An IKK/NF-κB Activation/p53 Deletion Sequence Drives Liver Carcinogenesis and Tumor Differentiation

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Generation of Inducible Ikk2ca Trp53−/− Mice to Study the Consequences of Inflammation and p53 Loss
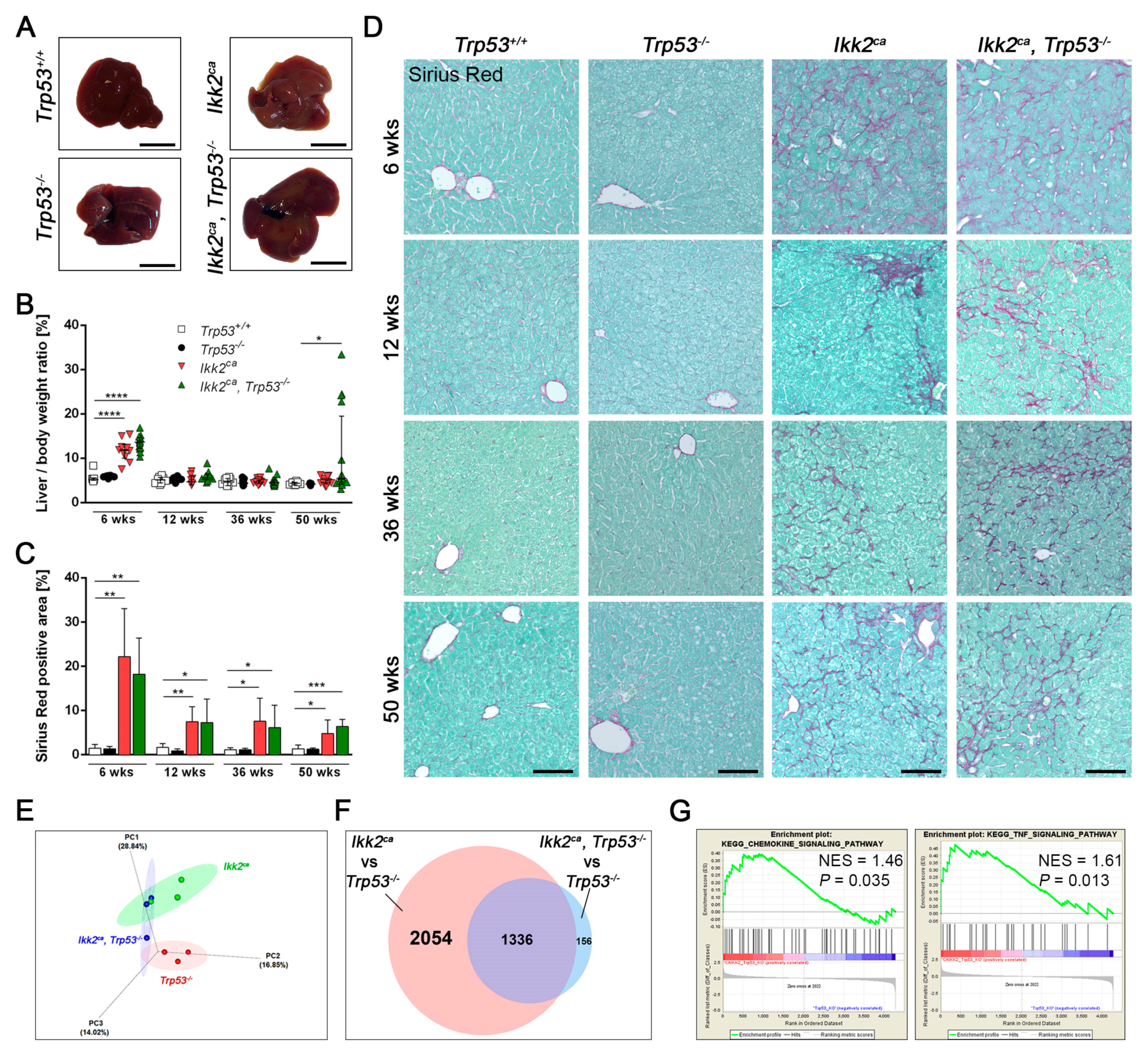
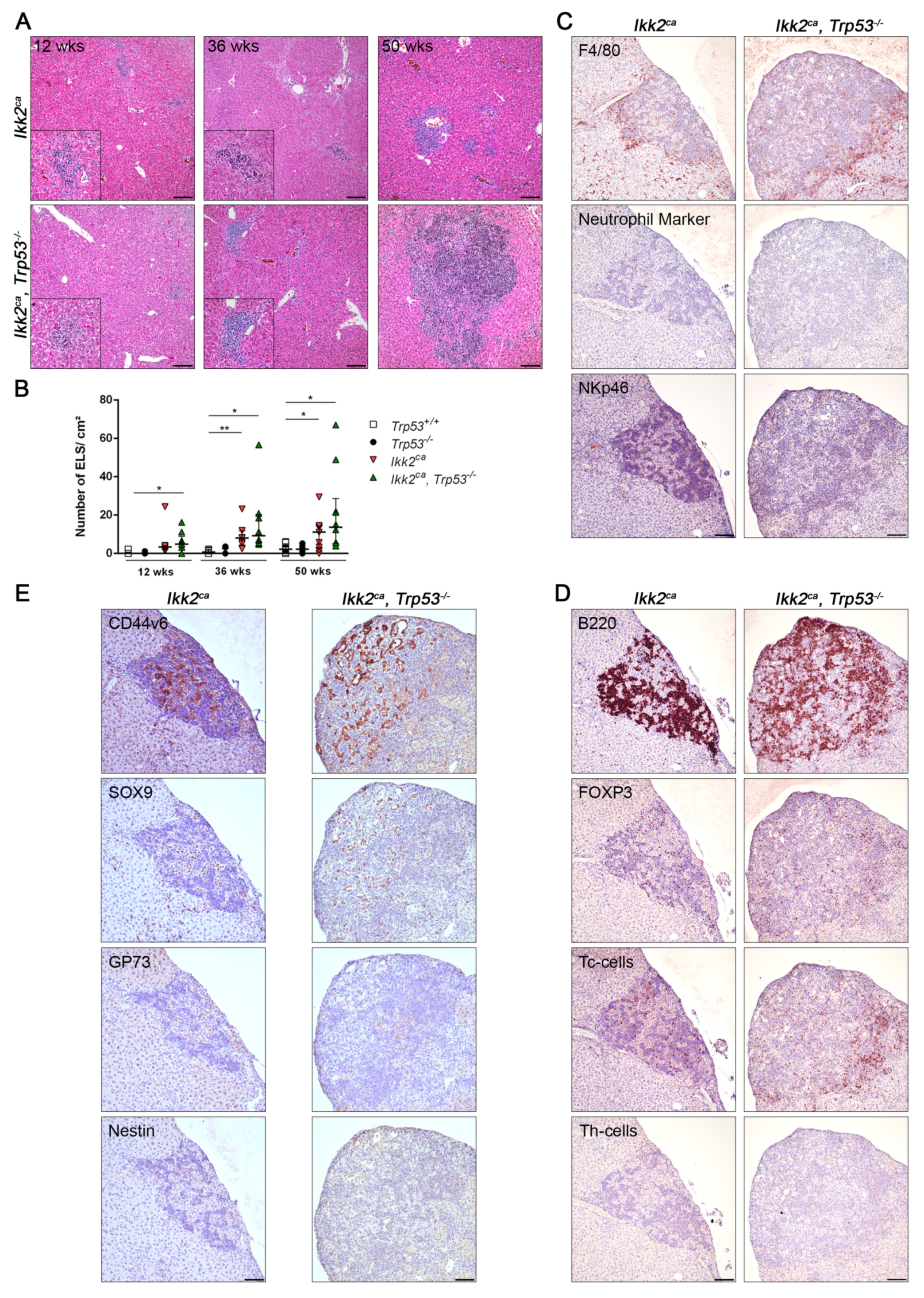
2.2. IKK2ca Expression Leads to Chronic Inflammation and Fibrosis Formation Independent of the p53 Status
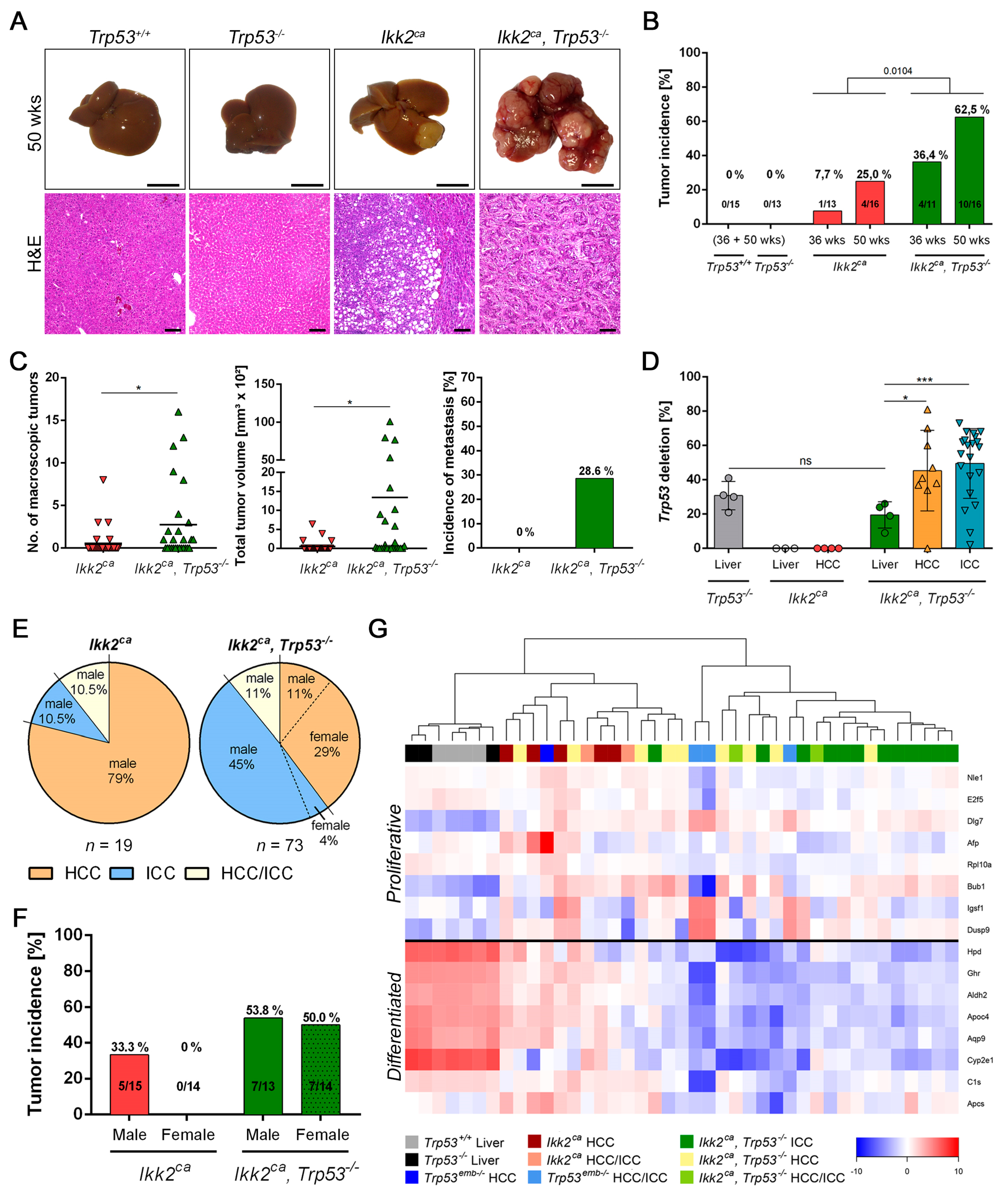
2.3. Trp53 Deletion Leads to Increased Tumor Burden and Shift in Tumor Differentiation
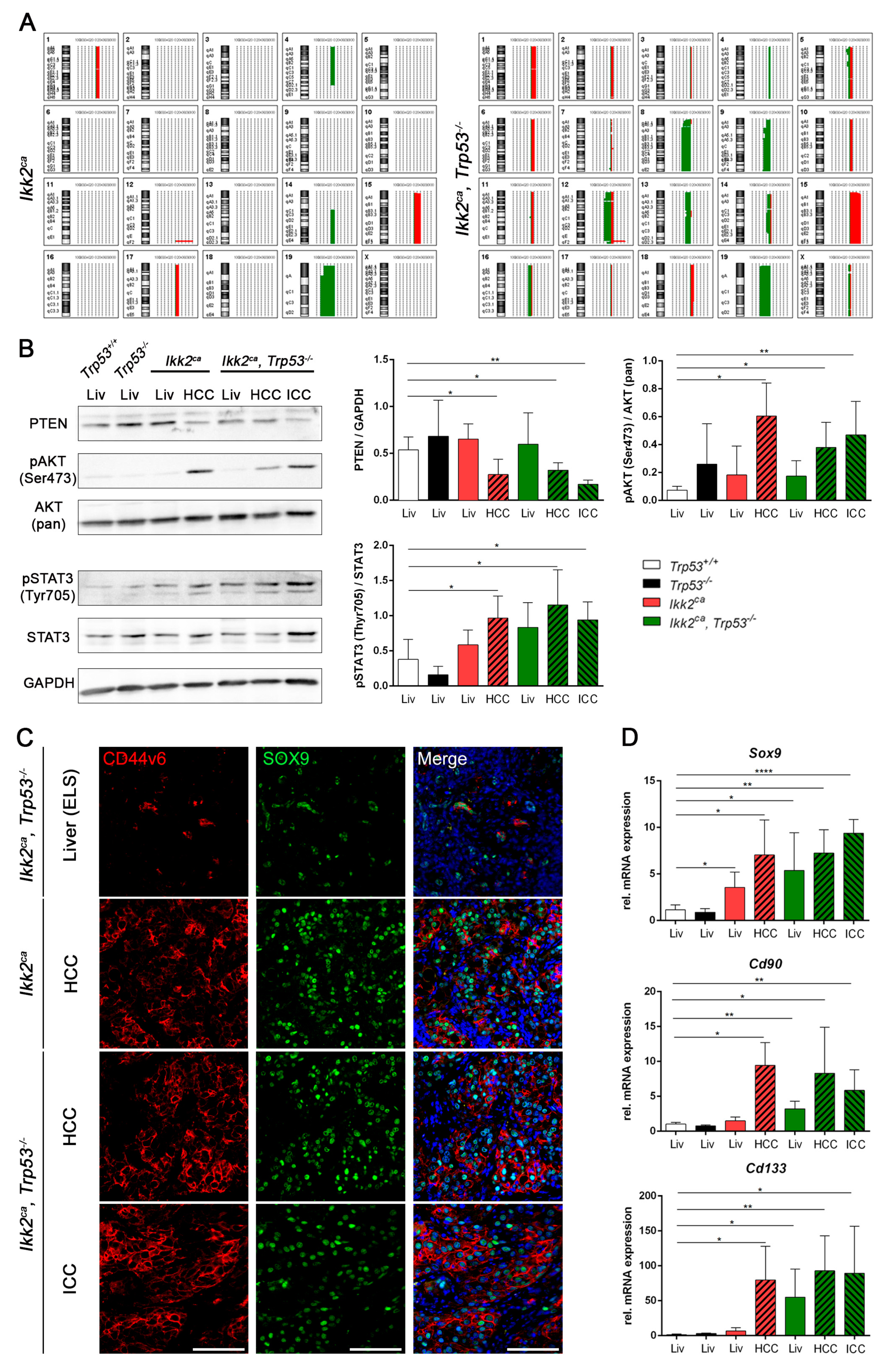
2.4. Liver Progenitor Marker Persist in Liver Tumors of IKK2ca Expressing Mice
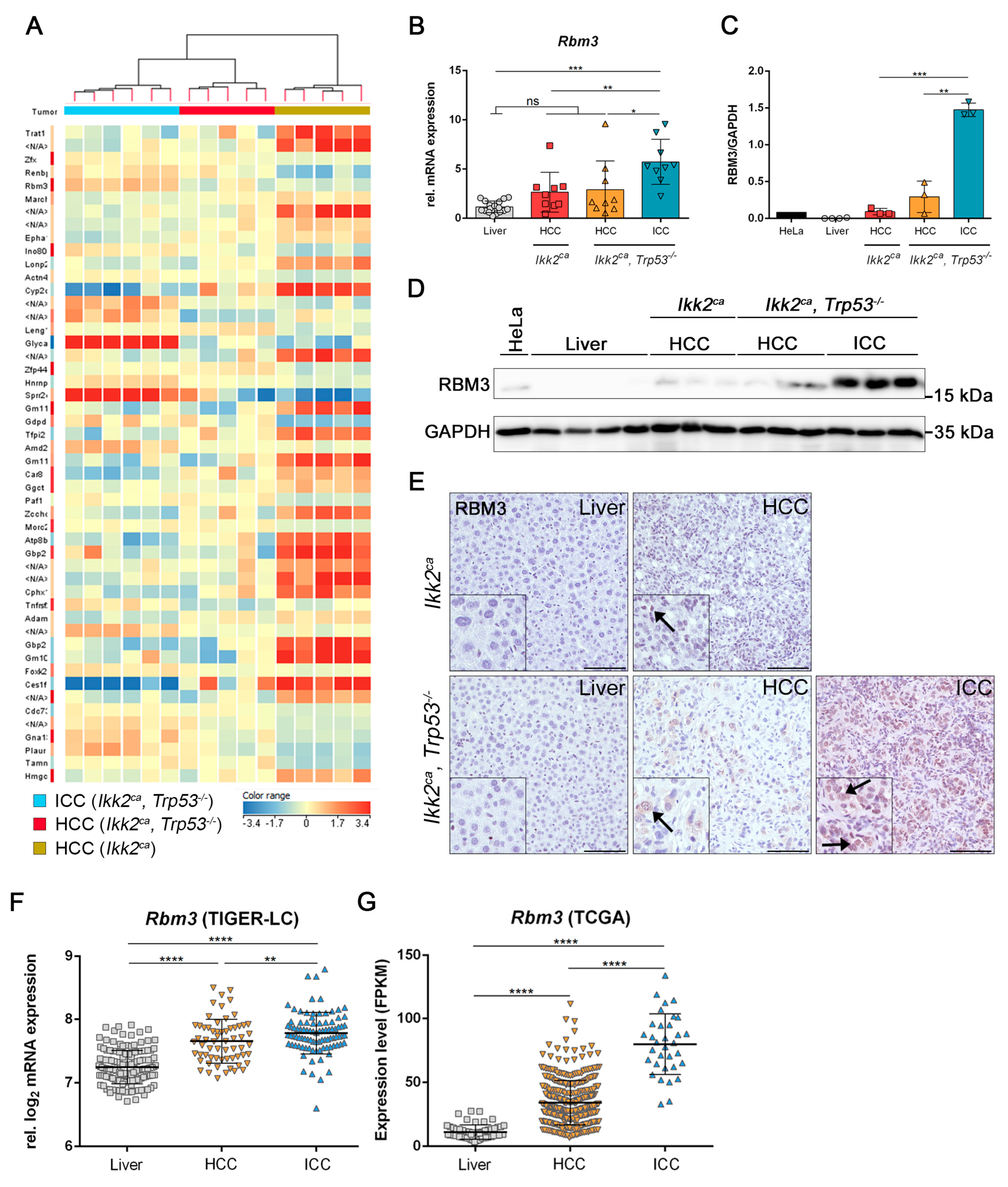
2.5. Differentially Regulated Genes in Liver Carcinogenesis of Ikk2ca Expressing Mice
3. Discussion
4. Material and Methods
4.1. Mouse Model
4.2. Liver Histology
4.3. Immunohistochemistry and Immunofluorescence
4.4. Genomic DNA Isolation and Analysis of Trp53 Deletion Rate
4.5. Western Blotting Analysis
4.6. RNA Isolation and cDNA Synthesis
4.7. Quantitative Real-time PCR and 16-gene Set Analysis
4.8. Serum Parameters
4.9. Electrophoretic Mobility Shift Assay (EMSA)
4.10. Array-based Comparative Genomic Hybridization (aCGH)
4.11. Gene Expression Analysis
4.12. Gene Expression Analysis of Human Data
