Glioblastoma: Microenvironment and Niche Concept

 and
and 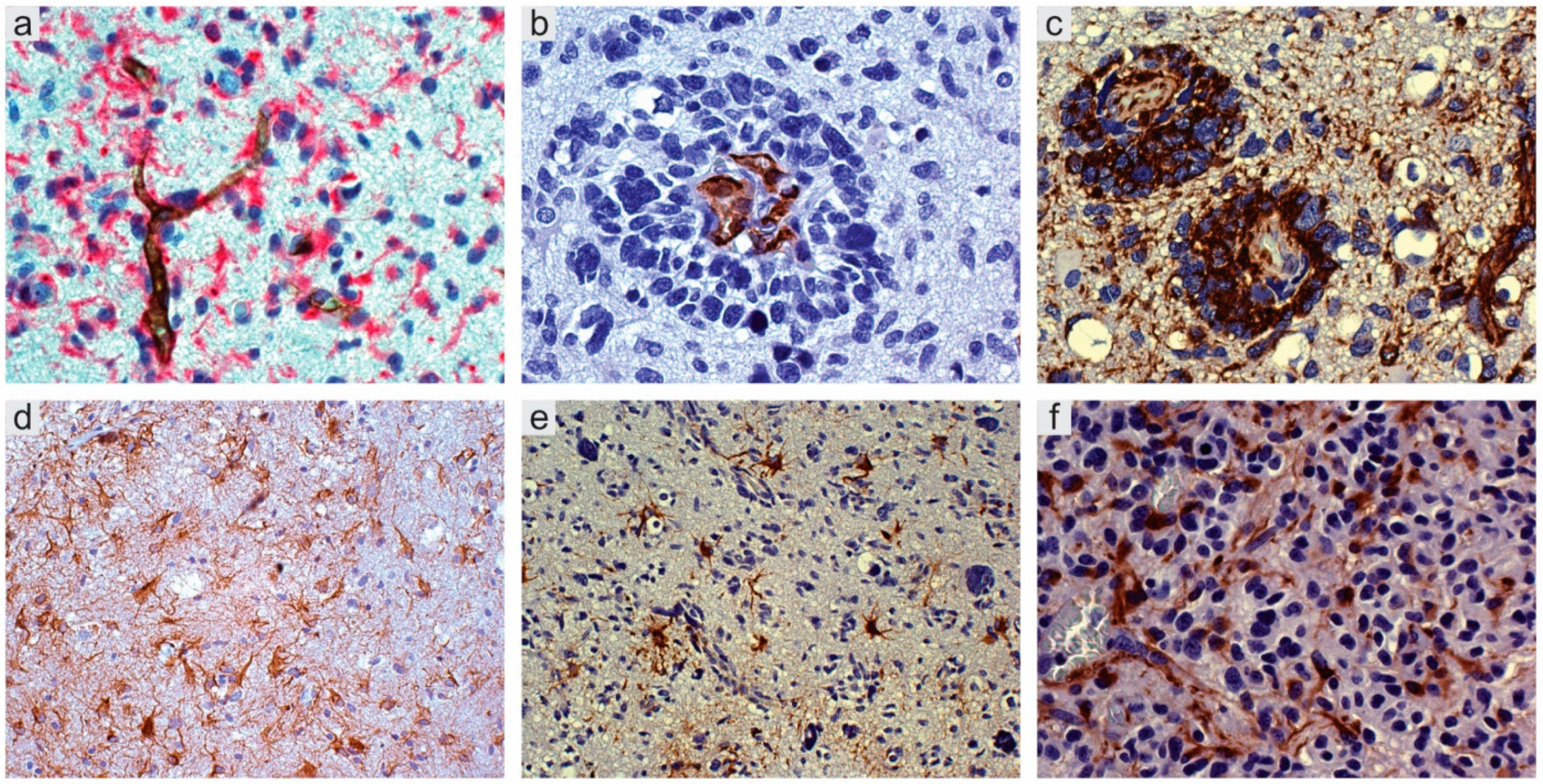{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
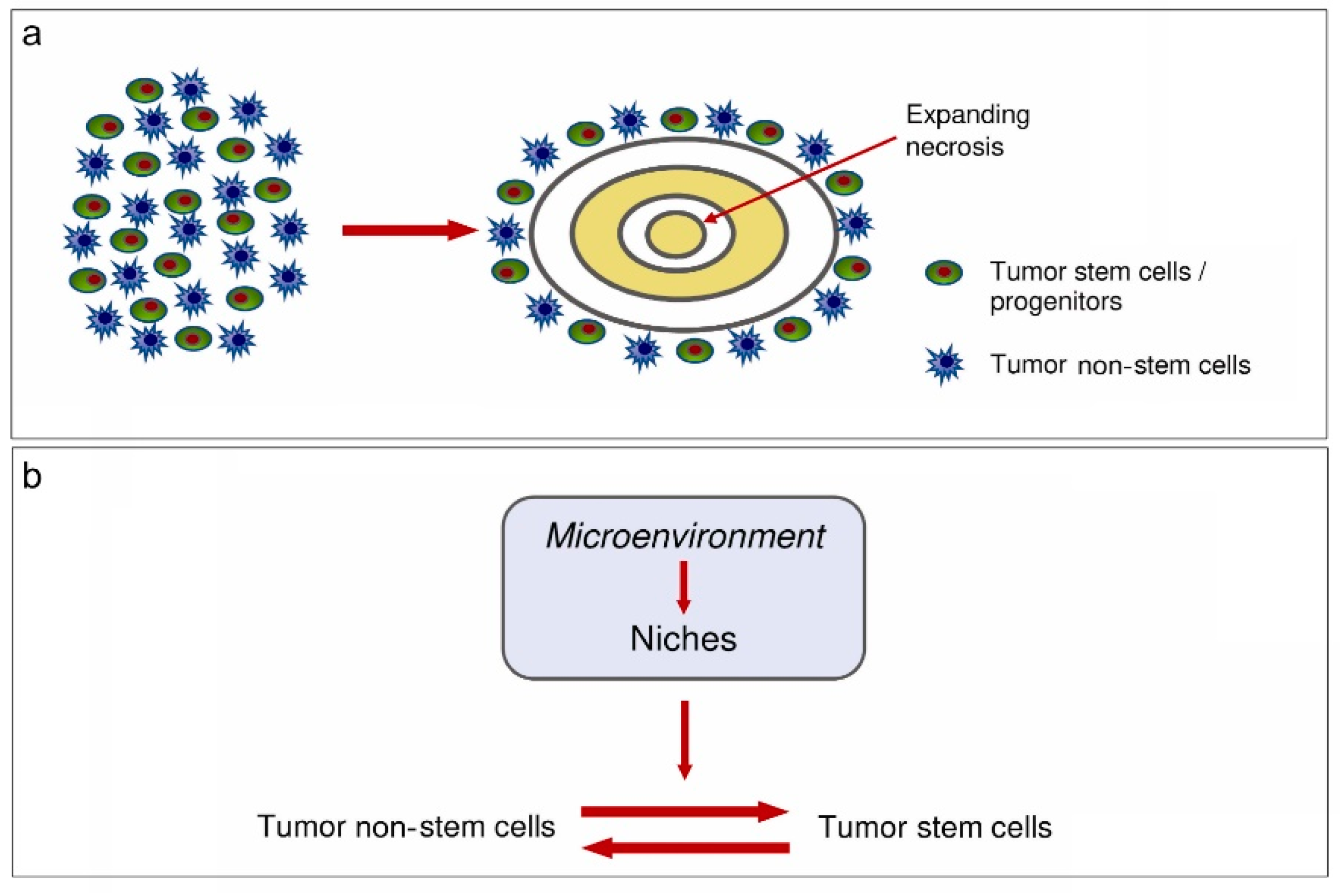
Abstract
:1. Introduction
2. Pathology of Niches
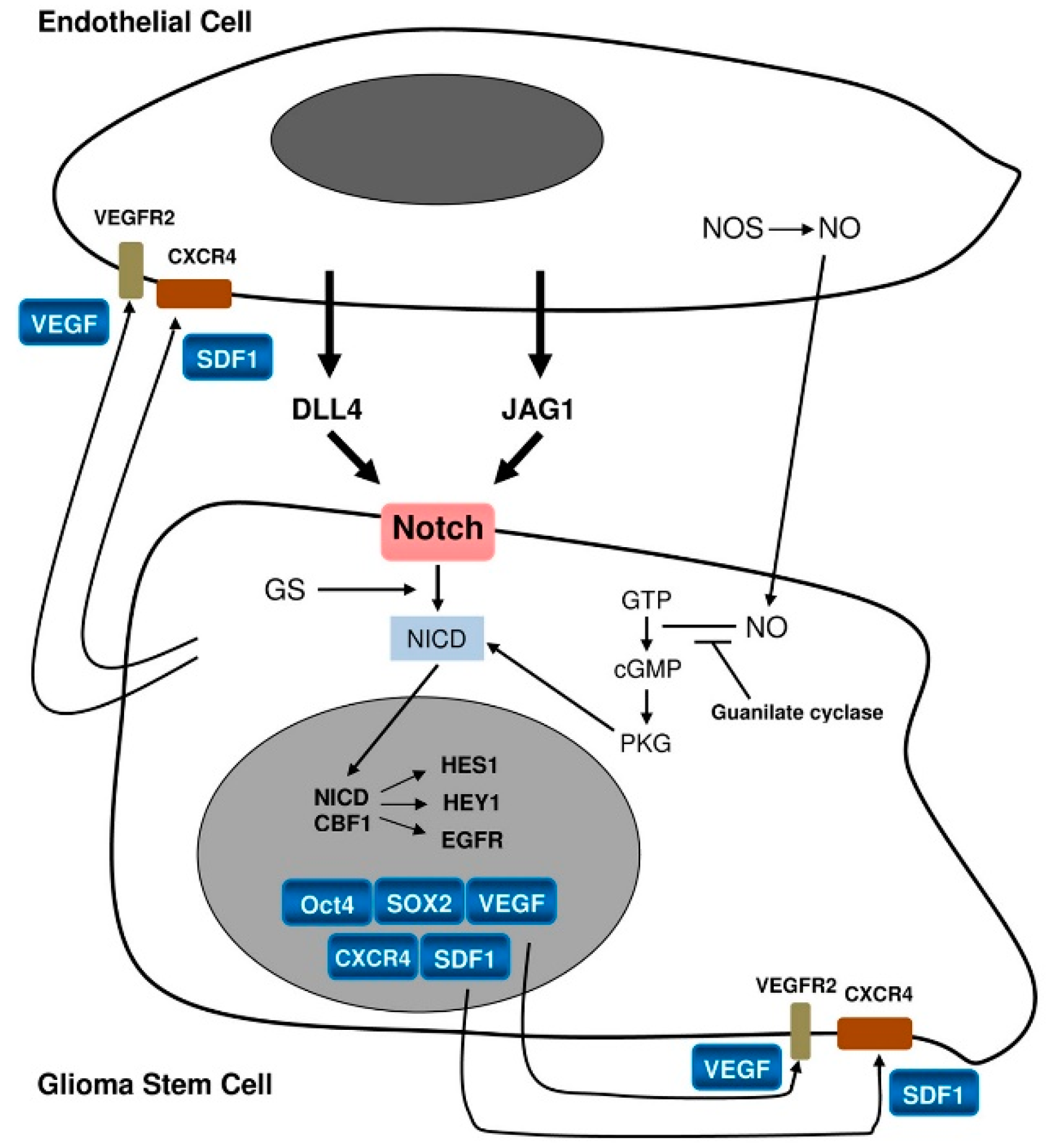
2.1. Perivascular Niches
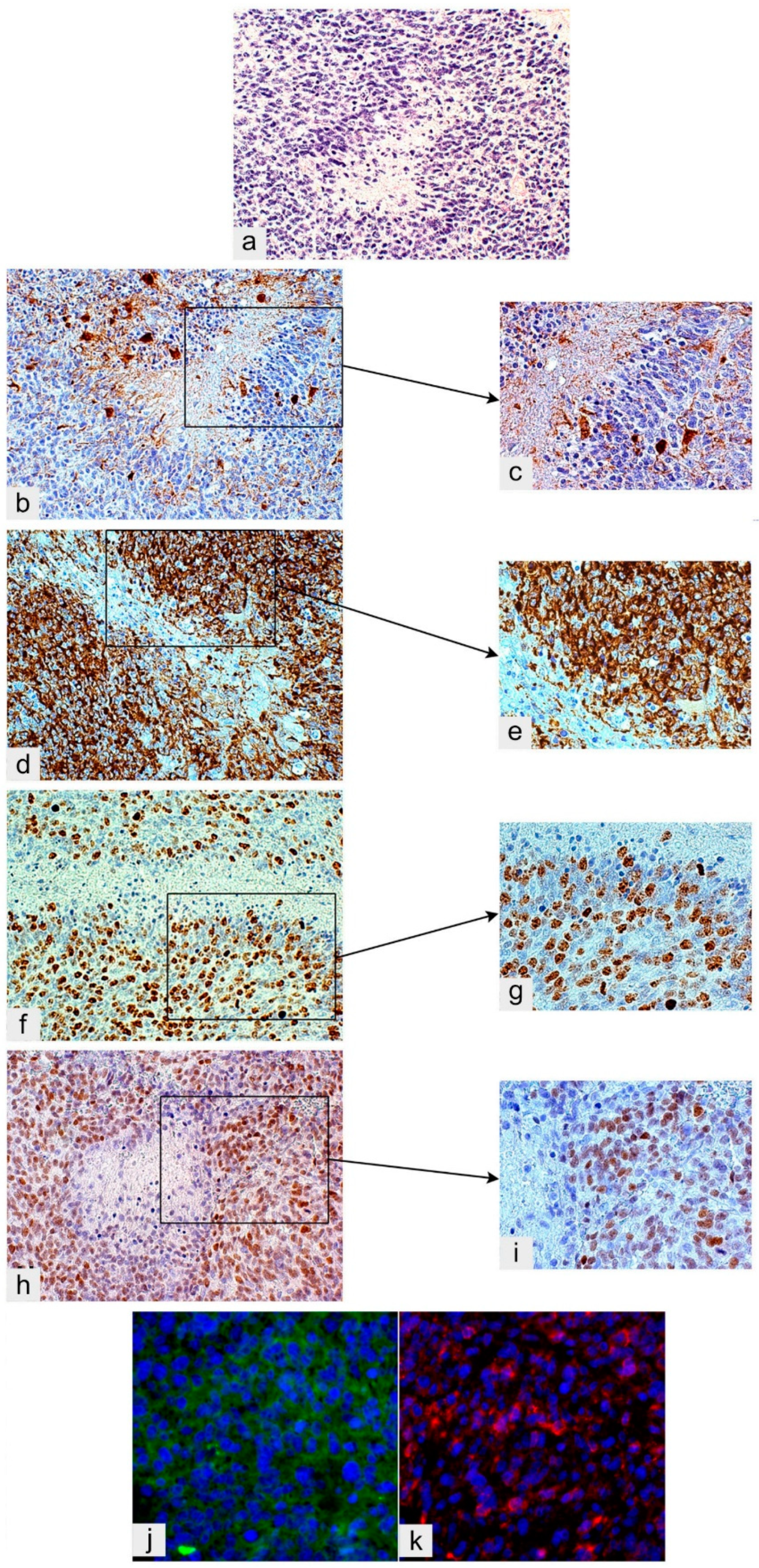
2.2. Perinecrotic Niches
3. Tumor Microenvironment (TME)
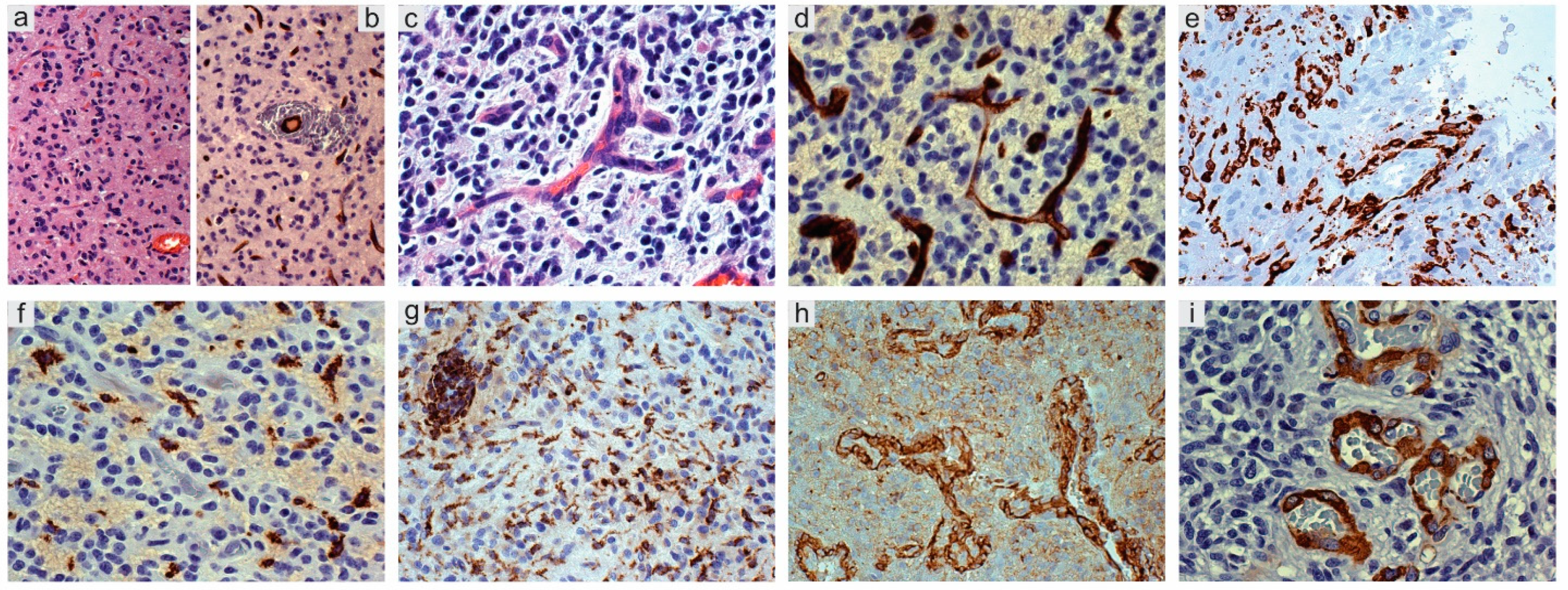
4. Glioma-Associated Microglia/Macrophages (GAMs)—Inflammatory Microenvironment
5. Pericytes
6. Reactive Astrocytes
7. Conclusions
Funding
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Holland, E.C. Progenitor cells and glioma formation. Curr. Opin. Neurol. 2001, 14, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Mellai, M.; Annovazzi, L.; Casalone, C.; Cassoni, P. Tumor Microenvironment: Perivascular and Perinecrotic Niches in Gliomas. In Molecular Considerations and Evolving Surgical Management Issues in the Treatment of Patients with a Brain Tumor, 1st ed.; Morgan, L.R., Ed.; InTech: Rjieka, Croatia, 2015; pp. 49–82. ISBN 978-953-51-2031-5. [Google Scholar]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Assanah, M.; Lochhead, R.; Ogden, A.; Bruce, J.; Goldman, J.J.; Canoll, P. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J. Neurosci. 2006, 26, 6781–6790. [Google Scholar] [CrossRef] [PubMed]
- Siebzehnrubl, F.A.; Reynolds, B.A.; Vescovi, A.; Steindler, D.A.; Deleyrolle, L.P. The origins of glioma: E Pluribus Unum? Glia 2011, 59, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.; Cadusseau, J.; Varlet, P.; Surena, A.L.; de Faria, G.P.; Dias-Morais, A.; Auger, N.; Léonard, N.; Daudigeos, E.; Dantas-Barbosa, C.; et al. Astrocytes reverted to a neural progenitor-like state with transforming growth factor alpha are sensitized to cancerous transformation. Stem Cells 2009, 27, 2373–2382. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.J.; Steindler, D.A. Common astrocytic programs during brain development, injury and cancer. Trends Neurosci. 2009, 32, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Buffo, A.; Rite, I.; Tripathi, P.; Lepier, A.; Colak, D.; Horn, A.P.; Mori, T.; Götz, M. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc. Natl. Acad. Sci. USA 2008, 105, 3581–3586. [Google Scholar] [CrossRef] [Green Version]
- Vescovi, A.L.; Galli, R.; Reynolds, B.A. Brain tumour stem cells. Nat. Rev. Cancer 2006, 6, 425–436. [Google Scholar] [CrossRef]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Gene Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, D.; Mellai, M.; Annovazzi, L.; Caldera, V.; Piazzi, A.; Denysenko, T.; Melcarne, A. Stem cell niches in glioblastoma: A neuropathological view. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Annovazzi, L.; Mazzucco, M.; Mellai, M. The microenvironment in gliomas: Phenotypic expressions. Cancers 2015, 7, 2352–2359. [Google Scholar] [CrossRef]
- Schiffer, D.; Annovazzi, L.; Mellai, M. A comprehensive view of tumor stem cells and their regulation by the microenvironment in glioblastoma. Neurol. Sci. 2017, 38, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Pallini, R.; Ricci-Vitiani, L.; Banna, G.L.; Signore, M.; Lombardi, D.; Todaro, M.; Stassi, G.; Martini, M.; Maira, G.; Larocca, L.M.; et al. Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 8205–8212. [Google Scholar] [CrossRef]
