; Boureux, A.; Valle, V.D.; Poirel, H.; Quang, C.T.; Mauchauffe, M.; Berthou, C.; Lessard, M.; Berger, R.; Ghysdael, J.; et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 1997, 278, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Tolentino, J.H.; Hazlehurst, L.A. Role of STAT3 in transformation and drug resistance in CML. Front. Oncol. 2012, 2, 30. [Google Scholar] [CrossRef] [PubMed]
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Jain, P.; Veletic, I.; Ferrajoli, A.; Burger, J.; O’Brien, S.; Bose, P.; et al. Constitutive phosphorylation of STAT3 by the CK2-BLNK-CD5 complex. Mol. Cancer Res. 2017, 15, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Ladewig, E.; Schiavoni, G.; Penson, A.; Fortini, E.; Pettirossi, V.; Wang, Y.; Rosseto, A.; Venanzi, A.; Vlasevska, S.; et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood 2018, 131, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, J.; Qian, J.; Li, H.; Romaguera, J.E.; Kwak, L.W.; Wang, M.; Yi, Q. Role of the microenvironment in mantle cell lymphoma: IL-6 is an important survival factor for the tumor cells. Blood 2012, 120, 3783–3792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinnider, B.F.; Kapp, U.; Mak, T.W. The role of interleukin 13 in classical Hodgkin lymphoma. Leuk. Lymphoma 2002, 43, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Alas, S.; Bonavida, B. Inhibition of constitutive STAT3 activity sensitizes resistant Non-Hodgkin’s lymphoma and multiple myeloma to chemotherapeutic drug-mediated apoptosis. Clin. Cancer Res. 2003, 9, 316–326. [Google Scholar] [PubMed]
- Piazza, F.; Manni, S.; Semenzato, G. Novel players in multiple myeloma pathogenesis: Role of protein kinases CK2 and GSK3. Leuk. Res. 2013, 37, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.A.; Grandis, J.R. STAT3 signaling: Anticancer strategies and challenges. Mol. Interv. 2011, 11, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, D.H.; Ahn, H.N.; Song, Y.S.; Lee, Y.J.; Ryu, J.H. Activation of estrogen receptor by bavachin from Psoralea corylifolia. Biomol. Ther. 2012, 20, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Sung, B.; Ahn, K.S.; Aggarwal, B.B. Butein suppresses constitutive and inducible signal transducer and activator of transcription (STAT) 3 activation and STAT3-regulated gene products through the induction of a protein tyrosine phosphatase SHP-1. Mol. Pharmacol. 2009, 75, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Padmavathi, G.; Rathnakaram, S.R.; Monisha, J.; Bordoloi, D.; Roy, N.K.; Kunnumakkara, A.B. Potential of butein, a tetrahydroxychalcone to obliterate cancer. Phytomedicine 2015, 22, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Padmavathi, G.; Roy, N.K.; Bordoloi, D.; Arfuso, F.; Mishra, S.; Sethi, G.; Bishayee, A.; Kunnumakkara, A.B. Butein in health and disease: A comprehensive review. Phytomedicine 2017, 25, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ruan, X.; Zhang, J.; Zhao, Q. Celastrol induces cell apoptosis and inhibits the expression of the AML1-ETO/C-KIT oncoprotein in t (8;21) leukemia. Molecules 2016, 21, 574. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Li, F.; Shanmugam, M.K.; Kannaiyan, R.; Goh, J.N.; Wong, K.F.; Wang, W.; Khin, E.; Tergaonkar, V.; Kumar, A.P.; et al. Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of STAT3/JAK2 signaling cascade in vitro and in vivo. Cancer Prev. Res. 2012, 5, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.T.; Li, K.; Liu, S.L.; Chu, K.H.; Toh, M.; Xie, W.D. Cucurbitacin B inhibits STAT3 and the Raf/Mek/Erk pathway in leukemia cell line K562. Cancer Lett. 2010, 289, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Chiu, M.H.; Nie, R.L.; Cordell, G.A.; Qiu, S.X. Cucurbitacins and cucurbitane glycosides: Structures and biological activities. Nat. Prod. Rep. 2005, 22, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Luca, C.; Daniela, B. STAT 3 as a target for cancer drug discovery. Curr. Med. Chem. 2008, 15, 834–843. [Google Scholar]
- Samudio, I.; Konopleva, M.; Safe, S.; McQueen, T.; Andreeff, M. Guggulsterones induce apoptosis and differentiation in acute myeloid leukemia: Identification of isomer-specific antileukemic activities of the pregnadienedione structure. Mol. Cancer Ther. 2005, 4, 1982–1992. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.S.; Sethi, G.; Sung, B.; Goel, A.; Ralhan, R.; Aggarwal, B.B. Guggulsterone, a farnesoid X receptor antagonist, inhibits constitutive and inducible STAT3 activation through induction of a protein tyrosine phosphatase SHP-1. Cancer Res. 2008, 68, 4406–4415. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Katiyar, S.K. Honokiol, an active compound of magnolia plant, inhibits growth, and progression of cancers of different organs. Adv. Exp. Med. Biol. 2016, 928, 245–265. [Google Scholar] [PubMed]
