Sestrins as a Therapeutic Bridge between ROS and Autophagy in Cancer

 ,
, 

Abstract
:1. ROS in Health and Disease
1.1. Regulation of ROS Levels in Physiology
1.2. A Prominent Example of the Cell Conundrum: ROS in Cancer Biology
1.3. Dysregulated Nutrient/Growth Homeostasis Signaling as a Source of ROS
2. Autophagy and Cancer
2.1. Macroautophagy
2.2. Dual Role of Autophagy in Cancer
3. Sestrins at the Crossroads of ROS, Oxidative Stress and Autophagy
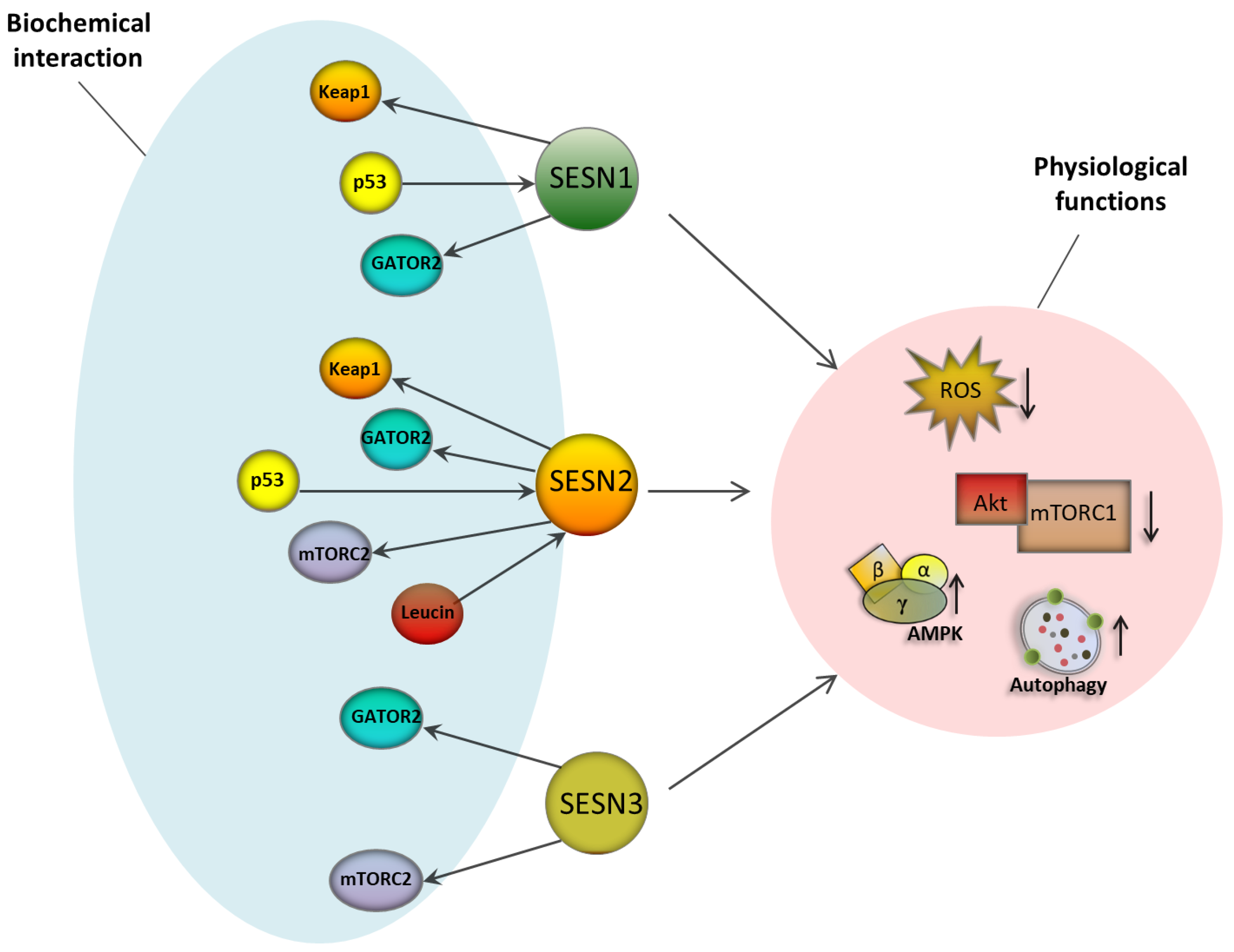
3.1. The Sestrin Protein Family
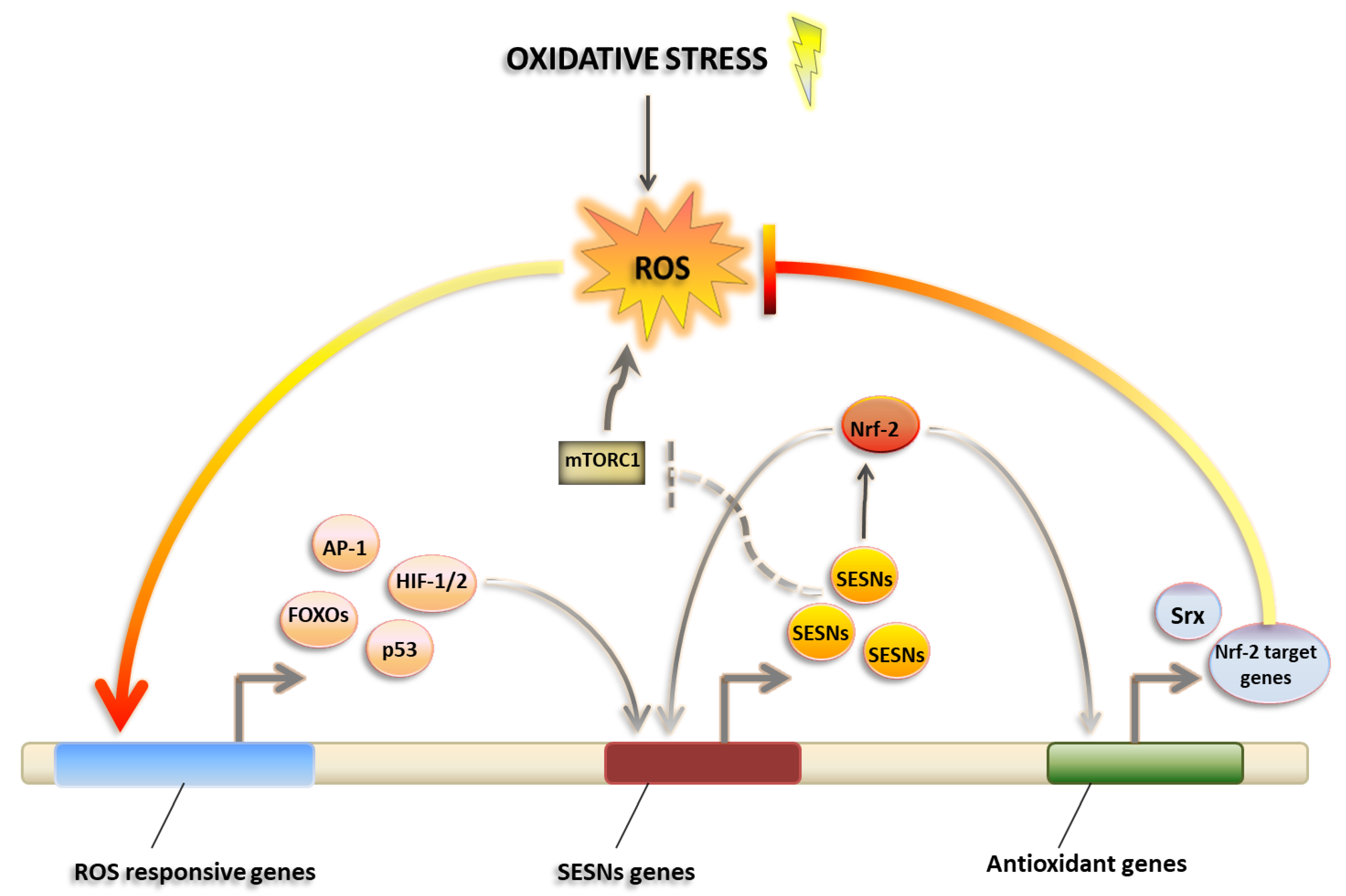
3.2. Sestrins as Master Regulators of ROS Management
3.3. Sestrins and Nutrient Sensing: An Entry Point to Intervene Autophagy?
4. Sestrins and the Therapeutical Management of Cancer
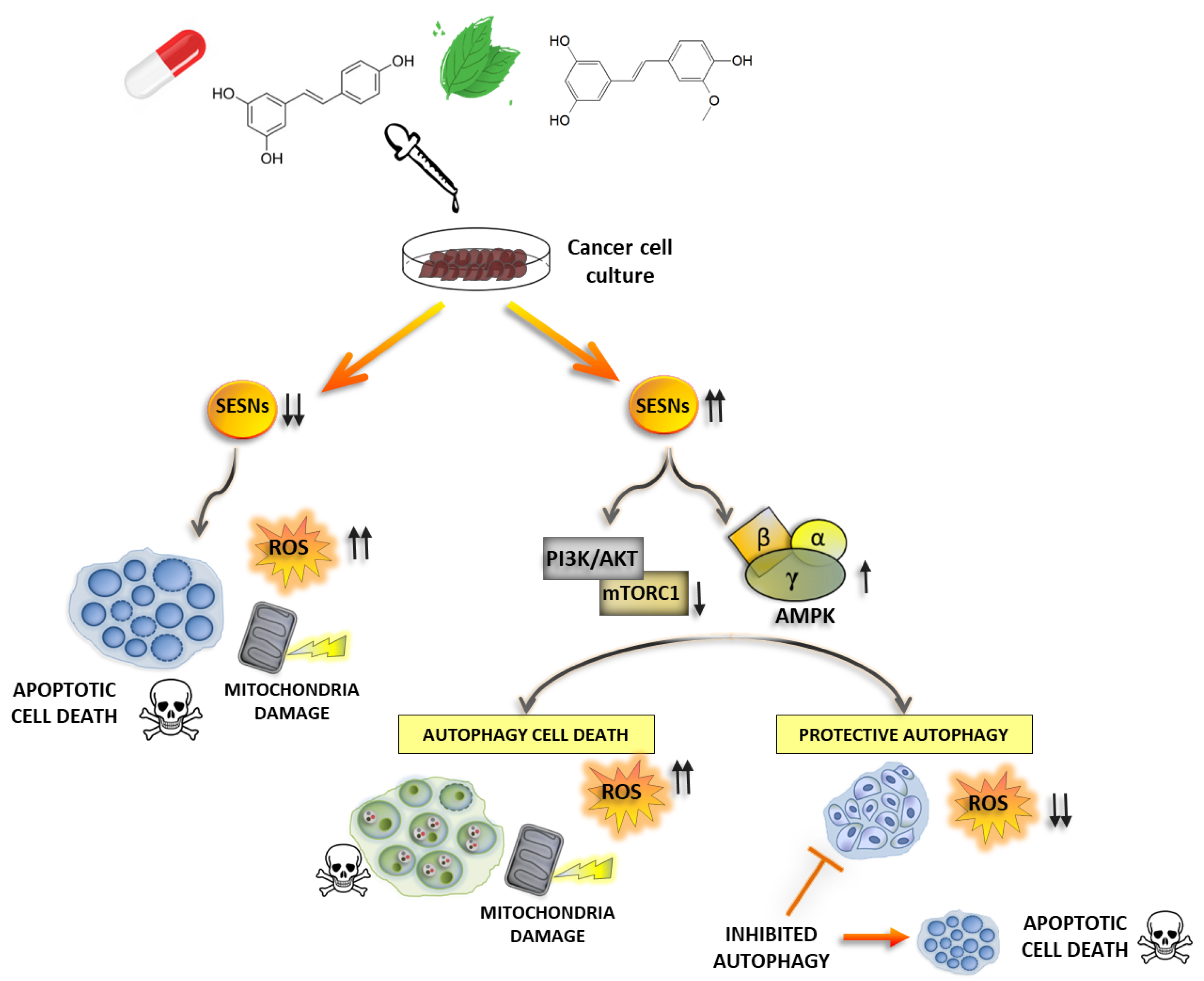
4.1. Modulation of SESNs by Natural Compounds
4.2. Other Therapeutics Acting on SESNs Expression
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bazhin, A.V.; Philippov, P.P.; Karakhanova, S. Reactive Oxygen Species in Cancer Biology and Anticancer Therapy. Oxid. Med. Cell. Longev. 2016, 2016, 4197815. [Google Scholar] [CrossRef] [PubMed]
