The Circulating Transcriptome as a Source of Biomarkers for Melanoma

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
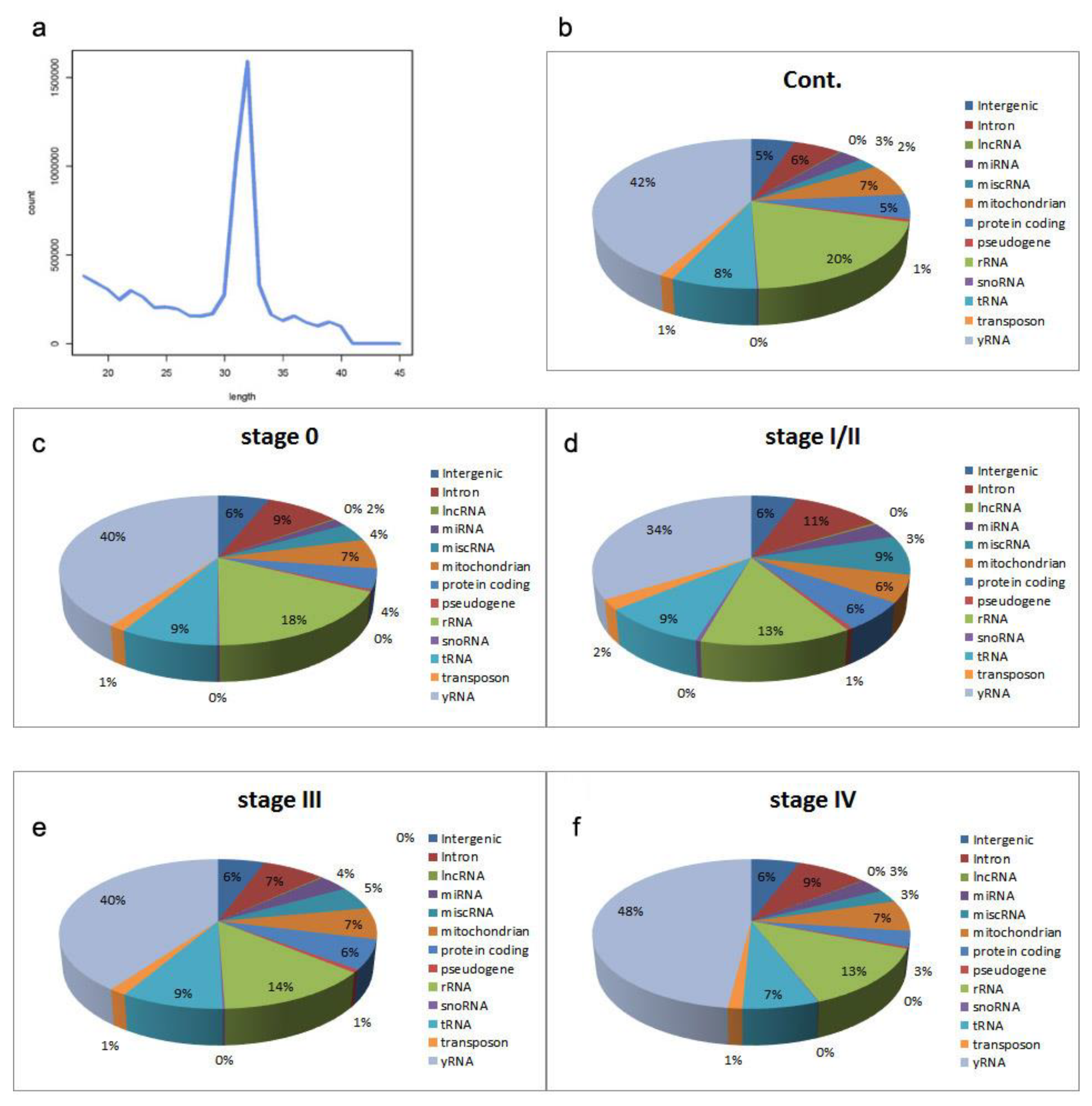
2.1. Sequencing the Circulating Transcriptome of Melanoma Patients
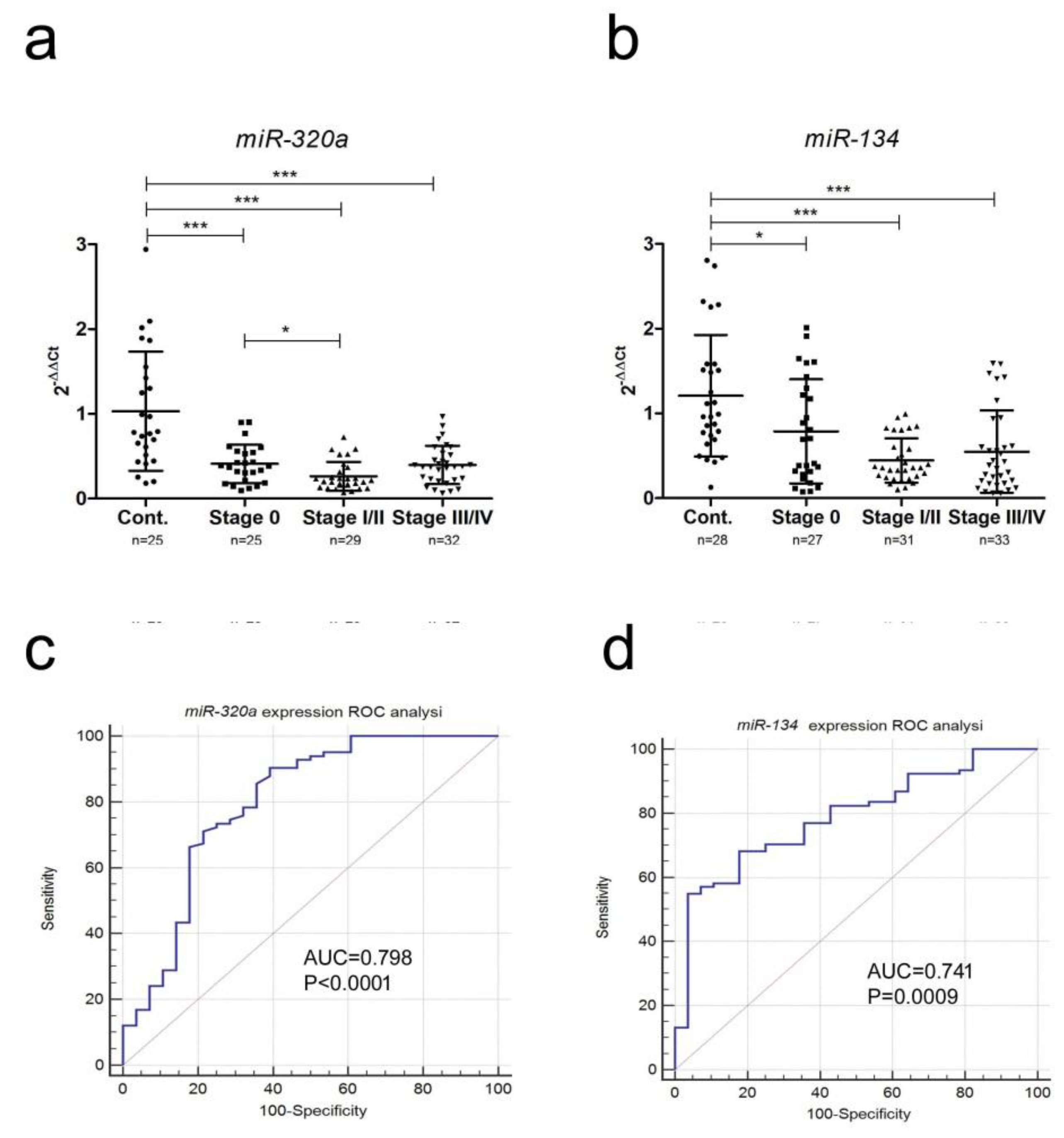
2.2. miRNA Expression
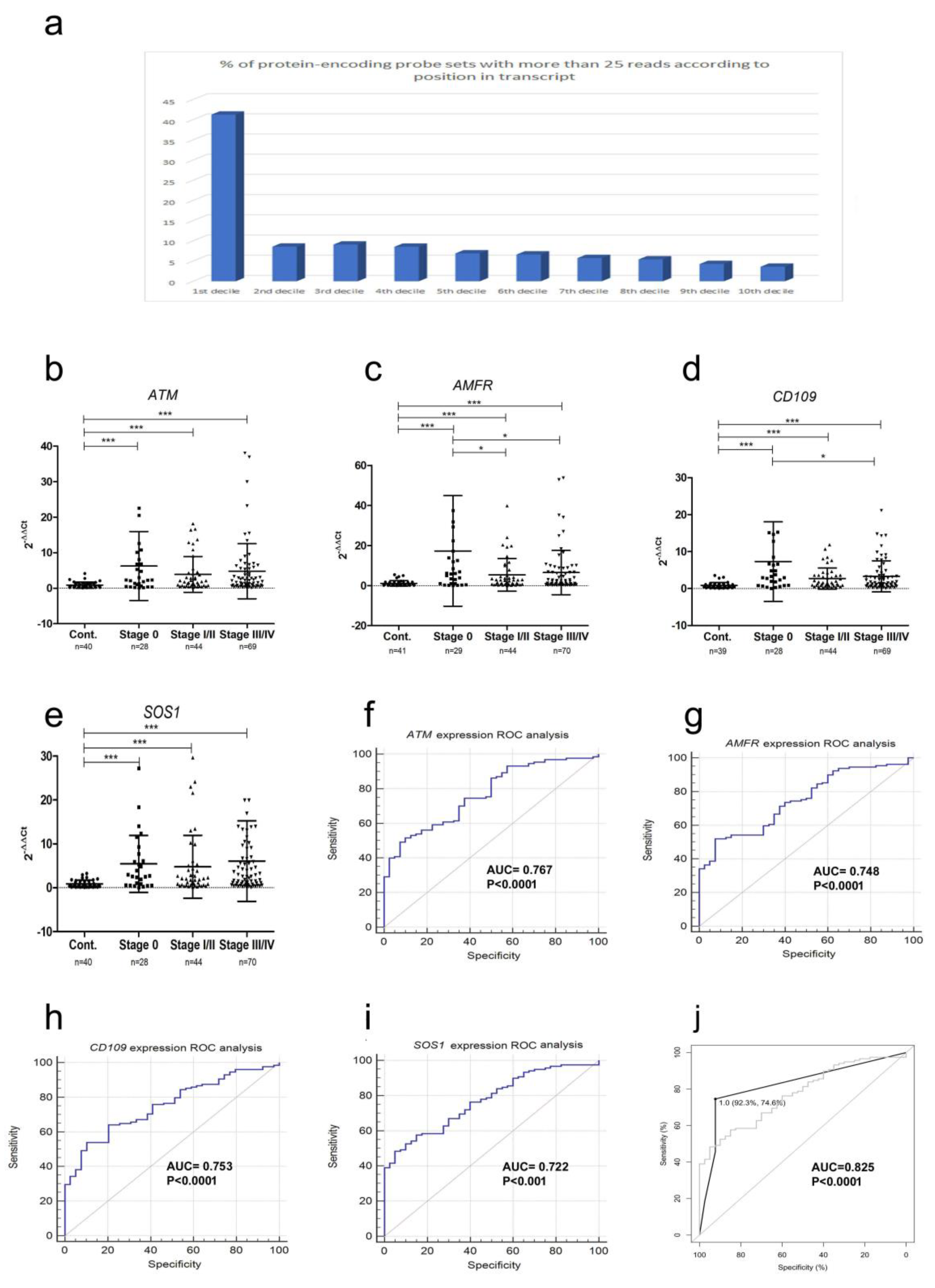
2.3. mRNA Fragment Expression
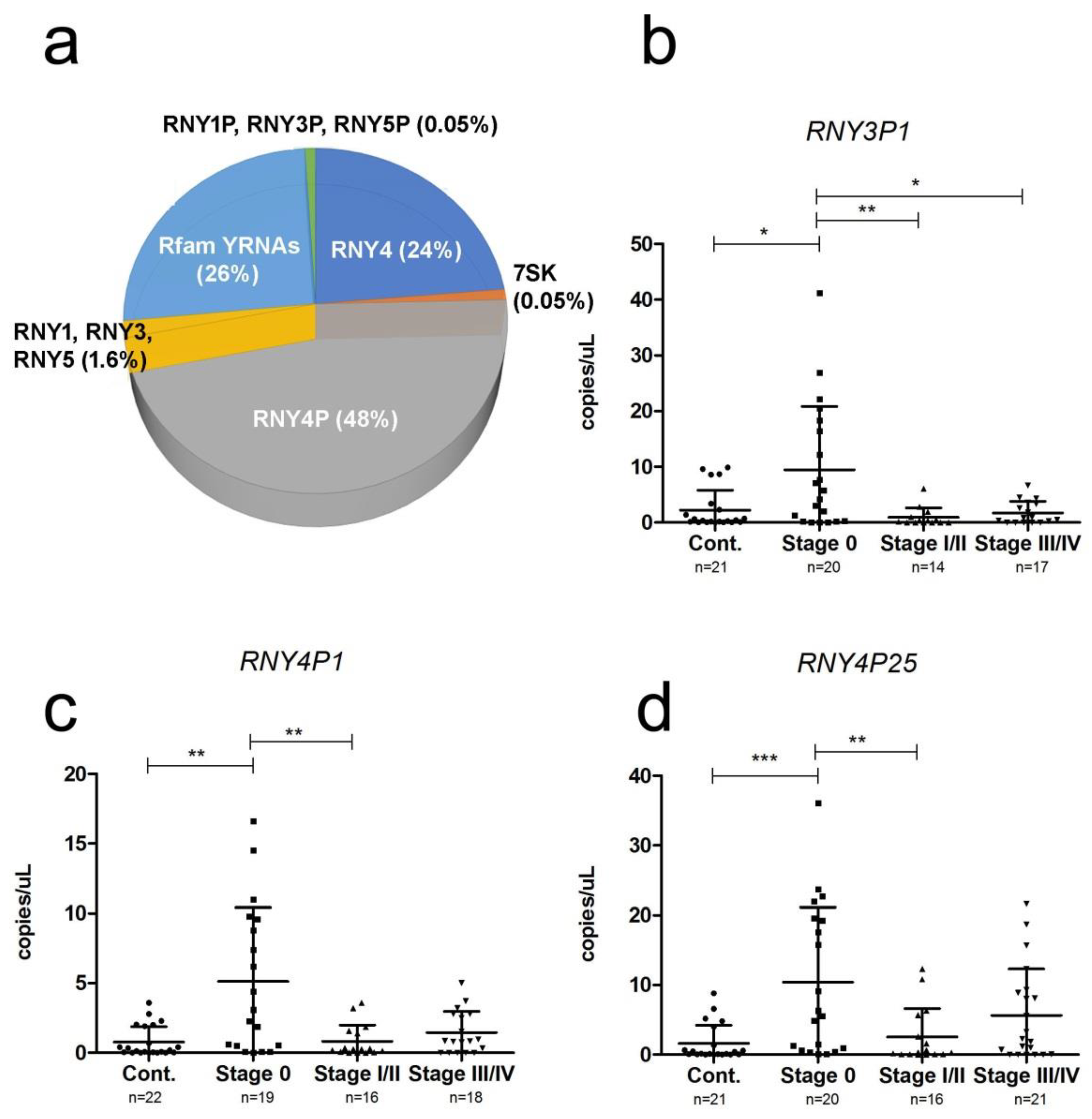
2.4. YRNA Expression
3. Discussion
4. Materials and Methods
4.1. Patient Cohorts
4.2. Library Construction and Next-Generation Sequencing
4.3. Bioinformatic Analysis
4.4. qRT-PCR (mRNA and miRNA) and ddPCR(yRNAs)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Chromosome Position | Length | p-Value | Cont. | St. 0 | St. I/II | St. III | St. IV |
|---|---|---|---|---|---|---|---|---|
| CD109 | 6:74446106–74446231 | 125 | 0.00103 | 39 | 102 | 126 | 207 | 318 |
| ARHGAP | 4:148744047–148744108 | 61 | 0.0018 | 45 | 9 | 11 | 10 | 8 |
| SOS1 | 2:39237725–39237844 | 119 | 0.00196 | 23 | 16 | 130 | 121 | 139 |
| ATM | 11:108099905–108100050 | 145 | 0.00316 | 5 | 20 | 8 | 7 | 105 |
| AMFR | 16:56396751–56396855 | 104 | 0.00329 | 11 | 32 | 59 | 59 | 121 |
| KCNIP3 | 2:95976103–95976544 | 441 | 0.00362 | 1 | 14 | 22 | 16 | 28 |
| POLE | 12:133248801–133248908 | 107 | 0.00456 | 0 | 36 | 11 | 12 | 72 |
| KLHL7 | 7:23213634–23214040 | 406 | 0.00479 | 0 | 6 | 10 | 8 | 40 |
| ZMYM4 | 1:35824626–35825047 | 421 | 0.00547 | 54 | 23 | 25 | 27 | 22 |
| GTDC1 | 2:144966170–144966371 | 211 | 0.00632 | 58 | 15 | 16 | 21 | 14 |
| RFWD2 | 1:176132951–176133027 | 76 | 0.00651 | 27 | 11 | 10 | 13 | 12 |
| ARHGAP10 | 4:148778704–148779039 | 335 | 0.00672 | 31 | 10 | 13 | 11 | 13 |
| RHBDD1 | 2:227778924–227779353 | 429 | 0.00774 | 71 | 63 | 39 | 33 | 28 |
| HNRNPM | 19:8527413–8527473 | 60 | 0.00798 | 42 | 15 | 20 | 17 | 19 |
| DYNC2H1 | 11:102988360–102988592 | 232 | 0.00835 | 0 | 15 | 17 | 15 | 60 |
| ITIH5 | 10:7613668–7614061 | 393 | 0.00885 | 0 | 11 | 12 | 6 | 32 |
| TNFA1P2 | 14:103592664–103593029 | 365 | 0.00974 | 29 | 28 | 84 | 76 | 60 |
| SLC40A1 | 2:190430080–190430325 | 245 | 0.00996 | 21 | 20 | 31 | 28 | 34 |
