Increased Mortality in SDHB but Not in SDHD Pathogenic Variant Carriers

 ,
,  ,
, 
Abstract
:1. Introduction
2. Subjects and Methods
2.1. Eligibility Criteria
2.2. Clinical Characteristics
2.3. Mortality and Survival
3. Results
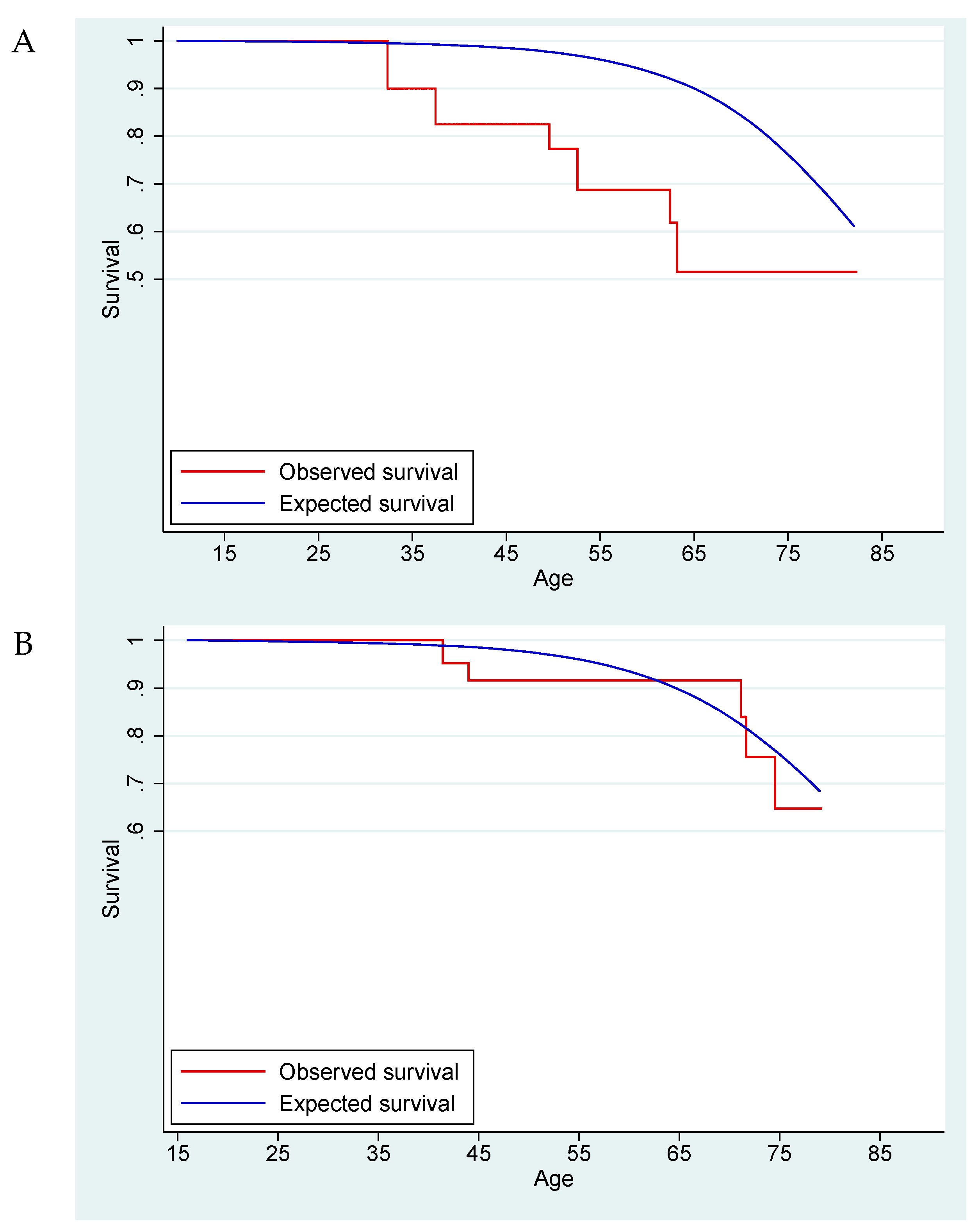
Mortality and SMR
4. Discussion
5. Conclusion
Author Contributions
Funding
Conflicts of Interest
Appendix A. SDHB Variants and SDHD Variants
| DNA Mutation | Predicted Protein Change | Number of Subjects (%) |
| Exon 3 deletion | p.? | 59 (30.7) |
| c.423 + 1G > A | p.? | 45 (23.4) |
| c.654G > A | p.(Trp218*) | 19 (9.9) |
| c.653G > C | p.(Trp218Ser) | 11 (5.7) |
| c.574T > C | p.(Cys192Arg) | 8 (4.2) |
| c.200 + 1G > A | p.? | 6 (3.1) |
| c.137G > A | p.(Arg46Gln) | 4 (2.1) |
| c.328A > C | p.(Thr110Pro) | 4 (2.1) |
| c.418G > T | p.(Val140Phe) | 4 (2.1) |
| c.725G > A | p.(Arg242His) | 3 (1.6) |
| c.649C > T | p.(Arg217Cys) | 3 (1.6) |
| c.590C > G | p.(Pro197Arg) | 3 (1.6) |
| c.686_725del | p.(Glu229fs) | 3 (1.6) |
| c.343C > T | p.(Arg115*) | 3 (1.6) |
| c.292T > C | p.(Cys98Arg) | 2 (1.0) |
| Deletion promoter and exon 1 | p.? | 1 (0.5) |
| Deletion promoter till exon 8 | p.0 | 2 (1.0) |
| Exon 2 deletion | p.? | 2 (1.0) |
| Exon 1 deletion | p.? | 2 (1.0) |
| c.713delT | p.(Phe238fs) | 1 (0.5) |
| c.727T > A | p.(Cys243Ser) | 1 (0.5) |
| c.761C > T | p.(Pro254Leu) | 1 (0.5) |
| c.626C > T | p.(Pro209Leu) | 1 (0.5) |
| c.380T > C | p.(Ile127Thr) | 1 (0.5) |
| c.325A > C | p.(Asn109His) | 1 (0.5) |
| c.1A > G | p.? | 1 (0.5) |
| c.119A > C | p.(Lys40Thr) | 1 (0.5) |
| c.274G > T | p.(Asp92Tyr) | 175 (74.7) |
| c.416T > C | p.(Leu139Pro) | 34 (14.6) |
| c.284T > C | p.(Leu95Pro) | 6 (2.6) |
| Deletion promoter, exon 1 and 2 | p.? | 4 (1.7) |
| c.242C > T | p.(Pro81Leu) | 3 (1.3) |
| c.337_340delGACT | p.(Asp113fs) | 2 (0.9) |
| c.122dupC | p.(Glu42fs) | 2 (0.9) |
| Exon 1. c.3G > C | p.(Met1Ile) | 1 (0.4) |
| Exon 2: c.169_169 + 9del10, splice donor mutation | p.? | 1 (0.4) |
| Intron 2 c.169_169 + 9del | p.? | 1 (0.4) |
