Glioblastoma’s Next Top Model: Novel Culture Systems for Brain Cancer Radiotherapy Research


Abstract
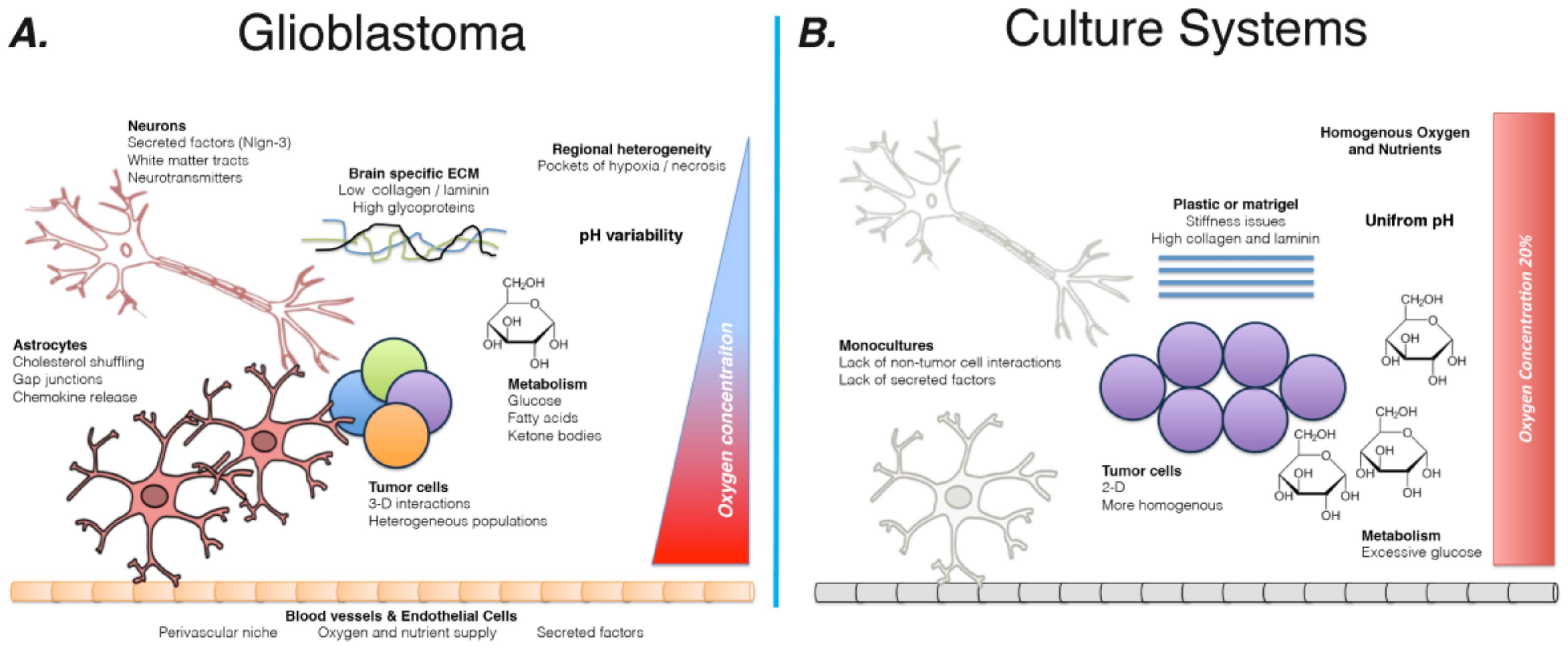
:1. Introduction
2. Limits of Current Pre-Clinical Systems
2.1. Current Practices for Pre-Clinical GBM Modeling
2.2. Tumor Intrinsic Factors
2.2.1. Inter and Intratumoral Genetic Heterogeneity
2.2.2. Cellular Plasticity
2.3. Microenvironmental Factors
2.3.1. Brain Extracellular Matrix
2.3.2. Regional Differences in the Tumor Microenvironment
2.3.3. Nutrient Availability and Metabolism
2.3.4. Non-Tumor Cells: Secreted Factors and Contact-Mediated Interactions
2.3.5. Dynamic Surroundings
3. Novel Strategies for GBM Cell Culture
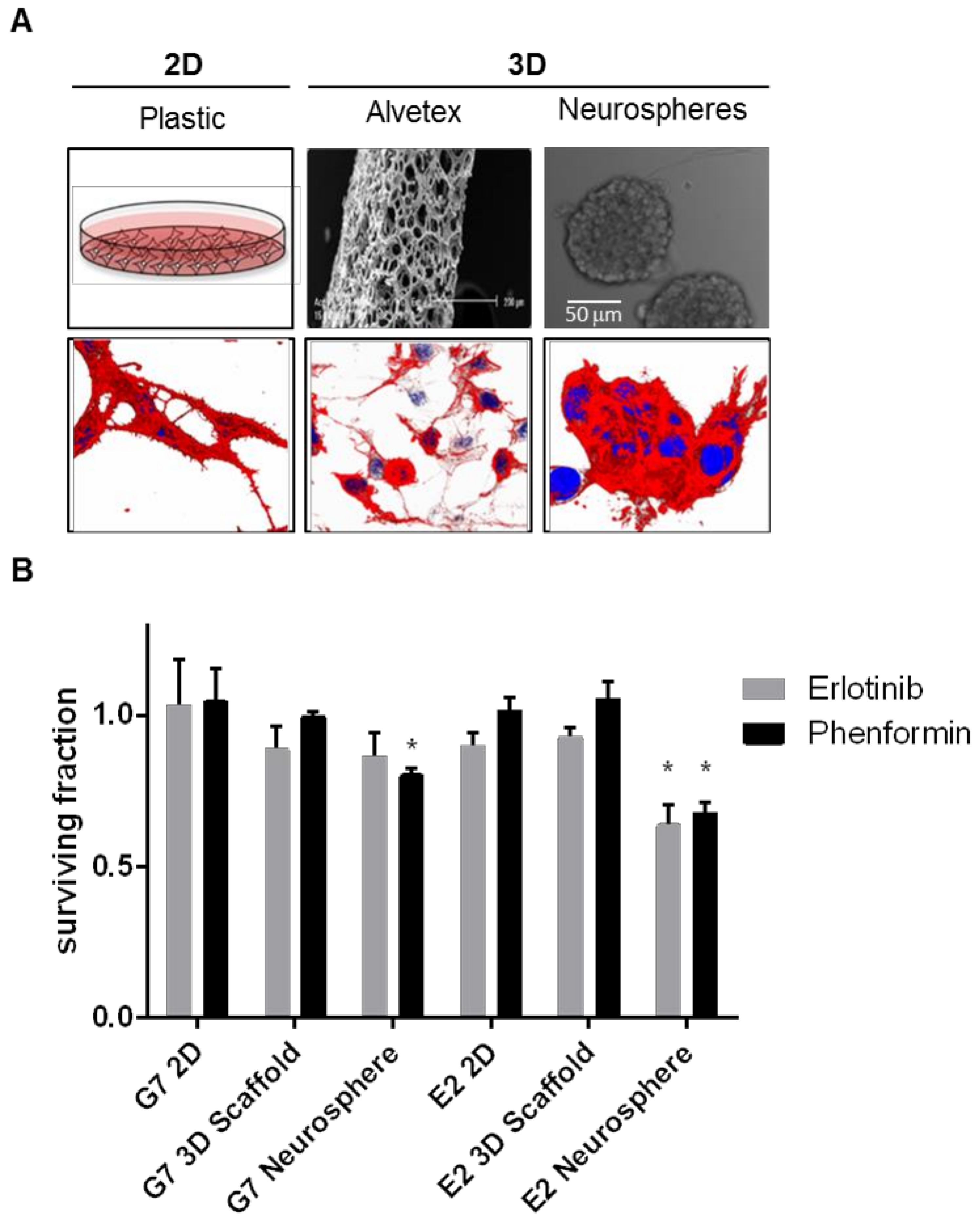
3.1. Suspension-Based Culture Models: Neurospheres and Organotypic Glioma Spheroids
3.2. Hydrogels
3.3. Matrigel-Coating for 2D Growth
3.4. Matrigel Plugs for 3D Growth
3.5. 3D Scaffolds
3.6. Microfluidic Systems
3.7. Brain Slices
3.8. Mini-Brains
3.9. Tumor Organoids
4. Conclusions, Caveats, and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Eskilsson, E.; Rosland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.W.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015, 17 (Suppl. 7), vii9–vii14. [Google Scholar] [CrossRef]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol. 2017, 19, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Goncalves, V.; Cardoso-Carneiro, D.; Valbom, I.; Cury, F.P.; Silva, V.A.; Granja, S.; Reis, R.M.; Baltazar, F.; Martinho, O. Metabolic alterations underlying Bevacizumab therapy in glioblastoma cells. Oncotarget 2017, 8, 103657–103670. [Google Scholar] [CrossRef] [PubMed]
- Marziali, G.; Signore, M.; Buccarelli, M.; Grande, S.; Palma, A.; Biffoni, M.; Rosi, A.; D’Alessandris, Q.G.; Martini, M.; Larocca, L.M.; et al. Metabolic/proteomic signature defines two glioblastoma subtypes with different clinical outcome. Sci. Rep. 2016, 6, 21557. [Google Scholar] [CrossRef] [PubMed]
- Caragher, S.P.; Sachdev, S.; Ahmed, A. Radiotherapy and Glioma Stem Cells: Searching for Chinks in Cellular Armor. Curr. Stem Cell. Rep. 2017, 3, 348–357. [Google Scholar] [CrossRef]
- Allen, M.; Bjerke, M.; Edlund, H.; Nelander, S.; Westermark, B. Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 2016, 8, 354re353. [Google Scholar] [CrossRef]
- Ledur, P.F.; Onzi, G.R.; Zong, H.; Lenz, G. Culture conditions defining glioblastoma cells behavior: What is the impact for novel discoveries? Oncotarget 2017, 8, 69185–69197. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zschenker, O.; Streichert, T.; Hehlgans, S.; Cordes, N. Genome-wide gene expression analysis in cancer cells reveals 3D growth to affect ECM and processes associated with cell adhesion but not DNA repair. PLoS ONE 2012, 7, e34279. [Google Scholar] [CrossRef] [PubMed]
- Luca, A.C.; Mersch, S.; Deenen, R.; Schmidt, S.; Messner, I.; Schafer, K.L.; Baldus, S.E.; Huckenbeck, W.; Piekorz, R.P.; Knoefel, W.T.; et al. Impact of the 3D Microenvironment on Phenotype, Gene Expression, and EGFR Inhibition of Colorectal Cancer Cell Lines. PLoS ONE 2013, 8, e59689. [Google Scholar] [CrossRef] [PubMed]
- Pontes Soares, C.; Midlej, V.; de Oliveira, M.E.W.; Benchimol, M.; Costa, M.L.; Mermelstein, C. 2D and 3D-organized cardiac cells shows differences in cellular morphology, adhesion junctions, presence of myofibrils and protein expression. PLoS ONE 2012, 7, e38147. [Google Scholar]
- Storch, K.; Eke, I.; Borgmann, K.; Krause, M.; Richter, C.; Becker, K.; Schrock, E.; Cordes, N. Three-dimensional cell growth confers radioresistance by chromatin density modification. Cancer Res. 2010, 70, 3925–3934. [Google Scholar] [CrossRef]