| SETDB1 | 1:150902443–150902932 | 489 | 0.0106 | 1 | 20 | 23 | 17 | 60 |
| IGF2BP3 | 7: 23385559–23385780 | 221 | 0.0118 | 27 | 27 | 11 | 14 | 15 |
| DENND3 | 8:142152302–142153707 | 205 | 0.0118 | 0 | 13 | 32 | 4 | 3 |
| GAK | 4:866054–866461 | 407 | 0.014 | 0 | 11 | 16 | 10 | 40 |
| ARHGEF12 | 11: 120278447–120282546 | 99 | 0.015 | 72 | 78 | 52 | 39 | 44 |
| HINT3 | 6: 126298790–126301387 | 497 | 0.0157 | 76 | 66 | 8 | 19 | 11 |
| HTR1E | 6: 87647124–87647541 | 417 | 0.0158 | 41 | 95 | 106 | 75 | 92 |
| UXS1 | 2:106780123–106780166 | 43 | 0.0171 | 60 | 5 | 5 | 6 | 2 |
| MPP2 | 17: 41983448–41983519 | 71 | 0.0173 | 11 | 9 | 22 | 29 | 18 |
| NAIP | 5: 70308275–70308745 | 470 | 0.0183 | 0 | 10 | 41 | 5 | 2 |
| SNCA | 4:90757894–90758379 | 485 | 0.0185 | 73 | 44 | 49 | 49 | 41 |
| TRAPPC9 | 8:140922366–140922544 | 178 | 0.0185 | 28 | 68 | 76 | 53 | 59 |
| PISD | 22:32034352–32034488 | 136 | 0.0189 | 225 | 533 | 600 | 413 | 557 |
| SERPINB1 | 6: 2836090–2836257 | 233 | 0.0193 | 29 | 14 | 10 | 14 | 13 |
| GRN | 17:42429383–42429616 | 167 | 0.0193 | 0 | 7 | 25 | 2 | 2 |
| EIF4G3 | 1: 21231376–21231464 | 88 | 0.0195 | 49 | 45 | 38 | 33 | 33 |
| ILK | 11: 6624961–6625456 | 495 | 0.0196 | 28 | 49 | 46 | 59 | 47 |
| C14ORF38 | 14:60031765–60031993 | 240 | 0.0197 | 26 | 7 | 11 | 9 | 7 |
| PSAP | 10:73578788–73579028 | 228 | 0.0197 | 1 | 13 | 27 | 44 | 13 |
| TMEM104 | 17: 72835466–72835918 | 452 | 0.0212 | 46 | 35 | 11 | 19 | 16 |
| TAOK1 | 17:27849298–27849537 | 239 | 0.0217 | 42 | 9 | 12 | 6 | 11 |
| SIAE | 11: 124506788–124507098 | 310 | 0.022 | 26 | 22 | 6 | 12 | 7 |
| CDK5RAP2 | 9: 123165584–123165940 | 356 | 0.0229 | 4347 | 9583 | 12031 | 8114 | 9164 |
| TPT1 | 13: 45911208–45911614 | 406 | 0.0239 | 55 | 31 | 38 | 34 | 30 |
| SSH3 | 11:67070919–67071162 | 243 | 0.024 | 236 | 73 | 71 | 46 | 40 |
| SSB | 2:170667368–170667554 | 186 | 0.0247 | 0 | 8 | 28 | 7 | 1 |
| ZNF430 | 19:21216892–21216990 | 98 | 0.025 | 48 | 11 | 17 | 12 | 8 |
| HBA2 | 16:222846–223262 | 494 | 0.0266 | 266 | 141 | 135 | 177 | 134 |
| ZFPM2 | 8:106456508–106456609 | 101 | 0.0304 | 70 | 41 | 43 | 49 | 49 |
| RBM5 | 3:50145665–50145738 | 73 | 0.0308 | 60 | 19 | 16 | 28 | 16 |