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Affo, S.; Yu, L.X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2016, 12, 153–186. [Google Scholar] [CrossRef] [PubMed]
- Arsura, M.; Panta, G.R.; Bilyeu, J.D.; Cavin, L.G.; Sovak, M.A.; Oliver, A.A.; Factor, V.; Heuchel, R.; Mercurio, F.; Thorgeirsson, S.S.; et al. Transient activation of NF-kappaB through a TAK1/IKK kinase pathway by TGF-beta1 inhibits AP-1/SMAD signaling and apoptosis: Implications in liver tumor formation. Oncogene 2003, 22, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Maeda, S.; Chang, L.; Karin, M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA 2006, 103, 10544–10551. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.S.; Maeda, S.; He, G.; Karin, M.; Leffert, H.L. Targeted deletion of hepatocyte Ikkbeta confers growth advantages. Biochem. Biophys. Res. Commun. 2009, 380, 349–354. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Koppe, C.; Verheugd, P.; Gautheron, J.; Reisinger, F.; Kreggenwinkel, K.; Roderburg, C.; Quagliata, L.; Terracciano, L.; Gassler, N.; Tolba, R.H.; et al. IκB kinaseα/β control biliary homeostasis and hepatocarcinogenesis in mice by phosphorylating the cell-death mediator receptor-interacting protein kinase 1. Hepatology 2016, 64, 1217–1231. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K.; Rao, M.S. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G852–G858. [Google Scholar] [CrossRef]