- Hehlgans, S.; Lange, I.; Eke, I.; Cordes, N. 3D cell cultures of human head and neck squamous cell carcinoma cells are radiosensitized by the focal adhesion kinase inhibitor TAE226. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2009, 92, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Poschau, M.; Dickreuter, E.; Singh-Muller, J.; Zscheppang, K.; Eke, I.; Liersch, T.; Cordes, N. EGFR and beta1-integrin targeting differentially affect colorectal carcinoma cell radiosensitivity and invasion. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2015, 116, 510–516. [Google Scholar] [CrossRef]
- Gomez-Roman, N.; Stevenson, K.; Gilmour, L.; Hamilton, G.; Chalmers, A.J. A novel 3D human glioblastoma cell culture system for modeling drug and radiation responses. Neuro Oncol. 2017, 19, 229–241. [Google Scholar] [CrossRef]
- Rich, J.N.; Reardon, D.A.; Peery, T.; Dowell, J.M.; Quinn, J.A.; Penne, K.L.; Wikstrand, C.J.; Van Duyn, L.B.; Dancey, J.E.; McLendon, R.E.; et al. Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol. 2004, 22, 133–142. [Google Scholar] [CrossRef]
- Prados, M.D.; Schold Sc, S.C., Jr; Fine, H.A.; Jaeckle, K.; Hochberg, F.; Mechtler, L.; Fetell, M.R.; Phuphanich, S.; Feun, L.; Janus, T.J.; et al. A randomized, double-blind, placebo-controlled, phase 2 study of RMP-7 in combination with carboplatin administered intravenously for the treatment of recurrent malignant glioma. Neuro Oncol. 2003, 5, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Giannini, C.; Sarkaria, J.N.; Saito, A.; Uhm, J.H.; Galanis, E.; Carlson, B.L.; Schroeder, M.A.; James, C.D. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 2005, 7, 164–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fael Al-Mayhani, T.M.; Ball, S.L.; Zhao, J.W.; Fawcett, J.; Ichimura, K.; Collins, P.V.; Watts, C. An efficient method for derivation and propagation of glioblastoma cell lines that conserves the molecular profile of their original tumours. J. Neurosci. Methods 2009, 176, 192–199. [Google Scholar] [CrossRef]
- Buckley, R.C. Tissue Culture Studies of the Glioblastoma Multiforme. Am. J. Pathol. 1929, 5, 467–472.5. [Google Scholar] [PubMed]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z.; et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 2018, 22, 514–528.e5. [Google Scholar] [CrossRef]
- Lee, G.; Auffinger, B.; Guo, D.; Hasan, T.; Deheeger, M.; Tobias, A.L.; Kim, J.Y.; Atashi, F.; Zhang, L.; Lesniak, M.S.; et al. Dedifferentiation of Glioma Cells to Glioma Stem-like Cells By Therapeutic Stress-induced HIF Signaling in the Recurrent GBM Model. Mol. Cancer Ther. 2016, 15, 3064–3076. [Google Scholar] [CrossRef] [Green Version]
- Vlashi, E.; Chen, A.M.; Boyrie, S.; Yu, G.; Nguyen, A.; Brower, P.A.; Hess, C.B.; Pajonk, F. Radiation-Induced Dedifferentiation of Head and Neck Cancer Cells Into Cancer Stem Cells Depends on Human Papillomavirus Status. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 1198–1206. [Google Scholar] [CrossRef] [Green Version]
- Olmez, I.; Shen, W.; McDonald, H.; Ozpolat, B. Dedifferentiation of patient-derived glioblastoma multiforme cell lines results in a cancer stem cell-like state with mitogen-independent growth. J. Cell. Mol. Med. 2015, 19, 1262–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahan, P.; Martinez Gala, J.; Delmas, C.; Monferran, S.; Malric, L.; Zentkowski, D.; Lubrano, V.; Toulas, C.; Cohen-Jonathan Moyal, E.; Lemarie, A. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: Possible involvement in radioresistance. Cell Death Dis. 2014, 5, e1543. [Google Scholar] [CrossRef] [PubMed]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef] [PubMed]