- Piccirillo, S.G.; Combi, R.; Cajola, L.; Patrizi, A.; Redaelli, S.; Bentivegna, A.; Baronchelli, S.; Maira, G.; Pollo, B.; Mangiola, A.; et al. Distinct pools of cancer stem-like cells coexist within human glioblastomas and display different tumorigenicity and independent genomic evolution. Oncogene 2009, 28, 1807–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Persano, L.; Della Puppa, A.; Frasson, C.; Sarto, E.; Scienza, R.; D’avella, D.; Basso, G.; et al. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells 2010, 28, 851–862. [Google Scholar] [CrossRef]
- Persano, L.; Rampazzo, E.; Della Puppa, A.; Pistollato, F.; Basso, G. The three-layer concentric model of glioblastoma: Cancer stem cells, microenvironmental regulation, and therapeutic implications. Sci. World J. 2011, 11, 1829–1841. [Google Scholar] [CrossRef] [PubMed]
- Valentini, M.C.; Mellai, M.; Annovazzi, L.; Melcarne, A.; Denysenko, T.; Cassoni, P.; Casalone, C.; Maurella, C.; Grifoni, S.; Fania, P.; et al. Comparison among conventional and advanced MRI, (18)F-FDG PET/CT, phenotype and genotype in glioblastoma. Oncotarget 2017, 8, 91636–91653. [Google Scholar] [CrossRef]
- Palmer, T.D.; Willhoite, A.R.; Gage, F.H. Vascular niche for adult hippocampal neurogenesis. J. Comp. Neurol. 2000, 425, 479–494. [Google Scholar] [CrossRef]
- Veeravagu, A.; Bababeygy, S.R.; Kalani, M.Y.; Hou, L.C.; Tse, V. The cancer stem cell-vascular niche complex in brain tumor formation. Stem Cells Dev. 2008, 17, 859–867. [Google Scholar] [CrossRef]
- Mirzadeh, Z.; Merkle, F.T.; Soriano-Navarro, M.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell 2008, 3, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Wang, Y.; Kokovay, E.; Lin, G.; Chuang, S.M.; Goderie, S.K.; Roysam, B.; Temple, S. Adult SVZ stem cells lie in a vascular niche: A quantitative analysis of niche cell-cell interactions. Cell Stem Cell 2008, 3, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Lorger, M. Tumor microenvironment in the brain. Cancers 2012, 4, 218–243. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Filatova, A.; Acker, T.; Garvalov, B.K. The cancer stem cell niche(s): The crosstalk between glioma stem cells and their microenvironment. Biochim. Biophys. Acta 2013, 1830, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Ho, I.A.W.; Shim, W.S.N. Contribution of the microenvironmental niche to glioblastoma heterogeneity. Biomed. Res. Int. 2017, 9634172, 13. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, D.; Mellai, M.; Bovio, E.; Bisogno, I.; Casalone, C.; Annovazzi, L. Glioblastoma niches: From the concept to the phenotypical reality. Neurol. Sci. 2018, 39, 1161–1168. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef] [PubMed]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef] [PubMed]