- Bunik, V.I.; Mkrtchyan, G.; Grabarska, A.; Oppermann, H.; Daloso, D.; Araujo, W.L.; Juszczak, M.; Rzeski, W.; Bettendorff, L.; Fernie, A.R.; et al. Inhibition of Mitochondrial 2-Oxoglutarate Dehydrogenase Impairs Viability of Cancer Cells in a Cell-Specific Metabolism-Dependent Manner. Oncotarget 2016, 7, 26400–26421. [Google Scholar] [CrossRef] [PubMed]
- Simmen, T.; Lynes, E.M.; Gesson, K.; Thomas, G. Oxidative Protein Folding in the Endoplasmic Reticulum: Tight Links to the Mitochondria-Associated Membrane (MAM). Biochim. Biophys. 2010, 1798, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell. Phys. 2016, 231, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox Signaling and Unfolded Protein Response Coordinate Cell Fate Decisions under ER Stress. Redox Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of Misfolded Proteins Prevents ER-Derived Oxidative Stress and Cell Death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wise, D.R.; Diehl, J.A.; Simon, M.C. Hypoxic Reactive Oxygen Species Regulate the Integrated Stress Response and Cell Survival. J. Biol. Chem. 2008, 283, 31153–31162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConkey, D.J. The Integrated Stress Response and Proteotoxicity in Cancer Therapy. Biochem. Biophys. Res. Commun. 