- Pavlyukov, M.S.; Yu, H.; Bastola, S.; Minata, M.; Shender, V.O.; Lee, Y.; Zhang, S.; Wang, J.; Komarova, S.; Wang, J.; et al. Apoptotic Cell-Derived Extracellular Vesicles Promote Malignancy of Glioblastoma Via Intercellular Transfer of Splicing Factors. Cancer Cell 2018, 34, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romero, N.; Gonzalez-Tejedo, C.; Carrion-Navarro, J.; Esteban-Rubio, S.; Rackov, G.; Rodriguez-Fanjul, V.; Oliver-De La Cruz, J.; Prat-Acin, R.; Peris-Celda, M.; Blesa, D.; et al. Cancer stem cells from human glioblastoma resemble but do not mimic original tumors after in vitro passaging in serum-free media. Oncotarget 2016, 7, 65888–65901. [Google Scholar] [CrossRef]
- Ben-David, U.; Ha, G.; Tseng, Y.Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [Green Version]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug. Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell. 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milwid, R.M.; Frascoli, F.; Steben, M.; Heffernan, J.M. HPV Screening and Vaccination Strategies in an Unscreened Population: A Mathematical Modeling Study. Bull. Math. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Brain extracellular matrix. Glycobiology 1996, 6, 489–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, T.J.; De Leon, E.; Griffin, K.R.; Stringer, B.W.; Day, B.W.; Fabry, B.; Cooper-White, J.; O’Neill, G.M. Differential response of patient-derived primary glioblastoma cells to environmental stiffness. Sci. Rep. 2016, 6, 23353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miroshnikova, Y.A.; Mouw, J.K.; Barnes, J.M.; Pickup, M.W.; Lakins, J.N.; Kim, Y.; Lobo, K.; Persson, A.I.; Reis, G.F.; McKnight, T.R.; et al. Tissue mechanics promote IDH1-dependent HIF1alpha-tenascin C feedback to regulate glioblastoma aggression. Nat. Cell. Biol. 2016, 18, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Bellail, A.C.; Hunter, S.B.; Brat, D.J.; Tan, C.; Van Meir, E.G. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell Biol. 2004, 36, 1046–1069. [Google Scholar] [CrossRef]
- Sunkin, S.M.; Ng, L.; Lau, C.; Dolbeare, T.; Gilbert, T.L.; Thompson, C.L.; Hawrylycz, M.; Dang, C. Allen Brain Atlas: An integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 2013, 41, D996–D1008. [Google Scholar] [CrossRef]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebelt, A.; Dzieciol, J.; Guzinska-Ustymowicz, K.; Lemancewicz, D.; Zimnoch, L.; Czykier, E. Angiogenesis in gliomas. Folia Histochem. Cytobiol. 2008, 46, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Spence, A.M.; Muzi, M.; Swanson, K.R.; O’Sullivan, F.; Rockhill, J.K.; Rajendran, J.G.; Adamsen, T.C.; Link, J.M.; Swanson, P.E.; Yagle, K.J.; et al. Regional hypoxia in glioblastoma multiforme quantified with [18F]fluoromisonidazole positron emission tomography before radiotherapy: Correlation with time to progression and survival. Clin. Cancer Res. 2008, 14, 2623–2630. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.M.; Judy, K.D.; Dunphy, I.; Jenkins, W.T.; Nelson, P.T.; Collins, R.; Wileyto, E.P.; Jenkins, K.; Hahn, S.M.; Stevens, C.W.; et al. Comparative measurements of hypoxia in human brain tumors using needle electrodes and EF5 binding. Cancer Res. 2004, 64, 1886–1892. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, S.M.; Judy, K.D.; Dunphy, I.; Jenkins, W.T.; Hwang, W.T.; Nelson, P.T.; Lustig, R.A.; Jenkins, K.; Magarelli, D.P.; Hahn, S.M.; et al. Hypoxia is important in the biology and aggression of human glial brain tumors. Clin. Cancer Res. 2004, 10, 8177–8184. [Google Scholar] [CrossRef]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-Driven Glycolysis Is a Therapeutic Target in Glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef]
- Egler, V.; Korur, S.; Failly, M.; Boulay, J.L.; Imber, R.; Lino, M.M.; Merlo, A. Histone deacetylase inhibition and blockade of the glycolytic pathway synergistically induce glioblastoma cell death. Clin. Cancer Res. 2008, 14, 3132–3140. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef]