- Westphal, M.; Lamszus, K. The neurobiology of gliomas: From cell biology to the development of therapeutic approaches. Nat. Rev. Neurosci. 2011, 12, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, P.; Johansson, B.R.; Levéen, P.; Betsholtz, C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997, 277, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D. Brain Tumors. Biology, Pathology and Clinical References, 2nd ed.; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1997; pp. 1–695. [Google Scholar]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Rong, Y.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. ‘Pseudopalisading’ necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef]
- Brat, D.J.; Castellano-Sanchez, A.A.; Hunter, S.B.; Pecot, M.; Cohen, C.; Hammond, E.H.; Devi, S.N.; Kaur, B.; Van Meir, E.G. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004, 64, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Chiò, A.; Giordana, M.T.; Mauro, A.; Migheli, A.; Vigliani, M.C. The vascular response to tumor infiltration in malignant gliomas. Morphometric and reconstruction study. Acta Neuropathol. 1989, 77, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Kargiotis, O.; Rao, J.S.; Kyritsis, A.P. Mechanisms of angiogenesis in gliomas. J. Neurooncol. 2006, 78, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; Wormer, J.R.; Kakar, H.; Breznik, B.; van der Swaan, B.; Hulsbos, R.; Tigchelaar, W.; Tonar, Z.; Khurshed, M.; Molenaar, R.J.; et al. Periarteriolar Glioblastoma Stem Cell Niches Express Bone Marrow Hematopoietic Stem Cell Niche Proteins. J. Histochem. Cytochem. 2018, 66, 155–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aderetti, D.A.; Hira, V.V.V.; Molenaar, R.J.; van Noorden, C.J.F. The hypoxic peri-arteriolar glioma stem cell niche, an integrated concept of five types of niches in human glioblastoma. Biochim. Biophys. Acta 2018, 1869, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Breznik, B.; Limbaeck Stokin, C.; Kos, J.; Khurshed, M.; Hira, V.V.V.; Bošnjak, R.; Lah, T.T.; Van Noorden, C.J.F. Cysteine cathepsins B, X and K expression in peri-arteriolar glioblastoma stem cell niches. J. Mol. Histol. 2018, 49, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Annovazzi, L.; Mazzucco, M.; Mellai, M. The origin of circumscribed necroses and perinecrotic niches in glioblastoma multiforme: An additional hypothesis. Integr. Cancer Sci. Ther. 2015, 2, 75–78. [Google Scholar] [CrossRef]
- Christensen, K.; Schrøder, H.D.; Kristensen, B.W. CD133 identifies perivascular niches in grade II-IV astrocytomas. J. Neurooncol. 2008, 90, 157–170. [Google Scholar] [CrossRef]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; von Stechow, L.; Schänzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nistér, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef]