2017, 482, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M.; Dalyot, I.; Engelberg, D.; Levitzki, A. Enhanced ROS Production in Oncogenically Transformed Cells Potentiates C-Jun N-Terminal Kinase and P38 Mitogen-Activated Protein Kinase Activation and Sensitization to Genotoxic Stress. Mol. Cell. Biol. 2001, 21, 6913–6926. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. The DNA Damage Response and Cancer Therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Rodríguez-Vargas, J.M.; Oliver-Pozo, F.J.; Dantzer, F. PARP1 and Poly(ADP-Ribosyl)Ation Signaling during Autophagy in Response to Nutrient Deprivation. Oxid. Med. Cell. Longev. 2019, 2019, 2641712. [Google Scholar] [CrossRef]
- Tao, J.; Wu, Q.Y.; Ma, Y.C.; Chen, Y.L.; Zou, C.G. Antioxidant Response is a Protective Mechanism against Nutrient Deprivation in C. Elegans. Sci. Rep. 2017, 7, 43547. [Google Scholar] [CrossRef] [PubMed]
- Remacle, J.; Raes, M.; Toussaint, O.; Renard, P.; Rao, G. Low Levels of Reactive Oxygen Species as Modulators of Cell Function. Mutat. Res. DNAging 1995, 316, 103–122. [Google Scholar] [CrossRef]