| Specific SDHD variant unknown (tested elsewhere) | unknown | 3 (1.3) |
References
- Cascon, A.; Comino-Mendez, I.; Curras-Freixes, M.; de Cubas, A.A.; Contreras, L.; Richter, S.; Peitzsch, M.; Mancikova, V.; Inglada-Perez, L.; Perez-Barrios, A.; et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J. Natl. Cancer Inst. 2015, 107, djv053. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Hensen, E.F.; van Duinen, N.; Jansen, J.C.; Corssmit, E.P.; Tops, C.M.; Romijn, J.A.; Vriends, A.H.; van der Mey, A.G.; Cornelisse, C.J.; Devilee, P.; et al. High prevalence of founder mutations of the succinate dehydrogenase genes in the Netherlands. Clin. Genet. 2012, 81, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Van der Tuin, K.; Mensenkamp, A.R.; Tops, C.M.J.; Corssmit, E.P.M.; Dinjens, W.N.; van de Horst-Schrivers, A.N.; Jansen, J.C.; de Jong, M.M.; Kunst, H.P.M.; Kusters, B.; et al. Clinical Aspects of SDHA-Related Pheochromocytoma and Paraganglioma: A Nationwide Study. J. Clin. Endocrinol. Metab. 2018, 103, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.A.; Ascher, D.B.; Pires, D.E.V.; Barnes, D.R.; Vialard, L.; Casey, R.T.; Bradshaw, N.; Adlard, J.; Aylwin, S.; Brennan, P.; et al. Tumour risks and genotype–phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J. Med. Genet. 2018, 55, 384–394. [Google Scholar] [PubMed]
- Rijken, J.A.; Niemeijer, N.D.; Jonker, M.A.; Eijkelenkamp, K.; Jansen, J.C.; van Berkel, A.; Timmers, H.J.L.M.; Kunst, H.P.M.; Bisschop, P.H.L.T.; Kerstens, M.N.; et al. The penetrance of paraganglioma and pheochromocytoma in SDHB germline mutation carriers. Clin. Genet. 2018, 93, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD mutations. JAMA 2004, 292, 943–951. [Google Scholar] [CrossRef]
- Kunst, H.P.; Rutten, M.H.; de Mönnink, J.P.; Hoefsloot, L.H.; Timmers, H.J.L.M.; Marres, H.A.M.; Jansen, J.C.; Kremer, H.; Bayley, J.-P.; Cremers, C.W.R.J. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin. Cancer Res. 2011, 17, 247–254. [Google Scholar] [CrossRef]