- Evans, S.M.; Judy, K.D.; Dunphy, I.; Jenkins, W.T.; Hwang, W.T.; Nelson, P.T.; Lustig, R.A.; Jenkins, K.; Magarelli, D.P.; Hahn, S.M.; et al. Hypoxia is important in the biology and aggression of human glial brain tumors. Clin. Cancer Res. 2004, 10, 8177–8184. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Becher, O.J.; Rosenblum, M.K.; Pandolfi, P.P.; Manova-Odorova, K.; Holland, E.C. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008, 22, 436–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Neaves, W.B. Normal stem cells and cancer stem cells: The niche matters. Cancer Res. 2006, 66, 4553–4557. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Lin, A.; Mahairaki, V.; Matsui, W.; Eberhart, C.G. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am. J. Pathol. 2010, 177, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.T.; Brem, S. Antiangiogenesis treatment for glioblastoma multiforme: Challenges and opportunities. J. Natl. Compr. Cancer Netw. 2008, 6, 515–522. [Google Scholar] [CrossRef]
- Fischer, U.; Radermacher, J.; Mayer, J.; Mehraein, Y.; Meese, E. Tumor hypoxia: Impact on gene amplification in glioblastoma. Int. J. Oncol. 2008, 33, 509–515. [Google Scholar] [CrossRef]
- Irshad, K.; Mohapatra, S.K.; Srivastava, C.; Garg, H.; Mishra, S.; Dikshit, B.; Sarkar, C.; Gupta, D.; Chandra, P.S.; Chattopadhyay, P.; et al. A combined gene signature of hypoxia and Notch pathway in human glioblastoma and its prognostic relevance. PLoS ONE 2015, 10, e0118201. [Google Scholar] [CrossRef]
- Gordan, J.D.; Bertout, J.A.; Hu, C.J.; Diehl, J.A.; Simon, M.C. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef]
- Binello, E.; Germano, I.M. Targeting glioma stem cells: A novel framework for brain tumors. Cancer Sci. 2011, 102, 1958–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp. Cell Res. 2012, 318, 2417–2426. [Google Scholar] [CrossRef]
- Jawhari, S.; Ratinaud, M.H.; Verdier, M. Glioblastoma, hypoxia and autophagy: A survival-prone ‘ménage-à-trois’. Cell Death Dis. 2016, 7, e2434. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Mellai, M.; Corona, C.; Casalone, C.; Annovazzi, L. Gliobastoma: Equilibrium and Interconvesion between Tumor Non-Stem Cells and Tumor Stem Cells. Biomed. J. Sci. Tech. Res. 2018, 8, 1–6. [Google Scholar] [CrossRef]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro Oncol. 2012, 14, 958–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrusiewicz, K.; Ellert-Miklaszewska, A.; Lipko, M.; Sielska, M.; Frankowska, M.; Kaminska, B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS ONE 2011, 6, e23902. [Google Scholar] [CrossRef] [PubMed]
- Glass, R.; Synowitz, M. CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol. 2014, 128, 347–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 9, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, S.; Müller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006, 8, 261–279. [Google Scholar] [CrossRef]
- Parney, I.F.; Waldron, J.S.; Parsa, A.T. Flow cytometry and in vitro analysis of human glioma-associated macrophages. J. Neurosurg. 2009, 110, 572–582. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Gabrusiewicz, K.; Heimberger, A. The controversial role of microglia in malignant gliomas. Clin. Dev. Immunol. 2013, 285246. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Tay, T.L.; Wolf, Y.; Jung, S. Microglia: Unique and common features with other tissue macrophages. Acta Neuropathol. 2014, 128, 319–331. [Google Scholar] [CrossRef]
- Müller, A.; Brandenburg, S.; Turkowski, K.; Müller, S.; Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 2015, 137, 278–288. [Google Scholar] [CrossRef]