| ITGB1 | 10:33218750–33218972 | 222 | 0.0312 | 80 | 37 | 29 | 45 | 32 |
| ZCCHC17 | 1: 31821676–31821821 | 145 | 0.0329 | 15 | 17 | 20 | 23 | 26 |
| PRKG2 | 4: 82136085–82136218 | 133 | 0.0332 | 40 | 49 | 20 | 29 | 26 |
| IFI6 | 1: 27992572–27992986 | 414 | 0.0341 | 0 | 0 | 46 | 1 | 2 |
| MX2 | 21: 42774561–42776800 | 458 | 0.0363 | 0 | 0 | 31 | 1 | 1 |
| PSMD2 | 3:184020467–184020611 | 144 | 0.0366 | 0 | 2 | 12 | 28 | 1 |
| HERC6 | 4:89363186–89364063 | 477 | 0.0376 | 0 | 3 | 54 | 4 | 1 |
| PPBP | 4:74853673–74853914 | 241 | 0.0387 | 685 | 259 | 301 | 403 | 260 |
| PKN2 | 1:89206671–89206971 | 300 | 0.0389 | 66 | 27 | 27 | 22 | 15 |
| TNIP1 | 5: 150415143–150415268 | 125 | 0.039 | 7 | 5 | 36 | 16 | 14 |
| ARGLU1 | 13: 107209096–107210043 | 347 | 0.0396 | 0 | 11 | 42 | 5 | 6 |
| LRRN2 | 1:204654448–204654861 | 413 | 0.0399 | 13 | 43 | 29 | 25 | 32 |
| ACACB | 12:109625804–109625967 | 163 | 0.0401 | 19 | 46 | 43 | 30 | 40 |
| EIF5B | 2: 99980108–99980325 | 217 | 0.0415 | 39 | 14 | 22 | 18 | 15 |
| FRA10AC1 | 10:95441237–95441355 | 118 | 0.0419 | 33 | 6 | 6 | 5 | 2 |
| RASA1 | 5:86627165–86627317 | 152 | 0.0437 | 35 | 10 | 15 | 17 | 16 |
| CHD9 | 16:53301839–53302038 | 199 | 0.0441 | 41 | 10 | 11 | 20 | 13 |
| TPM1 | 15:63351762–63351879 | 117 | 0.0443 | 38 | 12 | 18 | 19 | 12 |
| ACVR2 | 2:148657313–148657467 | 154 | 0.0462 | 26 | 21 | 16 | 10 | 14 |
| MCTP1 | 5:94224580–94224677 | 250 | 0.0475 | 6 | 12 | 20 | 19 | 25 |
| HOXC6 | 12:54404873–54407570 | 97 | 0.0481 | 26 | 62 | 71 | 45 | 49 |
| ATF4 | 22:39916183–39917676 | 267 | 0.0487 | 26 | 28 | 18 | 18 | 18 |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, M.A. Epidemiology, etiology, and control of melanoma. Med. Health R. I. 2001, 84, 234–236. [Google Scholar] [PubMed]
- Forsea, A.M.; Del Marmol, V.; de Vries, E.; Bailey, E.E.; Geller, A.C. Melanoma incidence and mortality in Europe: New estimates, persistent disparities. Br. J. Dermatol. 2012, 167, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Mercado, M.; Manterola, L.; Larrea, E.; Goicoechea, I.; Arestin, M.; Armesto, M.; Otaegui, D.; Lawrie, C.H. The circulating transcriptome as a source of non-invasive cancer biomarkers: Concepts and controversies of non-coding and coding RNA in body fluids. J. Cell. Mol. Med. 2015, 19, 2307–2323. [Google Scholar] [CrossRef] [PubMed]