- Hensen, E.F.; Jansen, J.C.; Siemers, M.D.; Oosterwijk, J.C.; Vriends, A.H.; Corssmit, E.P.; Bayley, J.-P.; van der Mey, A.G.; Cornelisse, C.J.; Devilee, P. The Dutch founder mutation SDHD.D92Y shows a reduced penetrance for the development of paragangliomas in a large multigenerational family. Eur. J. Hum. Genet. 2010, 18, 62–66. [Google Scholar] [CrossRef]
- Benn, D.E.; Gimenez-Roqueplo, A.P.; Reilly, J.R.; Bertherat, J.; Burgess, J.; Byth, K.; Croxson, M.; Dahia, P.L.; Elston, M.; Gimm, O.; et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 827–836. [Google Scholar] [CrossRef]
- Amar, L.; Baudin, E.; Burnichon, N.; Peyrard, S.; Silvera, S.; Bertherat, J.; Bertagna, X.; Schlumberger, M.; Jeunemaitre, X.; Gimenez-Roqueplo, A.; et al. Succinate Dehydrogenase B Gene Mutations Predict Survival in Patients with Malignant Pheochromocytomas or Paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 3822–3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hulsteijn, L.T.; Heesterman, B.; Jansen, J.C.; Bayley, J.P.; Hes, F.J.; Corssmit EPMDekkers, O.M. No evidence for increased mortality in SDHD variant carriers compared with the general population. Eur. J. Hum. Genet. 2015, 23, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Niemeijer, N.D.; Rijken, J.A.; Eijkelenkamp, K.; van der Horst-Schrivers, A.N.A.; Kerstens, M.N.; Tops, C.M.J.; van Berkel, A.; Timmers, H.J.L.M.; Kunst, H.P.M.; Leemans, C.R.; et al. The phenotype of SDHB germline mutation carriers; a nationwide study. Eur. J. Endocrinol. 2017, 177, 115–125. [Google Scholar] [CrossRef]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.L.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef] [Green Version]
- Dutch Guideline for Detecting Hereditary Tumors. 2010. Available online: https://www.stoet.nl (accessed on 17 March 2017).
- Dutch Guidelines for Oncology Care. 2016. Available online: http://www.oncoline.nl/familiair-paraganglioom (accessed on 17 March 2017).