- Zhu, W.; Carney, K.E.; Pigott, V.M.; Falgoust, M.N.; Clark, P.A.; Kuo, J.S.; Sun, D. Glioma-mediated microglial activation promotes glioma proliferation and migration: Roles of Na+/H+ exchanger isoform 1. Carcinogenesis 2016, 37, 839–851. [Google Scholar] [CrossRef]
- Szulzewsky, F.; Arora, S.; de Witte, L.; Ulas, T.; Markovic, D.; Schultze, J.L.; Holland, E.C.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Human glioblastoma associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia 2016, 64, 1416–1436. [Google Scholar] [CrossRef]
- Placone, A.L.; Quiñones-Hinojosa, A.; Searson, P.C. The role of astrocytes in the progression of brain cancer: Complicating the picture of the tumor microenvironment. Tumour Biol. 2016, 37, 61–69. [Google Scholar] [CrossRef]
- Kennedy, B.C.; Showers, C.R.; Anderson, D.E.; Anderson, L.; Canoll, P.; Bruce, J.N.; Anderson, R.C. Tumor-associated macrophages in glioma: Friend or foe? J. Oncol. 2013, 486912. [Google Scholar] [CrossRef]
- Morimura, T.; Neuchrist, C.; Kitz, K.; Budka, H.; Scheiner, O.; Kraft, D.; Lassmann, H. Monocyte subpopulations in human gliomas: Expression of Fc and complement receptors and correlation with tumor proliferation. Acta Neuropathol. 1990, 80, 287–294. [Google Scholar] [CrossRef]
- Schiffer, D.; Mellai, M.; Bovio, E.; Annovazzi, L. The neuropathological basis to the functional role of microglia/macrophages in gliomas. Neurol. Sci. 2017, 38, 1571–1577. [Google Scholar] [CrossRef]
- Feng, X.; Szulzewsky, F.; Yerevanian, A.; Chen, Z.; Heinzmann, D.; Rasmussen, R.D.; Alvarez-Garcia, V.; Kim, Y.; Wang, B.; Tamagno, I.; et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget 2015, 6, 15077–15094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils promote the malignant glioma phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Okada, H. Myeloid-derived suppressor cells (MDSCs) in gliomas and glioma-development. Immunol. Investig. 2012, 41, 658–679. [Google Scholar] [CrossRef] [PubMed]
- Badie, B.; Schartner, J.M. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery 2000, 46, 957–961. [Google Scholar] [PubMed]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Gabrusiewicz, B.; Rodriguez, J.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1, e85841. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Xiao, H.; Xu, M.; Ye, X.; Hu, J.; Li, F.; Li, M.; Luo, C.; Yu, S.; Bian, X.; et al. Glioma-initiating cells: A predominant role in microglia/macrophages tropism to glioma. J. Neuroimmunol. 2011, 232, 75–82. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef]
- Markovic, D.S.; Glass, R.; Synowitz, M.; Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef]
- Tartour, E.; Pere, H.; Maillere, B.; Terme, N.; Merillon, N.; Taieb, J.; Sandoval, F.; Quintin-Colonna, F.; Lacerda, K.; Karadimou, A.; et al. Angiogenesis and immunity: A bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer Metastasis Rev. 2011, 30, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Ku, M.C.; Markovic, D.; Dzaye, O.D.; Lehnardt, S.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Glioma-associated microglial MMP9 expression is upregulated by TLR2 signaling and sensitive to minocycline. Int. J. Cancer 2014, 135, 2569–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–11125. [Google Scholar] [CrossRef]