- Criego, A.B.; Tkac, I.; Kumar, A.; Thomas, W.; Gruetter, R.; Seaquist, E.R. Brain glucose concentrations in healthy humans subjected to recurrent hypoglycemia. J. Neurosci. Res. 2005, 82, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, K.; Boyd, N.H.; Anderson, J.C.; Willey, C.D.; Hjelmeland, A.B. Kinomic profiling of glioblastoma cells reveals PLCG1 as a target in restricted glucose. Biomark. Res. 2018, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volterra, A.; Meldolesi, J. Astrocytes, from brain glue to communication elements: The revolution continues. Nat. Rev. Neurosci. 2005, 6, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Villa, G.R.; Hulce, J.J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.L.; Gu, Y.; Lum, K.M.; Masui, K.; Yang, H.; et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, B.H.; Fair, J.M.; Jamal, M.; Camphausen, K.; Tofilon, P.J. Astrocytes enhance the invasion potential of glioblastoma stem-like cells. PLoS ONE 2013, 8, e54752. [Google Scholar] [CrossRef]
- Rath, B.H.; Wahba, A.; Camphausen, K.; Tofilon, P.J. Coculture with astrocytes reduces the radiosensitivity of glioblastoma stem-like cells and identifies additional targets for radiosensitization. Cancer Med. 2015, 4, 1705–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashima, G.; Suzuki, R.; Asai, J.; Fujimoto, T. Immunohistochemical analysis of reactive astrocytes around glioblastoma: An immunohistochemical study of postmortem glioblastoma cases. Clin. Neurol. Neurosurg. 2002, 104, 125–131. [Google Scholar] [CrossRef]
- Okolie, O.; Bago, J.R.; Schmid, R.S.; Irvin, D.M.; Bash, R.E.; Miller, C.R.; Hingtgen, S.D. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro Oncol. 2016, 18, 1622–1633. [Google Scholar] [CrossRef] [Green Version]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E.; et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156, 1002–1016. [Google Scholar] [CrossRef]
- Purves, D.; Augustine, G.J. Neuroscience, 5th ed.; Sinauer Associates: Sunderland, MA, USA, 2012; pp. xvi; 759p. [Google Scholar]
- Pinheiro, P.S.; Mulle, C. Presynaptic glutamate receptors: Physiological functions and mechanisms of action. Nat. Rev. Neurosci. 2008, 9, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Barres, B.A. Neuroscience: Glia—More than just brain glue. Nature 2009, 457, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef] [Green Version]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schunemann, D.P.; Grivicich, I.; Regner, A.; Leal, L.F.; de Araujo, D.R.; Jotz, G.P.; Fedrigo, C.A.; Simon, D.; da Rocha, A.B. Glutamate promotes cell growth by EGFR signaling on U-87MG human glioblastoma cell line. Pathol. Oncol. Res. 2010, 16, 285–293. [Google Scholar] [CrossRef]
- Caragher, S.P.; Hall, R.R., 3rd; Ahsan, R.; Ahmed, A.U. Monoamines in Glioblastoma: Complex biology with therapeutic potential. Neuro Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592. [Google Scholar] [CrossRef]
- Folkins, C.; Shaked, Y.; Man, S.; Tang, T.; Lee, C.R.; Zhu, Z.; Hoffman, R.M.; Kerbel, R.S. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res. 2009, 69, 7243–7251. [Google Scholar] [CrossRef]
- Plate, K.H.; Breier, G.; Weich, H.A.; Mennel, H.D.; Risau, W. Vascular endothelial growth factor and glioma angiogenesis: Coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int. J. Cancer 1994, 59, 520–529. [Google Scholar] [CrossRef]