- Suissa, S. Immortal time bias in pharmaco-epidemiology. Am. J. Epidemiol. 2008, 167, 492–499. [Google Scholar] [CrossRef]
- Statistics Netherlands. Available online: https://www.cbs.nl/ (accessed on 17 March 2017).
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Oudijk, L.; Gaal, J.; de Krijger, R.R. The Role of Immunohistochemistry and Molecular Analysis of Succinate Dehydrogenase in the Diagnosis of Endocrine and Non-Endocrine Tumors and Related Syndromes. Endocr. Pathol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Timmers, H.J.; Kozupa, A.; Eisenhofer, G.; Raygada, M.; Adams, K.T.; Solis, D.; Lenders, J.W.; Pacak, K. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Srirangalingam, U.; Walker, L.; Khoo, B.; MacDonald, F.; Gardner, D.; Wilkin, T.J.; Skelly, R.H.; George, E.; Spooner, D.; Monson, J.P.; et al. Clinical manifestations of familial paraganglioma and phaeochromocytomas in succinate dehydrogenase B (SDH-B) gene mutation carriers. Clin. Endocrinol. 2008, 69, 587–596. [Google Scholar] [CrossRef]
- Amar, L.; Bertherat, J.; Baudin, E.; Ajzenberg, C.; Bressac-de Paillerets, B.; Chabre, O.; Chamontin, B.; Delemer, B.; Giraud, S.; Murat, A.; et al. Genetic testing in pheochromocytoma or functional paraganglioma. J. Clin. Oncol. 2005, 23, 8812–8818. [Google Scholar] [CrossRef]
- Van Hulsteijn, L.T.; Dekkers, O.M.; Hes, F.J.; Smit, J.W.; Corssmit, E.P. Risk of malignant paraganglioma in SDHB-mutation and SDHD-mutation carriers: A systematic review and meta-analysis. J. Med. Genet. 2012, 49, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Hulsteijn, L.T.; den Dulk, A.C.; Hes, F.J.; Bayley, J.P.; Jansen, J.C.; Corssmit, E.P.M. No difference in phenotype of the main Dutch SDHD founder mutations. Clin. Endocrinol. 2013, 79, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Kapetanakis, S.; Chourmouzi, D.; Gkasdaris, G.; Katsaridis, V.; Eleftheriadis, E.; Givissis, P. Functional extra-adrenal paraganglioma of the retroperitoneum giving thoracolumbar spine metastases after a five-year disease-free follow-up: A rare malignant condition with challenging management. Pan Afr. Med. J. 2017, 28, 94. [Google Scholar] [CrossRef] [PubMed]
- Valadea, S.; Chazeraina, P.; Khaninea, V.; Lazardb, T.; Baudinc, E.; Zizaa, J.M. Late bone metastases of a pheochromocytoma. Rev. Med. Interne 2010, 31, 772–775. [Google Scholar]
- Papaspyrou, K.; Mann, W.J.; Amedee, R.G. Management of head and neck paragangliomas: Review of 120 patients. Head Neck 2009, 31, 381–387. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Clinical Characteristics | SDHB n = 192 | SDHD n = 232 |
|---|---|---|
| Male (%)/female (%) | 81 (42.2)/111 (57.8) | 123 (53.0)/109 (47.0) |
| Mean age at genetic testing | 46 years (range 9–77) | 44 years (range 16–73) |
| HNPGL (%) | 53 (27.6) | 198 (85.3) |
| sPGL (%) | 26 (13.5) | 18 (7.8) |
| Pheochromocytoma (%) | 4 (2.1) | 16 (6.9) |
| Malignant PGL (%) | 14 (7.3) | 4 (1.7) |
| Unaffected (%) | 110 (57.3) | 30 (12.9) |