- Domenis, R.; Cesselli, D.; Toffoletto, B.; Bourkoula, E.; Caponnetto, F.; Manini, I.; Beltrami, A.P.; Ius, T.; Skrap, M.; Di Loreto, C.; et al. Systemic T cells immunosuppression of glioma stem cell-derived exosomes is mediated by monocytic myeloid-derived suppressor cells. PLoS ONE 2017, 12, e0169932. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Nakano, I. Proneural-mesenchymal transformation of glioma stem cells: Do therapies cause evolution of target in glioblastoma? Future Oncol. 2014, 10, 1527–1530. [Google Scholar] [CrossRef] [PubMed]
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014, 14, 357–369. [Google Scholar] [CrossRef]
- da Fonseca, A.C.; Badie, B. Microglia and macrophages in malignant gliomas: Recent discoveries and implications for promising therapies. Clin. Dev. Immunol. 2013, 264124. [Google Scholar] [CrossRef]
- Poon, C.C.; Sarkar, S.; Ying, W.; Kelly, J.P. Glioblastoma-associated microglia and macrophages: Targets for therapy to improve prognosis. Brain 2017, 140, 1548–1560. [Google Scholar] [CrossRef]
- Butterfield, L.H. Dendritic cells in cancer immunotherapy clinical trials: Are we making progress? Front. Immunol. 2013, 4, 454. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Mellman, I. Cell biology of antigen processing in vitro and in vivo. Clin. Dev. Immunol. 2005, 23, 975–1028. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.; Delamarre, L. Dendritic cell-targeted vaccines. Front. Immunol. 2014, 5, 255. [Google Scholar] [CrossRef] [PubMed]
- Mineharu, Y.; Castro, M.G.; Lowenstein, P.R.; Sakai, N.; Miyamoto, S. Dendritic Cell-based Immunotherapy for glioma: Multiple regimens and implications in clinical trials. Neurol. Med. Chir 2013, 53, 741–754. [Google Scholar] [CrossRef]
- Yang, L.; Guo, G.; Niu, X.Y.; Liu, J. Dendritic cell-based immunotherapy treatment for glioblastoma multiforme. Biomed. Res. Int. 2015, 717530. [Google Scholar] [CrossRef]
- Nava, S.; Lisini, D.; Pogliani, S.; Dossena, M.; Bersano, A.; Pellegatta, S.; Parati, E.; Finocchiaro, G.; Frigerio, S. Safe and reproducible preparation of functional dendritic cells for immunotherapy in glioblastoma patients. Stem Cells Transl. Med. 2015, 4, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Schaller, T.H.; Sampson, J.H. Advances and challenges: Dendritic cell vaccination strategies for glioblastoma. Expert Rev. Vaccines 2017, 16, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, G.; Pellegatta, S. Immunotherapy with dendritic cells loaded with glioblastoma stem cells: From preclinical to clinical studies. Cancer Immunol. Immunother. 2016, 65, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Franklin, M.J.; Dudek, A.Z. Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am. J. Hematol. 2010, 85, 7593–7598. [Google Scholar] [CrossRef]