- Plate, K.H.; Breier, G.; Weich, H.A.; Risau, W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Hovinga, K.E.; Stalpers, L.J.; van Bree, C.; Donker, M.; Verhoeff, J.J.; Rodermond, H.M.; Bosch, D.A.; van Furth, W.R. Radiation-enhanced vascular endothelial growth factor (VEGF) secretion in glioblastoma multiforme cell lines—A clue to radioresistance? J. Neurooncol. 2005, 74, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Penfield, W. Microglia and the Process of Phagocytosis in Gliomas. Am. J. Pathol. 1925, 1, 77–90.15. [Google Scholar] [PubMed]
- Markovic, D.S.; Glass, R.; Synowitz, M.; Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska, A.; Kwiatkowska, A.; Slomnicki, L.; Dembinski, M.; Master, A.; Sliwa, M.; Franciszkiewicz, K.; Chouaib, S.; Kaminska, B. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion—An inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene 2008, 27, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Zhai, H.; Heppner, F.L.; Tsirka, S.E. Microglia/macrophages promote glioma progression. Glia 2011, 59, 472–485. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Tunici, P.; Bissola, L.; Lualdi, E.; Pollo, B.; Cajola, L.; Broggi, G.; Sozzi, G.; Finocchiaro, G. Genetic alterations and in vivo tumorigenicity of neurospheres derived from an adult glioblastoma. Mol. Cancer 2004, 3, 25. [Google Scholar] [CrossRef]
- Bjerkvig, R.; Tonnesen, A.; Laerum, O.D.; Backlund, E.O. Multicellular tumor spheroids from human gliomas maintained in organ culture. J. Neurosurg. 1990, 72, 463–475. [Google Scholar] [CrossRef] [PubMed]
- De Witt Hamer, P.C.; Van Tilborg, A.A.; Eijk, P.P.; Sminia, P.; Troost, D.; Van Noorden, C.J.; Ylstra, B.; Leenstra, S. The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene 2008, 27, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Narayan, R.S.; Fedrigo, C.A.; Brands, E.; Dik, R.; Stalpers, L.J.; Baumert, B.G.; Slotman, B.J.; Westerman, B.A.; Peters, G.J.; Sminia, P. The allosteric AKT inhibitor MK2206 shows a synergistic interaction with chemotherapy and radiotherapy in glioblastoma spheroid cultures. BMC Cancer 2017, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.S.; Aaberg-Jessen, C.; Andersen, C.; Schroder, H.D.; Kristensen, B.W. Glioma spheroids obtained via ultrasonic aspiration are viable and express stem cell markers: A new tissue resource for glioma research. Neurosurgery 2013, 73, 868–886, discussion 886. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lin, H.; Rauch, J.; Deleyrolle, L.P.; Reynolds, B.A.; Viljoen, H.J.; Zhang, C.; Zhang, C.; Gu, L.; Van Wyk, E.; et al. Scalable Culturing of Primary Human Glioblastoma Tumor-Initiating Cells with a Cell-Friendly Culture System. Sci. Rep. 2018, 8, 3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisemann, T.; Costa, B.; Strelau, J.; Mittelbronn, M.; Angel, P.; Peterziel, H. An advanced glioma cell invasion assay based on organotypic brain slice cultures. BMC Cancer 2018, 18, 103. [Google Scholar] [CrossRef] [PubMed]
- Sliwa, M.; Markovic, D.; Gabrusiewicz, K.; Synowitz, M.; Glass, R.; Zawadzka, M.; Wesolowska, A.; Kettenmann, H.; Kaminska, B. The invasion promoting effect of microglia on glioblastoma cells is inhibited by cyclosporin A. Brain 2007, 130, 476–489. [Google Scholar] [CrossRef] [Green Version]
- Marques-Torrejon, M.A.; Gangoso, E.; Pollard, S.M. Modelling glioblastoma tumour-host cell interactions using adult brain organotypic slice co-culture. Dis. Model. Mech. 2018, 11, dmm031435. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [PubMed]
- Kaaijk, P.; Troost, D.; Sminia, P.; Hulshof, M.C.; van der Kracht, A.H.; Leenstra, S.; Bosch, D.A. Hypofractionated radiation induces a decrease in cell proliferation but no histological damage to organotypic multicellular spheroids of human glioblastomas. Eur. J. Cancer 1997, 33, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Sminia, P.; Acker, H.; Eikesdal, H.P.; Kaaijk, P.; Enger, P.; Slotman, B.; Bjerkvig, R. Oxygenation and response to irradiation of organotypic multicellular spheroids of human glioma. Anticancer Res. 2003, 23, 1461–1466. [Google Scholar] [PubMed]