- Mandrekar, P.; Szabo, G. Signalling pathways in alcohol-induced liver inflammation. J. Hepatol. 2009, 50, 1258–1266. [Google Scholar] [CrossRef] [Green Version]
- Hösel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Zedler, U.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J.; et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009, 50, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Larrea, E.; Sola, I.; Majano, P.L.; Jiménez, C.; Civeira, M.P.; Prieto, J. Nuclear factor-kappa B in the liver of patients with chronic hepatitis C: Decreased RelA expression is associated with enhanced fibrosis progression. Hepatology 2001, 34, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Kosters, A.; Karpen, S.J. The role of inflammation in cholestasis: Clinical and basic aspects. Semin. Liver Dis. 2010, 30, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.H.; Schwabe, R.F.; Bataller, R.; Russo, M.P.; Jobin, C.; Brenner, D.A. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003, 37, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Hellerbrand, C.; Jobin, C.; Iimuro, Y.; Licato, L.; Sartor, R.B.; Brenner, D.A. Inhibition of NFkappaB in activated rat hepatic stellate cells by proteasome inhibitors and an IkappaB super-repressor. Hepatology 1998, 27, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239. [Google Scholar] [CrossRef]
- Woo, H.G.; Wang, X.W.; Budhu, A.; Kim, Y.H.; Kwon, S.M.; Tang, Z.Y.; Sun, Z.; Harris, C.C.; Thorgeirsson, S.S. Association of TP53 mutations with stem cell-like gene expression and survival of patients with hepatocellular carcinoma. Gastroenterology 2011, 140, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.F.; Lechel, A.; Obenauf, A.C.; Begus-Nahrmann, Y.; Kraus, J.M.; Hoffmann, E.M.; Duda, J.; Eshraghi, P.; Hartmann, D.; Liss, B.; et al. Disruption of Trp53 in livers of mice induces formation of carcinomas with bilineal differentiation. Gastroenterology 2012, 142, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Leithäuser, F.; Gul, S.; Fiedler, K.; Güldiken, N.; Espenlaub, S.; Holzmann, K.H.; Hipp, N.; Sindrilaru, A.; Luedde, T.; et al. Hepatic activation of IKK/NFκB signaling induces liver fibrosis via macrophage-mediated chronic inflammation. Hepatology 2012, 56, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Ohrnberger, S.; Thavamani, A.; Braeuning, A.; Lipka, D.B.; Kirilov, M.; Geffers, R.; Autenrieth, S.E.; Römer, M.; Zell, A.; Bonin, M.; et al. Dysregulated serum response factor triggers formation of hepatocellular carcinoma. Hepatology 2015, 61, 979–989. [Google Scholar] [CrossRef]
- Cairo, S.; Armengol, C.; De Reyniès, A.; Wei, Y.; Thomas, E.; Renard, C.A.; Goga, A.; Balakrishnan, A.; Semeraro, M.; Gresh, L.; et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 2008, 14, 471–484. [Google Scholar] [CrossRef]