- Harrell, C.R.; Simovic Markovic, B.; Fellabaum, C.; Arsenijevic, A.; Djonov, V.; Volarevic, V. Molecular mechanisms underlying therapeutic potential of pericytes. J. Biomed. Sci. 2018, 25, 21. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Stallcup, W.B.; You, W.K.; Kucharova, K.; Cejudo-Martin, P.; Yotsumoto, F. NG2 proteoglycan-dependent contributions of pericytes and macrophages to brain tumor vascularization and progression. Microcirculation 2016, 23, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Stallcup, W.B. NG2 proteoglycan enhances brain tumor progression by promoting beta–1 Integrin activation in both Cis and Trans orientations. Cancers 2017, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Hasan, M.N.; Maniar, S.; Jia, W.; Sun, D. Reactive astrocytes in glioblastoma multiforme. Mol. Neurobiol. 2018, 55, 6927–6938. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Zengotita, M.; Yachnis, A.T. Gliosis Versus Glioma? Don’t Grade Until You Know. Adv. Anat. Pathol. 2012, 19, 239–249. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.R.; Howarth, C.; Sibson, N.R. The role of astrocytes in CNS tumors: Pre-clinical models and novel imaging approaches. Front. Cell Neurosci. 2013, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Barbero, S.; Bajetto, A.; Bonavia, R.; Porcile, C.; Piccioli, P.; Pirani, P.; Ravetti, J.L.; Zona, G.; Spaziante, R.; Florio, T.; et al. Expression of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1 in human brain tumors and their involvement in glial proliferation in vitro. Ann. N. Y. Acad. Sci. 2002, 973, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Biasoli, D.; Sobrinho, M.F.; da Fonseca, A.C.C.; de Matos, G.G.; Romão, L.; de Moraes Maciel, R.; Rehen, S.K.; Moura-Neto, V.; Borges, H.L.; Lima, F.R.; et al. Glioblastoma cells inhibit astrocytic p53-expression favoring cancer malignancy. Oncogenesis 2014, 3, e123. [Google Scholar] [CrossRef]
- Lin, Q.; Liu, Z.; Ling, F.; Xu, G. Astrocytes protect glioma cells from chemotherapy and upregulate survival genes via gap junctional communication. Mol. Med. Rep. 2016, 13, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jin, X.; Sohn, Y.W.; Jin, X.; Jeon, H.Y.; Kim, E.J.; Ham, S.W.; Jeon, H.M.; Chang, S.Y.; Oh, S.Y.; et al. Tumoral RANKL activates astrocytes that promote glioma cell invasion through cytokine signaling. Cancer Lett. 2014, 353, 194–200. [Google Scholar] [CrossRef]
- Gielen, P.R.; Aftab, Q.; Ma, N.; Chen, V.C.; Hong, X.; Lozinsky, S.; Naus, C.C.; Sin, W.C. Connexin43 confers temozolomide resistance in human glioma cells by modulating the mitochondrial apoptosis pathway. Neuropharmacology 2013, 75, 539–548. [Google Scholar] [CrossRef]
- Bharti, R.; Dey, G.; Mandal, M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: A snapshot of IL-6 mediated involvement. Cancer Lett. 2016, 375, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xia, T.; Wang, D.; Huang, B.; Zhao, P.; Wang, J.; Qu, X.; Li, X. Human astrocytes secrete IL-6 to promote glioma migration and invasion through upregulation of cytomembrane MMP14. Oncotarget 2016, 7, 62425–62438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, X.; Sin, W.C.; Harris, A.L.; Naus, C.C. Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget 2015, 6, 15566–15577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Emdad, L.; Bacolod, M.D.; Kegelman, T.P.; Shen, X.N.; Alzubi, M.A.; Das, S.K.; Sarkar, D.; Fisher, P.B. Astrocyte elevated gene-1 interacts with Akt isoform 2 to control glioma growth, survival, and pathogenesis. Cancer Res. 2014, 4, 7321–7332. [Google Scholar] [CrossRef] [PubMed]