- De Witt Hamer, P.C.; Leenstra, S.; Van Noorden, C.J.; Zwinderman, A.H. Organotypic glioma spheroids for screening of experimental therapies: How many spheroids and sections are required? Cytometry A 2009, 75, 528–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedron, S.; Becka, E.; Harley, B.A. Regulation of glioma cell phenotype in 3D matrices by hyaluronic acid. Biomaterials 2013, 34, 7408–7417. [Google Scholar] [CrossRef] [PubMed]
- Pedron, S.; Becka, E.; Harley, B.A. Spatially gradated hydrogel platform as a 3D engineered tumor microenvironment. Adv. Mater. 2015, 27, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Florczyk, S.J.; Wang, K.; Jana, S.; Wood, D.L.; Sytsma, S.K.; Sham, J.; Kievit, F.M.; Zhang, M. Porous chitosan-hyaluronic acid scaffolds as a mimic of glioblastoma microenvironment ECM. Biomaterials 2013, 34, 10143–10150. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.S.; Dejesus, J.; Short, A.R.; Otero, J.J.; Sarkar, A.; Winter, J.O. Glioblastoma behaviors in three-dimensional collagen-hyaluronan composite hydrogels. ACS Appl. Mater. Interfaces 2013, 5, 9276–9284. [Google Scholar] [CrossRef]
- Sarkar, S.; Nuttall, R.K.; Liu, S.; Edwards, D.R.; Yong, V.W. Tenascin-C stimulates glioma cell invasion through matrix metalloproteinase-12. Cancer Res. 2006, 66, 11771–11780. [Google Scholar] [CrossRef]
- Xiao, W.; Sohrabi, A.; Seidlits, S.K. Integrating the glioblastoma microenvironment into engineered experimental models. Future Sci. OA 2017, 3, FSO189. [Google Scholar] [CrossRef]
- Hughes, C.S.; Postovit, L.M.; Lajoie, G.A. Matrigel: A complex protein mixture required for optimal growth of cell culture. Proteomics 2010, 10, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hanggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. 2017, 19, 1316–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mennens, S.F.B.; Bolomini-Vittori, M.; Weiden, J.; Joosten, B.; Cambi, A.; van den Dries, K. Substrate stiffness influences phenotype and function of human antigen-presenting dendritic cells. Sci. Rep. 2017, 7, 17511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venugopal, B.; Mogha, P.; Dhawan, J.; Majumder, A. Cell density overrides the effect of substrate stiffness on human mesenchymal stem cells’ morphology and proliferation. Biomater. Sci. 2018, 6, 1109–1119. [Google Scholar] [CrossRef] [Green Version]
- Lv, D.; Yu, S.C.; Ping, Y.F.; Wu, H.; Zhao, X.; Zhang, H.; Cui, Y.; Chen, B.; Zhang, X.; Dai, J.; et al. A three-dimensional collagen scaffold cell culture system for screening anti-glioma therapeutics. Oncotarget 2016, 7, 56904–56914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, A.N.; Anderson, J.C.; Duarte, C.W.; Shevin, R.S.; Langford, C.P.; Singh, R.; Gillespie, G.Y.; Willey, C.D. Combinatorial Drug Testing in 3D Microtumors Derived from GBM Patient-Derived Xenografts Reveals Cytotoxic Synergy in Pharmacokinomics-informed Pathway Interactions. Sci. Rep. 2018, 8, 8412. [Google Scholar] [CrossRef]
- Rape, A.; Ananthanarayanan, B.; Kumar, S. Engineering strategies to mimic the glioblastoma microenvironment. Adv. Drug. Deliv. Rev. 2014, 79–80, 172–183. [Google Scholar] [CrossRef]
- Ananthanarayanan, B.; Kim, Y.; Kumar, S. Elucidating the mechanobiology of malignant brain tumors using a brain matrix-mimetic hyaluronic acid hydrogel platform. Biomaterials 2011, 32, 7913–7923. [Google Scholar] [CrossRef] [Green Version]