| Sex | Mutation | Predicted Protein Change | Location of PGL | Age at PGL Diagnosis (years) | Age at Diagnosis of Malignant Disease (years) | Age at Death (years) | Location of Metastases | Cause of Death |
|---|---|---|---|---|---|---|---|---|
| M | SDHB exon 3 deletion | p.? | Presacral | 28 | 28 | 32 | Bone | Progressive malignant PGL |
| F | SDHB c.654G > A | p.(Trp218*) | Bladder | 19 | 58 | 62 | Lymph nodes, bone | Progressive malignant PGL |
| F | SDHB exon 3 deletion | p.? | Para-vertebral abdominal | 33 | 33 | 37 | Lymph nodes, bone | Progressive malignant PGL |
| F | SDHB c.727T > A | p.(Cys243Ser) | Retroperitoneal (para-aortic) | 52 | 55 | 63 | Bone | Myocardial infarction, heart failure and acute respiratory distress syndrome |
| F | SDHB c.423 + 1G > A | p.? | n.a. | 49 | n.a. | 52 | n.a. | Respiratory insufficiency due to lung bleeding after chemoradiotherapy for lung cancer |
| F | SDHB c.423 + 1G > A | p.? | n.a. | 42 | n.a. | 49 | n.a. | Metastatic breast cancer |
| F | SDHD c.274G > T | p.(Asp92Tyr) | Bladder | 42 | 42 | 43 | Lymph nodes, bone marrow | Progressive malignant PGL |
| F | SDHD c.274G > T | p.(Asp92Tyr) | Mediastinal | 67 | 67 | 74 | Lymph nodes, bone | Unknown, however the patient was known to have progressive malignant PGL |
| F | SDHD c.274G > T | p.(Asp92Tyr) | Bilateral CBT, VBT | 55 | n.a. | 71 | n.a. | Cardiac arrest |
| F | SDHD c.242C > T | p.(Pro81Leu) | CBT | 38 | n.a. | 41 | n.a. | Breast cancer |
| M | SDHD c.274G > T | p.(Asp92Tyr) | CBT, jugular PGL, retroperitoneal | 52 | n.a. | 71 | n.a. | Prostate cancer |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rijken, J.A.; van Hulsteijn, L.T.; Dekkers, O.M.; Niemeijer, N.D.; Leemans, C.R.; Eijkelenkamp, K.; van der Horst-Schrivers, A.N.A.; Kerstens, M.N.; van Berkel, A.; Timmers, H.J.L.M.; et al. Increased Mortality in SDHB but Not in SDHD Pathogenic Variant Carriers. Cancers 2019, 11, 103. https://doi.org/10.3390/cancers11010103
Rijken JA, van Hulsteijn LT, Dekkers OM, Niemeijer ND, Leemans CR, Eijkelenkamp K, van der Horst-Schrivers ANA, Kerstens MN, van Berkel A, Timmers HJLM, et al. Increased Mortality in SDHB but Not in SDHD Pathogenic Variant Carriers. Cancers. 2019; 11(1):103. https://doi.org/10.3390/cancers11010103
Chicago/Turabian StyleRijken, Johannes A., Leonie T. van Hulsteijn, Olaf M. Dekkers, Nicolasine D. Niemeijer, C. René Leemans, Karin Eijkelenkamp, Anouk N.A. van der Horst-Schrivers, Michiel N. Kerstens, Anouk van Berkel, Henri J.L.M. Timmers, and et al. 2019. "Increased Mortality in SDHB but Not in SDHD Pathogenic Variant Carriers" Cancers 11, no. 1: 103. https://doi.org/10.3390/cancers11010103
APA StyleRijken, J. A., van Hulsteijn, L. T., Dekkers, O. M., Niemeijer, N. D., Leemans, C. R., Eijkelenkamp, K., van der Horst-Schrivers, A. N. A., Kerstens, M. N., van Berkel, A., Timmers, H. J. L. M., Kunst, H. P. M., Bisschop, P. H. L. T., Dreijerink, K. M. A., van Dooren, M. F., Hes, F. J., Jansen, J. C., Corssmit, E. P. M., & Hensen, E. F. (2019). Increased Mortality in SDHB but Not in SDHD Pathogenic Variant Carriers. Cancers, 11(1), 103. https://doi.org/10.3390/cancers11010103