- Lawrie, C.H.; Gal, S.; Dunlop, H.M.; Pushkaran, B.; Liggins, A.P.; Pulford, K.; Banham, A.H.; Pezzella, F.; Boultwood, J.; Wainscoat, J.S.; et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br. J. Haematol. 2008, 141, 672–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiiyama, R.; Fukushima, S.; Jinnin, M.; Yamashita, J.; Miyashita, A.; Nakahara, S.; Kogi, A.; Aoi, J.; Masuguchi, S.; Inoue, Y.; et al. Sensitive detection of melanoma metastasis using circulating microRNA expression profiles. Melanoma Res. 2013, 23, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E.B.; Shang, S.; de Miera, E.V.; Fog, J.U.; Teilum, M.W.; Ma, M.W.; Berman, R.S.; Shapiro, R.L.; Pavlick, A.C.; Hernando, E.; et al. Serum microRNAs as biomarkers for recurrence in melanoma. J. Transl. Med. 2012, 10, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savelyeva, A.V.; Kuligina, E.V.; Bariakin, D.N.; Kozlov, V.V.; Ryabchikova, E.I.; Richter, V.A.; Semenov, D.V. Variety of RNAs in Peripheral Blood Cells, Plasma, and Plasma Fractions. Biomed. Res. Int. 2017, 2017, 7404912. [Google Scholar] [CrossRef]
- Wong, B.C.; Lo, Y.M. Plasma RNA integrity analysis: Methodology and validation. Ann. N. Y. Acad. Sci. 2006, 1075, 174–178. [Google Scholar] [CrossRef]
- Pruijn, G.J.; Wingens, P.A.; Peters, S.L.; Thijssen, J.P.; van Venrooij, W.J. Ro RNP associated Y RNAs are highly conserved among mammals. Biochim. Biophys. Acta 1993, 1216, 395–401. [Google Scholar] [CrossRef]
- Stein, A.J.; Fuchs, G.; Fu, C.; Wolin, S.L.; Reinisch, K.M. Structural insights into RNA quality control: The Ro autoantigen binds misfolded RNAs via its central cavity. Cell 2005, 121, 529–539. [Google Scholar] [CrossRef]
- Christov, C.P.; Gardiner, T.J.; Szuts, D.; Krude, T. Functional requirement of noncoding Y RNAs for human chromosomal DNA replication. Mol. Cell. Biol. 2006, 26, 6993–7004. [Google Scholar] [CrossRef]