- Won, C.; Kim, B.H.; Yi, E.H.; Choi, K.J.; Kim, E.K.; Jeong, J.M.; Lee, J.H.; Jang, J.J.; Yoon, J.H.; Jeong, W.I.; et al. Signal transducer and activator of transcription 3-mediated CD133 up-regulation contributes to promotion of hepatocellular carcinoma. Hepatology 2015, 62, 1160–1173. [Google Scholar] [CrossRef]
- Finkin, S.; Yuan, D.; Stein, I.; Taniguchi, K.; Weber, A.; Unger, K.; Browning, J.L.; Goossens, N.; Nakagawa, S.; Gunasekaran, G.; et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat. Immunol. 2015, 16, 1235–1244. [Google Scholar] [CrossRef]
- Sakurai, T.; Kashida, H.; Komeda, Y.; Nagai, T.; Hagiwara, S.; Watanabe, T.; Kitano, M.; Nishida, N.; Fujita, J.; Kudo, M. Stress Response Protein RBM3 Promotes the Development of Colitis-associated Cancer. Inflamm. Bowel. Dis. 2017, 23, 57–65. [Google Scholar] [CrossRef]
- Pilotte, J.; Kiosses, W.; Chan, S.W.; Makarenkova, H.P.; Dupont-Versteegden, E.; Vanderklish, P.W. Morphoregulatory functions of the RNA-binding motif protein 3 in cell spreading, polarity and migration. Sci. Rep. 2018, 8, 7367. [Google Scholar] [CrossRef]