- Fisher, P.B. Activation of the nuclear factor kappaB pathway by astrocyte elevated gene-1: Implications for tumor progression and metastasis. Cancer Res. 2006, 66, 1509–1516. [Google Scholar]
- Sarkar, D.; Park, E.S.; Emdad, L.; Lee, S.G.; Su, Z.Z.; Fisher, P.B. Molecular basis of nuclear factor-kappaB activation by astrocyte elevated gene-1. Cancer Res. 2008, 8, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Duan, Y.; Wang, P.; Gao, R.; Chen, X.; Ou, Y.; Liang, M.; Wang, Z.; Yuan, Y.; Wang, L.; et al. DYT-40, a novel synthetic 2-styryl-5-nitroimidazole derivative, blocks malignant glioblastoma growth and invasion by inhibiting AEG-1 and NF-κB signaling pathways. Sci. Rep. 2016, 6, 27331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okolie, O.; Bago, J.R.; Schmid, R.S.; Irvin, D.M.; Bash, E.; Miller, C.R.; Hingtgen, S.D. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro Oncol. 2016, 18, 1622–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S. Cell death-based treatment of glioblastoma. Cell Death Dis. 2018, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Siddharth, K.J.; Zuniga, R. High Grade Glioma—Standard Approach, Obstacles and Future Directions. In Tumors of Central Nervous System. Primary and Secondary, 1st ed.; Morgan, L.R., Ed.; InTech: Rjieka, Croatia, 2014; pp. 3–29. ISBN 978-953-51-1576-2. [Google Scholar]
- Sikorski, C.W.; Lesniak, M.S. Immunotherapy for malignant glioma: Current approaches and future directions. Neurol. Res. 2005, 27, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.C.; Davis, A.A.; Wainwright, D.A. Immunotherapy for cancer in the central nervous system: Current and future directions. Oncoimmunology 2015, 5, e1082027. [Google Scholar] [CrossRef] [PubMed]
- Zloza, A.; Karolina Palucka, A.; Coussens, L.M.; Gotwals, P.J.; Headley, M.B.; Jaffee, E.M.; Lund, A.W.; Sharpe, A.H.; Sznol, M.; Wainwright, D.A.; et al. Workshop on challenges, insights, and future directions for mouse and humanized models in cancer immunology and immunotherapy: A report from the associated programs of the 2016 annual meeting for the Society for Immunotherapy of cancer. J. Immunother. Cancer 2017, 5, 77. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2019, 11, 5. https://doi.org/10.3390/cancers11010005
Schiffer D, Annovazzi L, Casalone C, Corona C, Mellai M. Glioblastoma: Microenvironment and Niche Concept. Cancers. 2019; 11(1):5. https://doi.org/10.3390/cancers11010005
Chicago/Turabian StyleSchiffer, Davide, Laura Annovazzi, Cristina Casalone, Cristiano Corona, and Marta Mellai. 2019. "Glioblastoma: Microenvironment and Niche Concept" Cancers 11, no. 1: 5. https://doi.org/10.3390/cancers11010005
APA StyleSchiffer, D., Annovazzi, L., Casalone, C., Corona, C., & Mellai, M. (2019). Glioblastoma: Microenvironment and Niche Concept. Cancers, 11(1), 5. https://doi.org/10.3390/cancers11010005