- Sim, S.; Wolin, S.L. Emerging roles for the Ro 60-kDa autoantigen in noncoding RNA metabolism. Wiley Interdiscip. Rev. RNA 2011, 2, 686–699. [Google Scholar] [CrossRef] [Green Version]
- Sole, C.; Tramonti, D.; Schramm, M.; Goicoechea, I.; Armesto, M.; Hernandez, L.I.; Manterola, L.; Fernandez-Mercado, M.; Mujika, K.; Tuneu, A.; Jaka, A.; et al. Biodonostia Research Institute; University of Oxford; University of Heidelberg; Oncology Institute of Gipuzkoa; Hospital Universitario Donostia; Centre for Genomic Regulation; Universitat Pompeu Fabra; CIBERESP; Hospital del Mar Research Institute; Stockholm University; Imperial College London; de Octubre Hospital; CIBERONC; IKERBASQUE. (Library optimization for sequencing of circulating transcriptome in melanoma patients) San Sebastian, Spain, unpublished work, 2018.
- Fuchs, R.T.; Sun, Z.; Zhuang, F.; Robb, G.B. Bias in ligation-based small RNA sequencing library construction is determined by adaptor and RNA structure. PLoS ONE 2015, 10, e0126049. [Google Scholar] [CrossRef]
- Sole, C.; Tramonti, D.; Schramm, M.; Goicoechea, I.; Armesto, M.; Hernandez, L.I.; Manterola, L.; Fernandez-Mercado, M.; Mujika, K.; Tuneu, A.; et al. Biodonostia Research Institute; University of Oxford; University of Heidelberg; Oncology Institute of Gipuzkoa; Hospital Universitario Donostia; Centre for Genomic Regulation; Universitat Pompeu Fabra; CIBERESP; Hospital del Mar Research Institute; Stockholm University; Imperial College London; de Octubre Hospital; CIBERONC; IKERBASQUE. miR-21-5p and miR-92b-3p detection in Melanoma. San Sebastian, Spain, unpublished work, 2018.
- Sole, C.; Tramonti, D.; Schramm, M.; Goicoechea, I.; Armesto, M.; Hernandez, L.I.; Manterola, L.; Fernandez-Mercado, M.; Mujika, K.; Tuneu, A.; et al. Biodonostia Research Institute; University of Oxford; University of Heidelberg; Oncology Institute of Gipuzkoa; Hospital Universitario Donostia; Centre for Genomic Regulation; Universitat Pompeu Fabra; CIBERESP; Hospital del Mar Research Institute; Stockholm University; Imperial College London; de Octubre Hospital; CIBERONC; IKERBASQUE. mRNA targets of miR-134-5p and miR-320a using predictive algorithms TargetScan and miRDB. San Sebastian, Spain, unpublished work, 2018.