- Jiguet Jiglaire, C.; Baeza-Kallee, N.; Denicolai, E.; Barets, D.; Metellus, P.; Padovani, L.; Chinot, O.; Figarella-Branger, D.; Fernandez, C. Ex vivo cultures of glioblastoma in three-dimensional hydrogel maintain the original tumor growth behavior and are suitable for preclinical drug and radiation sensitivity screening. Exp. Cell Res. 2014, 321, 99–108. [Google Scholar] [CrossRef]
- Wang, C.; Tong, X.; Yang, F. Bioengineered 3D brain tumor model to elucidate the effects of matrix stiffness on glioblastoma cell behavior using PEG-based hydrogels. Mol. Pharm. 2014, 11, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.E.; Pedron, S.; Harley, B.A.C. The Combined Influence of Hydrogel Stiffness and Matrix-Bound Hyaluronic Acid Content on Glioblastoma Invasion. Macromol. Biosci. 2017, 17, 1700018. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Perez, M.; Voytik-Harbin, S.L.; Rickus, J.L. Extracellular Matrix Properties Regulate the Migratory Response of Glioblastoma Stem Cells in Three-Dimensional Culture. Tissue Eng. Part A 2015, 21, 2572–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heffernan, J.M.; Overstreet, D.J.; Le, L.D.; Vernon, B.L.; Sirianni, R.W. Bioengineered Scaffolds for 3D Analysis of Glioblastoma Proliferation and Invasion. Ann. Biomed. Eng. 2015, 43, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Pedron, S.; Hanselman, J.S.; Schroeder, M.A.; Sarkaria, J.N.; Harley, B.A.C. Extracellular Hyaluronic Acid Influences the Efficacy of EGFR Tyrosine Kinase Inhibitors in a Biomaterial Model of Glioblastoma. Adv. Healthc. Mater. 2017, 6, 1700529. [Google Scholar] [CrossRef]
- Xiao, W.; Zhang, R.; Sohrabi, A.; Ehsanipour, A.; Sun, S.; Liang, J.; Walthers, C.M.; Ta, L.; Nathanson, D.A.; Seidlits, S.K. Brain-Mimetic 3D Culture Platforms Allow Investigation of Cooperative Effects of Extracellular Matrix Features on Therapeutic Resistance in Glioblastoma. Cancer Res. 2018, 78, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Akay, M.; Hite, J.; Avci, N.G.; Fan, Y.; Akay, Y.; Lu, G.; Zhu, J.J. Drug Screening of Human GBM Spheroids in Brain Cancer Chip. Sci. Rep. 2018, 8, 15423. [Google Scholar] [CrossRef] [PubMed]
- Plenz, D.; Kitai, S.T. Up and down states in striatal medium spiny neurons simultaneously recorded with spontaneous activity in fast-spiking interneurons studied in cortex-striatum-substantia nigra organotypic cultures. J. Neurosci. 1998, 18, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Bershteyn, M.; Kriegstein, A.R. Cerebral organoids in a dish: Progress and prospects. Cell 2013, 155, 19–20. [Google Scholar] [CrossRef]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wang, L.; Yu, H.; Yin, F.; Wang, Y.; Liu, H.; Jiang, L.; Qin, J. In situ generation of human brain organoids on a micropillar array. Lab Chip 2017, 17, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Quadrato, G.; Brown, J.; Arlotta, P. The promises and challenges of human brain organoids as models of neuropsychiatric disease. Nat. Med. 2016, 22, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Deeken, J.F.; Loscher, W. The blood-brain barrier and cancer: Transporters, treatment, and Trojan horses. Clin. Cancer Res. 2007, 13, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Broniscer, A.; Panetta, J.C.; O’Shaughnessy, M.; Fraga, C.; Bai, F.; Krasin, M.J.; Gajjar, A.; Stewart, C.F. Plasma and cerebrospinal fluid pharmacokinetics of erlotinib and its active metabolite OSI-420. Clin. Cancer Res. 2007, 13, 1511–1515. [Google Scholar] [CrossRef] [PubMed]
- Stone, H.B.; Bernhard, E.J.; Coleman, C.N.; Deye, J.; Capala, J.; Mitchell, J.B.; Brown, J.M. Preclinical Data on Efficacy of 10 Drug-Radiation Combinations: Evaluations, Concerns, and Recommendations. Transl. Oncol. 2016, 9, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Novel System | Summary | Pros | Cons | Key References |
|---|---|---|---|---|