- Nakatani, T.; Roy, G.; Fujimoto, N.; Asahara, T.; Ito, A. Sex hormone dependency of diethylnitrosamine-induced liver tumors in mice and chemoprevention by leuprorelin. Jpn. J. Cancer Res. 2001, 92, 249–256. [Google Scholar] [CrossRef]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Dieu-Nosjean, M.C.; Giraldo, N.A.; Kaplon, H.; Germain, C.; Fridman, W.H.; Sautès-Fridman, C. Tertiary lymphoid structures, drivers of the anti-tumor responses in human cancers. Immunol. Rev. 2016, 271, 260–275. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, J.; Petitprez, F.; Becht, E.; Laurent, A.; Hirsch, T.Z.; Rousseau, B.; Luciani, A.; Amaddeo, G.; Derman, J.; Charpy, C.; et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J. Hepatol. 2019, 70, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Webster, G.A.; Perkins, N.D. Transcriptional cross talk between NF-kappaB and p53. Mol. Cell Biol. 1999, 19, 3485–3495. [Google Scholar] [CrossRef] [PubMed]
- Ikenoue, T.; Terakado, Y.; Nakagawa, H.; Hikiba, Y.; Fujii, T.; Matsubara, D.; Noguchi, R.; Zhu, C.; Yamamoto, K.; Kudo, Y.; et al. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci. Rep. 2016, 6, 23899. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.K.; Fang, Z.; Jiang, T.Y.; Wan, Z.H.; Pan, Y.F.; Ma, Y.H.; Shi, Y.Y.; Tan, Y.X.; Dong, L.W.; Zhang, Y.J.; et al. Combination of Kras activation and PTEN deletion contributes to murine hepatopancreatic ductal malignancy. Cancer Lett. 2018, 421, 161–169. [Google Scholar] [CrossRef]