- Battle, T.E.; Arbiser, J.; Frank, D.A. The natural product honokiol induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia (B-CLL) cells. Blood 2005, 106, 690–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, L.; Yu, Z.; Wu, J.; Yu, K.; Hong, G.; Lu, Z.; Gao, S. Honokiol inhibits constitutive and inducible STAT3 signaling via PU.1-induced SHP1 expression in acute myeloid leukemia cells. Tohoku J. Exp. Med. 2015, 237, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, K.; Hideshima, T.; Hamasaki, M.; Raje, N.; Kumar, S.; Hideshima, H.; Shiraishi, N.; Yasui, H.; Roccaro, A.M.; Richardson, P.; et al. Honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and -independent apoptosis. Blood 2005, 106, 1794–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, C.; Arbiser, J.L.; Mori, N. Honokiol induces cell cycle arrest and apoptosis via inhibition of survival signals in adult T-cell leukemia. Biochim. Biophys. Acta 2012, 1820, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Li, H.; Xing, C.; Ye, H.; Feng, J.; Wu, J.; Lu, Z.; Fang, J.; Gao, S. Honokiol induces proteasomal degradation of AML1-ETO oncoprotein via increasing ubiquitin conjugase UbcH8 expression in leukemia. Biochem. Pharmacol. 2017, 128, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, W.; Sabbe, L.; Kaileh, M.; Haegeman, G.; Heyninck, K. Molecular insight in the multifunctional activities of Withaferin A. Biochem. Pharmacol. 2012, 84, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Yco, L.P.; Mocz, G.; Opoku-Ansah, J.; Bachmann, A.S. Withaferin A inhibits STAT3 and induces tumor cell death in neuroblastoma and multiple myeloma. Biochem. Insights 2014, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Ryan, D.; Kim, J.S.; Zhang, Y.; Chen, Z.; Haura, E.; Laudano, A.; Sebti, S.; Hamilton, A.D.; Jove, R. Phosphotyrosyl peptides block STAT3-mediated DNA binding activity, gene regulation, and cell transformation. J. Biol. Chem. 2001, 276, 45443–45455. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Kim, J.S.; Zhang, S.; Yuan, J.; Huang, M.; Glenn, M.; Haura, E.; Sebti, S.; Hamilton, A.D.; Jove, R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol. Cancer Ther. 2004, 3, 261–269. [Google Scholar] [PubMed]
- Hillion, J.; Belton, A.M.; Shah, S.N.; Turkson, J.; Jing, N.; Tweardy, D.J.; Di Cello, F.; Huso, D.L.; Resar, L.M. Nanoparticle delivery of inhibitory signal transducer and activator of transcription 3 G-quartet oligonucleotides blocks tumor growth in HMGA1 transgenic model of t-cell leukemia. Leuk. Lymphoma 2014, 55, 1194–1197. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Zhang, S.; Mora, L.B.; Burns, A.; Sebti, S.; Jove, R. A novel platinum compound inhibits constitutive STAT3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J. Biol. Chem. 2005, 280, 32979–32988. [Google Scholar] [CrossRef] [PubMed]
- Alas, S.; Bonavida, B. Rituximab inactivates signal transducer and activation of transcription 3 (STAT3) activity in B-Non-Hodgkin’s lymphoma through inhibition of the interleukin 10 autocrine/paracrine loop and results in down-regulation of Bcl-2 and sensitization to cytotoxic drugs. Cancer Res. 2001, 61, 5137–5144. [Google Scholar] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Wilks, A.F. The JAK kinases: Not just another kinase drug discovery target. Semin. Cell Dev. Biol. 2008, 19, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.P.; Kantarjian, H.M.; Jain, N.; Manshouri, T.; Thomas, D.A.; Garcia-Manero, G.; Kennedy, D.; Estrov, Z.; Cortes, J.; Verstovsek, S. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood 2010, 115, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Kasper, S.; Breitenbuecher, F.; Hoehn, Y.; Heidel, F.; Lipka, D.B.; Markova, B.; Huber, C.; Kindler, T.; Fischer, T. The kinase inhibitor LS104 induces apoptosis, enhances cytotoxic effects of chemotherapeutic drugs and is targeting the receptor tyrosine kinase FLT3 in acute myeloid leukemia. Leuk. Res. 2008, 32, 1698–1708. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.; Le Gouill, S.; Tai, Y.T.; Shringarpure, R.; Tassone, P.; Neri, P.; Podar, K.; Catley, L.; Hideshima, T.; Chauhan, D.; et al. Janus kinase inhibitor INCB20 has antiproliferative and apoptotic effects on human myeloma cells in vitro and in vivo. Mol. Cancer Ther. 2009, 8, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, A.; Vogt, M.; Monnigmann, M.; Clahsen, T.; Sommer, U.; Haan, S.; Poli, V.; Heinrich, P.C.; Muller-Newen, G. Nucleocytoplasmic shuttling of persistently activated STAT3. J. Cell Sci. 2007, 120, 3249–3261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahipal, A.; Malafa, M. Importins and exportins as therapeutic targets in cancer. Pharmacol. Ther. 2016, 164, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Krebs, D.L.; Hilton, D.J. Socs proteins: Negative regulators of cytokine signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Talpaz, M.; Shah, N.P.; Kantarjian, H.; Donato, N.; Nicoll, J.; Paquette, R.; Cortes, J.; O’Brien, S.; Nicaise, C.; Bleickardt, E.; et al. Dasatinib in imatinib-resistant philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2006, 354, 2531–2541. [Google Scholar] [CrossRef] [PubMed]
- Listed, N. Gleevec (STI-571) for chronic myeloid leukemia. Med. Lett. Drugs Ther. 2001, 43, 49–50. [Google Scholar]
- Ajayi, S.; Becker, H.; Reinhardt, H.; Engelhardt, M.; Zeiser, R.; Von Bubnoff, N.; Wasch, R. Ruxolitinib. Recent Results Cancer Res. 2018, 212, 119–132. [Google Scholar] [PubMed]
- Pemmaraju, N.; Kantarjian, H.; Kadia, T.; Cortes, J.; Borthakur, G.; Newberry, K.; Garcia-Manero, G.; Ravandi, F.; Jabbour, E.; Dellasala, S.; et al. A phase I/II study of the janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Beebe, J.D.; Liu, J.-Y.; Zhang, J.-T. Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Hematological Malignancy | Subtype(s) | Activation Mechanism(s) | References |
|---|---|---|---|
| Acute Leukaemias | ALL | BCR/abl fusion protein | [76] |
| TEL-JAK2 fusion protein | [77] | ||
| AML | Aberrant exogenous cytokine signaling | [75] | |
| Constitutive Protein Tyrosine Kinases (PTK) activation without exposure to exogenous cytokines | [75] | ||
| Activating STAT3 mutations | [21] | ||
| Hypermethylation, hence, silencing of negative regulators of STAT3 (PIAS3, SOCS3 and PTP) | [21] | ||
| Chronic Leukemias | CLL | Casein Kinase2(CK2)-B cell linker (BLNK)-CD5 complex causes constitutive phosphorylation | [79] |
| CML | JAK activation by bone marrow microenvironment | [78] | |
| Lymphomas | HL | Activating mutations in JAK1 and STAT3 | [80] |
| Autocrine secretion of IL13 by HRS cells | [82] | ||
| NHL | Autocrine and paracrine secretion of IL6 and IL13 | [81,83] | |
| Multiple Myeloma | - | Autocrine and paracrine secretion of IL6 and subsequent activation of JAK1 | [62] |
| Overexpression and hyperactivation of CK2 | [84] |
| Inhibitor | Indication | Status | References |
|---|---|---|---|
| Dasatinib (Tyrosine Kinase Inhibitor) | ALL, CML | FDA approved, specifically for Ph+ cases | [122] |
| Imatinib (Bcr-Abl Tyrosine Kinase Inhibitor) | CML, ALL | FDA approved, specifically for Ph+ cases | [123] |
| Ruxolitinib (JAK 1&2 inhibitor) | Myeloproliferative neoplasms | FDA approved for Myelofibrosis and as second line treatment for Polycythemia vera | [124] |
| AML | Phase I/II trial terminated due to lack of efficacy | [125] | |
| Pyrimethamine (Direct STAT3 SH2 domain inhibitor) | CLL | Phase I/II clinical trials | [126] |
| AZD 9150 (STAT3 Antisense oligonucleotide) | Lymphoma | Phase I dose escalation study completed | [108] |
| OPB 31121 (STAT3 SH2 domain inhibitor) | MM, NHL Leukemia | Trial terminated due to high toxicity and poor pharmacokinetics | [126] |
| OPB 51602 (STAT3 SH2 domain inhibitor) | MM, AML NHL, CML | Trial terminated due to inefficacy in hematological malignancies | [126] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arora, L.; Kumar, A.P.; Arfuso, F.; Chng, W.J.; Sethi, G. The Role of Signal Transducer and Activator of Transcription 3 (STAT3) and Its Targeted Inhibition in Hematological Malignancies. Cancers 2018, 10, 327. https://doi.org/10.3390/cancers10090327
Arora L, Kumar AP, Arfuso F, Chng WJ, Sethi G. The Role of Signal Transducer and Activator of Transcription 3 (STAT3) and Its Targeted Inhibition in Hematological Malignancies. Cancers. 2018; 10(9):327. https://doi.org/10.3390/cancers10090327
Chicago/Turabian StyleArora, Loukik, Alan Prem Kumar, Frank Arfuso, Wee Joo Chng, and Gautam Sethi. 2018. "The Role of Signal Transducer and Activator of Transcription 3 (STAT3) and Its Targeted Inhibition in Hematological Malignancies" Cancers 10, no. 9: 327. https://doi.org/10.3390/cancers10090327
APA StyleArora, L., Kumar, A. P., Arfuso, F., Chng, W. J., & Sethi, G. (2018). The Role of Signal Transducer and Activator of Transcription 3 (STAT3) and Its Targeted Inhibition in Hematological Malignancies. Cancers, 10(9), 327. https://doi.org/10.3390/cancers10090327