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. PanelomiX: A threshold-based algorithm to create panels of biomarkers. Transl. Proteom. 2013, 1, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Yao, J.; Wu, D.C.; Nottingham, R.M.; Mohr, S.; Hunicke-Smith, S.; Lambowitz, A.M. High-throughput sequencing of human plasma RNA by using thermostable group II intron reverse transcriptases. RNA 2016, 22, 111–128. [Google Scholar] [CrossRef]
- Danielson, K.M.; Rubio, R.; Abderazzaq, F.; Das, S.; Wang, Y.E. High Throughput Sequencing of Extracellular RNA from Human Plasma. PLoS ONE 2017, 12, e0164644. [Google Scholar] [CrossRef]
- Guo, Y.; Vickers, K.; Xiong, Y.; Zhao, S.; Sheng, Q.; Zhang, P.; Zhou, W.; Flynn, C.R. Comprehensive evaluation of extracellular small RNA isolation methods from serum in high throughput sequencing. BMC Genom. 2017, 18, 50. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Huang, X.; Woodcock, M.; Du, M.; Dittmar, R.; Wang, Y.; Tsai, S.; Kohli, M.; Boardman, L.; Patel, T.; et al. Plasma extracellular RNA profiles in healthy and cancer patients. Sci. Rep. 2016, 6, 19413. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Guo, W.; Li, S.; Dai, W.; Zhang, N.; Zhao, T.; Wang, H.; Ma, J.; Yi, X.; Ge, R.; et al. Serum miR-16: A Potential Biomarker for Predicting Melanoma Prognosis. J. Investig. Dermatol. 2016, 136, 985–993. [Google Scholar] [CrossRef]
- Armand-Labit, V.; Meyer, N.; Casanova, A.; Bonnabau, H.; Platzer, V.; Tournier, E.; Sansas, B.; Verdun, S.; Thouvenot, B.; Hilselberger, B.; et al. Identification of a Circulating MicroRNA Profile as a Biomarker of Metastatic Cutaneous Melanoma. Acta Derm-Venereol. 2016, 96, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldanha, G.; Potter, L.; Shendge, P.; Osborne, J.; Nicholson, S.; Yii, N.; Varma, S.; Aslam, M.I.; Elshaw, S.; Papadogeorgakis, E.; et al. Plasma microRNA-21 is associated with tumor burden in cutaneous melanoma. J. Investig. Dermatol. 2013, 133, 1381–1384. [Google Scholar] [CrossRef]
- Triozzi, P.L.; Achberger, S.; Aldrich, W.; Crabb, J.W.; Saunthararajah, Y.; Singh, A.D. Association of tumor and plasma microRNA expression with tumor monosomy-3 in patients with uveal melanoma. Clin. Epigenet. 2016, 8, 80. [Google Scholar] [CrossRef] [PubMed]
- Kozubek, J.; Ma, Z.; Fleming, E.; Duggan, T.; Wu, R.; Shin, D.G.; Dadras, S.S. In-depth characterization of microRNA transcriptome in melanoma. PLoS ONE 2013, 8, e72699. [Google Scholar] [CrossRef]
- Fang, Z.; Tang, J.; Bai, Y.; Lin, H.; You, H.; Jin, H.; Lin, L.; You, P.; Li, J.; Dai, Z.; et al. Plasma levels of microRNA-24, microRNA-320a, and microRNA-423-5p are potential biomarkers for colorectal carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 86. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Dong, Q.G.; Sun, L.P.; He, C.Y.; Yuan, Y. Expression of serum miR-20a-5p, let-7a, and miR-320a and their correlations with pepsinogen in atrophic gastritis and gastric cancer: A case-control study. BMC Clin. Pathol. 2013, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; Wang, Y.S.; Sun, Y.F.; Miao, L.X.; Wang, J.; Li, Y.S.; Liu, H.Y.; Liu, Q.L. Plasma microRNA-320, microRNA-let-7e and microRNA-21 as novel potential biomarkers for the detection of retinoblastoma. Biomed. Rep. 2014, 2, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Wagenseller, A.G.; Shada, A.; D’Auria, K.M.; Murphy, C.; Sun, D.; Molhoek, K.R.; Papin, J.A.; Dutta, A.; Slingluff, C.L., Jr. MicroRNAs induced in melanoma treated with combination targeted therapy of Temsirolimus and Bevacizumab. J. Transl. Med. 2013, 11, 218. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.Y.; Zhang, F.; Sun, C.C.; Li, S.J.; Li, G.; Gong, F.Y.; Bo, T.; He, J.; Hua, R.X.; Hu, W.D.; et al. miR-134: A Human Cancer Suppressor? Mol. Ther. Nucleic Acids 2017, 6, 140–149. [Google Scholar] [CrossRef]
- Venkatesan, N.; Kanwar, J.; Deepa, P.R.; Khetan, V.; Crowley, T.M.; Raguraman, R.; Sugneswari, G.; Rishi, P.; Natarajan, V.; Biswas, J.; et al. Clinico-Pathological Association of Delineated miRNAs in Uveal Melanoma with Monosomy 3/Disomy 3 Chromosomal Aberrations. PLoS ONE 2016, 11, e0146128. [Google Scholar] [CrossRef]
- Kopreski, M.S.; Benko, F.A.; Kwak, L.W.; Gocke, C.D. Detection of tumor messenger RNA in the serum of patients with malignant melanoma. Clin. Cancer Res. 1999, 5, 1961–1965. [Google Scholar]
- Reddi, K.K.; Holland, J.F. Elevated serum ribonuclease in patients with pancreatic cancer. Proc. Natl. Acad. Sci. USA 1976, 73, 2308–2310. [Google Scholar] [CrossRef]
- Sole, C.; Tramonti, D.; Schramm, M.; Goicoechea, I.; Armesto, M.; Hernandez, L.I.; Manterola, L.; Fernandez-Mercado, M.; Mujika, K.; Tuneu, A.; et al. Biodonostia Research Institute; University of Oxford; University of Heidelberg; Oncology Institute of Gipuzkoa; Hospital Universitario Donostia; Centre for Genomic Regulation; Universitat Pompeu Fabra; CIBERESP; Hospital del Mar Research Institute; Stockholm University; Imperial College London; de Octubre Hospital; CIBERONC; IKERBASQUE. Length profile in sequencing between healthy and melanoma patient samples. San Sebastian, Spain, unpublished work, 2018.