- Moon, S.H.; Kim, D.K.; Cha, Y.; Jeon, I.; Song, J.; Park, K.S. PI3K/Akt and Stat3 signaling regulated by PTEN control of the cancer stem cell population, proliferation and senescence in a glioblastoma cell line. Int. J. Oncol. 2013, 42, 921–928. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44⁺CD24⁻ stem cell-like breast cancer cells in human tumors. J. Clin. Invest. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Karnevi, E.; Dror, L.B.; Mardinoglu, A.; Elebro, J.; Heby, M.; Olofsson, S.E.; Nodin, B.; Eberhard, J.; Gallagher, W.; Uhlén, M.; et al. Translational study reveals a two-faced role of RBM3 in pancreatic cancer and suggests its potential value as a biomarker for improved patient stratification. Oncotarget 2017, 9, 6188–6200. [Google Scholar] [CrossRef] [Green Version]
- Marino, S.; Vooijs, M.; van Der Gulden, H.; Jonkers, J.; Berns, A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000, 14, 994–1004. [Google Scholar]
- Maier, H.J.; Marienfeld, R.; Wirth, T.; Baumann, B. Critical role of RelB serine 368 for dimerization and p100 stabilization. J. Biol. Chem. 2003, 278, 39242–39250. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaisaingmongkol, J.; Budhu, A.; Dang, H.; Rabibhadana, S.; Pupacdi, B.; Kwon, S.M.; Forgues, M.; Pomyen, Y.; Bhudhisawasdi, V.; Lertprasertsuke, N.; et al. TIGER LC Consortium. Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017, 32, 57–70. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svinarenko, M.; Katz, S.-F.; Tharehalli, U.; Mulaw, M.A.; Maier, H.J.; Sunami, Y.; Fischer, S.K.; Chen, Y.; Heurich, S.; Erkert, L.; et al. An IKK/NF-κB Activation/p53 Deletion Sequence Drives Liver Carcinogenesis and Tumor Differentiation. Cancers 2019, 11, 1410. https://doi.org/10.3390/cancers11101410
Svinarenko M, Katz S-F, Tharehalli U, Mulaw MA, Maier HJ, Sunami Y, Fischer SK, Chen Y, Heurich S, Erkert L, et al. An IKK/NF-κB Activation/p53 Deletion Sequence Drives Liver Carcinogenesis and Tumor Differentiation. Cancers. 2019; 11(10):1410. https://doi.org/10.3390/cancers11101410
Chicago/Turabian StyleSvinarenko, Michael, Sarah-Fee Katz, Umesh Tharehalli, Medhanie A. Mulaw, Harald J. Maier, Yoshiaki Sunami, Sarah K. Fischer, Yuexin Chen, Sabine Heurich, Lena Erkert, and et al. 2019. "An IKK/NF-κB Activation/p53 Deletion Sequence Drives Liver Carcinogenesis and Tumor Differentiation" Cancers 11, no. 10: 1410. https://doi.org/10.3390/cancers11101410
APA StyleSvinarenko, M., Katz, S. -F., Tharehalli, U., Mulaw, M. A., Maier, H. J., Sunami, Y., Fischer, S. K., Chen, Y., Heurich, S., Erkert, L., Tannapfel, A., Wirth, T., Schirmbeck, R., Seufferlein, T., & Lechel, A. (2019). An IKK/NF-κB Activation/p53 Deletion Sequence Drives Liver Carcinogenesis and Tumor Differentiation. Cancers, 11(10), 1410. https://doi.org/10.3390/cancers11101410