| Matrigel Plugs | Tumor cells are grow embedded in a 3D matrigel | Allow cells to grow in 3D Better approximation of the stiffness of the brain ECM ECM-derived growth factors better mirror how GFs are available to the tumor | Mono-culture Sarcoma ECM is not equivalent to the brain ECM (elevated collagen and laminin) | [15] |
| Neurospheres | Cells are grown in suspension, often in neural stem cell promoting media | Allow cells to grow in 3D Ensure that cells examined are tumorigenic, likely the key target for anti-glioma therapy | Mono-culture | [91] |
| Organotypic glioma spheroids | Cells from patient resections, grown in 3D cultures | Multiple cells types (including macrophages and endothelial cells) Maintenance of genetic hierarchy within tumor cells | Low throughput ££ | [92,93,94,95] |
| 3D Scaffolds | Cells grow in a matrix, allowing for 3D cell interactions | Allow cells to grow in 3D Easily scaled to high-throughput drug screening Can include brain-ECM specific components Better recapitulation of gene expression and invasion | Mono-culture Oxygen and nutrient availability is consistent across the entire population ££ | [18] |
| Microfluidic Systems | Cells are grown in hydrogel tubes, which are filled with circulating media | Time-dependent exposure to nutrients/more dynamic micronvironment Allow for the generation of many GSCs from a small initial sample | Mono-culture ££ | [96] |
| Brain Slices | Tumor cells are implanted into mouse brains | Expose tumor to all the neighboring non-tumor cells Great for examining invasion in the presence of white matter and secreted factors | Implantation is always problematic Mouse host with human cells Lack of circulation | [97,98,99] |
| Mini-brains (transduced) | Cerebral organoids are genetically modified to generate tumors | Expose tumor to all the neighboring non-tumor cells Real-time live cell imaging compatible Allow for examination of tumor initiation and early progression | ££ Time requirements Lack of vessel formation/endothelial cells Currently lack microglia Genetic engerieed tumors inherently involve selecting a driving mutation Not clear which cell type is being transduced during electroporation Still rely on matrigel seed | [100] |
| Mini-brains (implanted) | Cerebral organoids, once established, have patient derived cell injected into the cortex | High potential for personalization and precision medicine Human to human graft | ££ Time requirements Lack of vessel formation/endothelial cells Currently lack microglia Still rely on matrigel seed | [100,101] |
| Tumor Organoids | Tumor cells are used as the starting point for generation of a “cerebral” organoid | Maintain nutrient and oxygen gradients Maintain regional intratumoral heterogeneity Appear to promote preservation of cellular phenotype | Mono-culture ££ Time requirements Still rely on matrigel seed | [102] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caragher, S.; Chalmers, A.J.; Gomez-Roman, N. Glioblastoma’s Next Top Model: Novel Culture Systems for Brain Cancer Radiotherapy Research. Cancers 2019, 11, 44. https://doi.org/10.3390/cancers11010044
Caragher S, Chalmers AJ, Gomez-Roman N. Glioblastoma’s Next Top Model: Novel Culture Systems for Brain Cancer Radiotherapy Research. Cancers. 2019; 11(1):44. https://doi.org/10.3390/cancers11010044
Chicago/Turabian StyleCaragher, Seamus, Anthony J. Chalmers, and Natividad Gomez-Roman. 2019. "Glioblastoma’s Next Top Model: Novel Culture Systems for Brain Cancer Radiotherapy Research" Cancers 11, no. 1: 44. https://doi.org/10.3390/cancers11010044
APA StyleCaragher, S., Chalmers, A. J., & Gomez-Roman, N. (2019). Glioblastoma’s Next Top Model: Novel Culture Systems for Brain Cancer Radiotherapy Research. Cancers, 11(1), 44. https://doi.org/10.3390/cancers11010044