- Helfrich, I.; Edler, L.; Sucker, A.; Thomas, M.; Christian, S.; Schadendorf, D.; Augustin, H.G. Angiopoietin-2 levels are associated with disease progression in metastatic malignant melanoma. Clin. Cancer Res. 2009, 15, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.; Weber, J.S. Taming the wild-types: Targeting PAK1 in melanomas that lack BRAF mutations. J. Natl. Cancer Inst. 2013, 105, 591–592. [Google Scholar] [CrossRef]
- Lu, H.; Liu, S.; Zhang, G.; Bin, W.; Zhu, Y.; Frederick, D.T.; Hu, Y.; Zhong, W.; Randell, S.; Sadek, N.; et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 2017, 550, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Emori, M.; Tsukahara, T.; Murase, M.; Kano, M.; Murata, K.; Takahashi, A.; Kubo, T.; Asanuma, H.; Yasuda, K.; Kochin, V.; et al. High expression of CD109 antigen regulates the phenotype of cancer stem-like cells/cancer-initiating cells in the novel epithelioid sarcoma cell line ESX and is related to poor prognosis of soft tissue sarcoma. PLoS ONE 2013, 8, e84187. [Google Scholar] [CrossRef]
- Fang, S.; Ferrone, M.; Yang, C.; Jensen, J.P.; Tiwari, S.; Weissman, A.M. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2001, 98, 14422–14427. [Google Scholar] [CrossRef] [Green Version]
- Timar, J.; Raso, E.; Dome, B.; Ladanyi, A.; Banfalvi, T.; Gilde, K.; Raz, A. Expression and function of the AMF receptor by human melanoma in experimental and clinical systems. Clin. Exp. Metastasis 2002, 19, 225–232. [Google Scholar] [CrossRef]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M.; Hessmann, E.; Gaedcke, J.; Guthle, M.; Zenke, M.; et al. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nat. Commun. 2015, 6, 7677. [Google Scholar] [CrossRef]
- Halaban, R.; Krauthammer, M. RASopathy Gene Mutations in Melanoma. J. Investig. Dermatol. 2016, 136, 1755–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Cecere, R.; Philip, A. CD109 released from human bone marrow mesenchymal stem cells attenuates TGF-beta-induced epithelial to mesenchymal transition and stemness of squamous cell carcinoma. Oncotarget 2017, 8, 95632–95647. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, X.; Cui, Y.; Tang, Y.; Chen, W.; Li, S.; Yu, H.; Pan, Y.; Wang, C. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 2014, 41, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Garnier, J.P.; Letellier, S.; Cassinat, B.; Lebbe, C.; Kerob, D.; Baccard, M.; Morel, P.; Basset-Seguin, N.; Dubertret, L.; Bousquet, B.; et al. Clinical value of combined determination of plasma L-DOPA/tyrosine ratio, S100B, MIA and LDH in melanoma. Eur. J. Cancer 2007, 43, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Hendrick, J.P.; Wolin, S.L.; Rinke, J.; Lerner, M.R.; Steitz, J.A. Ro small cytoplasmic ribonucleoproteins are a subclass of La ribonucleoproteins: Further characterization of the Ro and La small ribonucleoproteins from uninfected mammalian cells. Mol. Cell. Biol. 1981, 1, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.T.; Langley, A.R.; Christov, C.P.; Kheir, E.; Shafee, T.; Gardiner, T.J.; Krude, T. Dynamic interaction of Y RNAs with chromatin and initiation proteins during human DNA replication. J. Cell Sci. 2011, 124, 2058–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christov, C.P.; Trivier, E.; Krude, T. Noncoding human Y RNAs are overexpressed in tumours and required for cell proliferation. Br. J. Cancer 2008, 98, 981–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, M.P.; Krude, T. Functional roles of non-coding Y RNAs. Int. J. Biochem. Cell Biol. 2015, 66, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhahbi, J.M.; Spindler, S.R.; Atamna, H.; Boffelli, D.; Mote, P.; Martin, D.I. 5′-YRNA fragments derived by processing of transcripts from specific YRNA genes and pseudogenes are abundant in human serum and plasma. Physiol. Genom. 2013, 45, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Dhahbi, J.M.; Spindler, S.R.; Atamna, H.; Boffelli, D.; Martin, D.I. Deep Sequencing of Serum Small RNAs Identifies Patterns of 5′ tRNA Half and YRNA Fragment Expression Associated with Breast Cancer. Biomark. Cancer 2014, 6, 37–47. [Google Scholar] [CrossRef]
- Victoria Martinez, B.; Dhahbi, J.M.; Nunez Lopez, Y.O.; Lamperska, K.; Golusinski, P.; Luczewski, L.; Kolenda, T.; Atamna, H.; Spindler, S.R.; Golusinski, W.; et al. Circulating small non-coding RNA signature in head and neck squamous cell carcinoma. Oncotarget 2015, 6, 19246–19263. [Google Scholar]
- Yeri, A.; Courtright, A.; Reiman, R.; Carlson, E.; Beecroft, T.; Janss, A.; Siniard, A.; Richholt, R.; Balak, C.; Rozowsky, J.; et al. Total Extracellular Small RNA Profiles from Plasma, Saliva, and Urine of Healthy Subjects. Sci. Rep. 2017, 7, 44061. [Google Scholar] [CrossRef] [PubMed]
- Nientiedt, M.; Schmidt, D.; Kristiansen, G.; Muller, S.C.; Ellinger, J. YRNA Expression Profiles are Altered in Clear Cell Renal Cell Carcinoma. Eur. Urol. Focus 2016. [Google Scholar] [CrossRef] [PubMed]
- Corrie, P.G.; Marshall, A.; Dunn, J.A.; Middleton, M.R.; Nathan, P.D.; Gore, M.; Davidson, N.; Nicholson, S.; Kelly, C.G.; Marples, M.; et al. Adjuvant bevacizumab in patients with melanoma at high risk of recurrence (AVAST-M): Preplanned interim results from a multicentre, open-label, randomised controlled phase 3 study. Lancet Oncol. 2014, 15, 620–630. [Google Scholar] [CrossRef]
- Lamble, S.; Batty, E.; Attar, M.; Buck, D.; Bowden, R.; Lunter, G.; Crook, D.; El-Fahmawi, B.; Piazza, P. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 2013, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, L.R.; Zweig, A.S.; Hinrichs, A.S.; Karolchik, D.; Kuhn, R.M.; Wong, M.; Sloan, C.A.; Rosenbloom, K.R.; Roe, G.; Rhead, B.; et al. The UCSC Genome Browser database: Extensions and updates 2013. Nucleic Acids Res. 2013, 41, D64–D69. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Moret, I.; Sanchez-Izquierdo, D.; Iborra, M.; Tortosa, L.; Navarro-Puche, A.; Nos, P.; Cervera, J.; Beltran, B. Assessing an improved protocol for plasma microRNA extraction. PLoS ONE 2013, 8, e82753. [Google Scholar] [CrossRef]
- Lardizabal, M.N.; Nocito, A.L.; Daniele, S.M.; Ornella, L.A.; Palatnik, J.F.; Veggi, L.M. Reference genes for real-time PCR quantification of microRNAs and messenger RNAs in rat models of hepatotoxicity. PLoS ONE 2012, 7, e36323. [Google Scholar] [CrossRef]
- Xiang, M.; Zeng, Y.; Yang, R.; Xu, H.; Chen, Z.; Zhong, J.; Xie, H.; Xu, Y.; Zeng, X. U6 is not a suitable endogenous control for the quantification of circulating microRNAs. Biochem. Biophys. Res. Commun. 2014, 454, 210–214. [Google Scholar] [CrossRef]
- Marabita, F.; de Candia, P.; Torri, A.; Tegner, J.; Abrignani, S.; Rossi, R.L. Normalization of circulating microRNA expression data obtained by quantitative real-time RT-PCR. Brief Bioinform. 2016, 17, 204–212. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.R.; DeLong, D.M.; Clarke-Pearson, D.L. Comparing the areas under two or more correlated receiver operating characteristic curves: A nonparametric approach. Biometrics 1988, 44, 837–845. [Google Scholar] [CrossRef] [PubMed]




| Stage | n | Age (Median) | Sex (M/F/(NK)) | |
|---|---|---|---|---|
| Control | 8 | 58 | 4/4 | |
| Pool 0 | 5 | 76 | 3/2 | |
| NGS cohort | Pool I/II | 8 | 63 | 4/4 |
| Pool III | 8 | 68 | 3/5 | |
| Pool IV | 8 | 59 | 3/5 | |
| Control | 47 | 54 | 18/23 | |
| mRNA | Stage 0 | 34 | 64 | 11/17/(1) |
| validation cohort | Stage I/II | 52 | 58 | 19/25 |
| Stage III/IV | 87 | 57.5 | 32/37/(2) | |
| Control | 28 | 58 | 12/14/(2) | |
| miRNA | Stage 0 | 29 | 51 | 6/19/(4) |
| validation cohort | Stage I/II | 33 | 60 | 10/14/(9) |
| Stage III/IV | 34 | 55 | 11/15/(8) | |
| Control | 22 | 57.5 | 11/11/ | |
| YRNA | Stage 0 | 20 | 51 | 11/5/(4) |
| validation cohort | Stage I/II | 17 | 48 | 9/8 |
| Stage III/IV | 21 | 61 | 8/13 | |
| Total | - | 426 | 58 | 175/221/(30) |
| miRNA | Cont. | Stage 0 | Stage I/II | Stage III | Stage IV | p-Value |
|---|---|---|---|---|---|---|
| miR-134-5p | 7474 | 7290 | 2293 | 4200 | 4876 | 0.0158 |
| miR-320a-3p | 117,411 | 76,952 | 56,705 | 86,933 | 74,154 | 0.0181 |
| miR-21-5p | 356 | 405 | 368 | 782 | 469 | 0.0183 |
| miR-92b-3p | 14 | 14 | 20 | 25 | 51 | 0.0232 |
| miR-98-5p | 9850 | 12,999 | 9124 | 14,243 | 13,627 | 0.0232 |
| miR-16-3p | 21 | 29 | 49 | 39 | 53 | 0.0277 |
| Let-7b | 385 | 507 | 326 | 267 | 396 | 0.0286 |
| miR-1827 | 58 | 28 | 7 | 7 | 11 | 0.0375 |
| miR-1180 | 107 | 145 | 49 | 45 | 42 | 0.0392 |
| miR-628 | 499 | 667 | 375 | 502 | 407 | 0.0496 |
| miR-486 | 4581.23 | 6014.45 | 1501.33 | 2704.89 | 1879.67 | 0.0497 |
| Probe | AUC | Sensitivity | Specificity | 95% CI | 0 * | I/II * | III/IV * |
|---|---|---|---|---|---|---|---|
| miR-320a-3p | 0.798 | 90% | 61% | 0.712–0.869 | 0.751 | 0.870 | 0.828 |
| miR-134-5p | 0.788 | 55% | 96% | 0.704–0.858 | 0.680 | 0.868 | 0.811 |
| ATM | 0.767 | 61% | 72% | 0.697–0.829 | 0.769 | 0.734 | 0.715 |
| AMFR | 0.748 | 52% | 92% | 0.676–0.812 | 0.822 | 0.709 | 0.641 |
| CD109 | 0.753 | 54% | 90% | 0.680–0.816 | 0.816 | 0.706 | 0.702 |
| SOS1 | 0.772 | 48% | 95% | 0.699–0.835 | 0.796 | 0.694 | 0.693 |
| Panel | 0.825 | 75% | 92% | - | - | - | - |
| Stage 0/I/II | Stage III/IV | Fold Change | p-Value | |
|---|---|---|---|---|
| RNY4P18 | 85 | 193 | 2.2 | 0.00033 |
| RNY3P1 | 86 | 210 | 2.4 | 0.00063 |
| RNY4P6 | 12,442 | 24,473 | 1.9 | 0.00072 |
| RNY4P1 | 68 | 146 | 2.1 | 0.00090 |
| RNY4P25 | 180 | 317 | 1.7 | 0.0032 |
| Cohort | n Samples | |
|---|---|---|
| mRNA | Oxford | 30 |
| AVAST-M | 83 | |
| Madrid | 51 | |
| San Sebastián | 67 | |
| miRNA | AVAST-M | 6 |
| Madrid | 35 | |
| San Sebastián | 83 | |
| yRNA | Madrid | 51 |
| San Sebastián | 67 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solé, C.; Tramonti, D.; Schramm, M.; Goicoechea, I.; Armesto, M.; Hernandez, L.I.; Manterola, L.; Fernandez-Mercado, M.; Mujika, K.; Tuneu, A.; et al. The Circulating Transcriptome as a Source of Biomarkers for Melanoma. Cancers 2019, 11, 70. https://doi.org/10.3390/cancers11010070
Solé C, Tramonti D, Schramm M, Goicoechea I, Armesto M, Hernandez LI, Manterola L, Fernandez-Mercado M, Mujika K, Tuneu A, et al. The Circulating Transcriptome as a Source of Biomarkers for Melanoma. Cancers. 2019; 11(1):70. https://doi.org/10.3390/cancers11010070
Chicago/Turabian StyleSolé, Carla, Daniela Tramonti, Maike Schramm, Ibai Goicoechea, María Armesto, Luiza I. Hernandez, Lorea Manterola, Marta Fernandez-Mercado, Karmele Mujika, Anna Tuneu, and et al. 2019. "The Circulating Transcriptome as a Source of Biomarkers for Melanoma" Cancers 11, no. 1: 70. https://doi.org/10.3390/cancers11010070
APA StyleSolé, C., Tramonti, D., Schramm, M., Goicoechea, I., Armesto, M., Hernandez, L. I., Manterola, L., Fernandez-Mercado, M., Mujika, K., Tuneu, A., Jaka, A., Tellaetxe, M., Friedländer, M. R., Estivill, X., Piazza, P., Ortiz-Romero, P. L., Middleton, M. R., & Lawrie, C. H. (2019). The Circulating Transcriptome as a Source of Biomarkers for Melanoma. Cancers, 11(1), 70. https://doi.org/10.3390/cancers11010070