- Li, J.; Huang, Q.; Long, X.; Guo, X.; Sun, X.; Jin, X.; Li, Z.; Ren, T.; Yuan, P.; Huang, X.; et al. Mitochondrial Elongation-Mediated Glucose Metabolism Reprogramming Is Essential for Tumor Cell Survival during Energy Stress. Oncogene 2017, 36, 4901–4912. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Wakimoto, H.; Iafrate, A.J.; Tanaka, S.; Loebel, F.; Lelic, N.; Wiederschain, D.; Bedel, O.; Deng, G.; Zhang, B.; et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 2015, 28, 773–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. P53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef]
- Cordani, M.; Butera, G.; Dando, I.; Torrens-Mas, M.; Butturini, E.; Pacchiana, R.; Oppici, E.; Cavallini, C.; Gasperini, S.; Tamassia, N.; et al. Mutant P53 Blocks SESN1/AMPK/PGC-1α/UCP2 Axis Increasing Mitochondrial O 2ˉ Production in Cancer Cells. Br. J. Cancer 2018, 119, 994–1008. [Google Scholar] [CrossRef]
- Erudaitius, D.; Mantooth, J.; Huang, A.; Soliman, J.; Doskey, C.M.; Buettner, G.R.; Rodgers, V.G.J. Calculated Cell-Specific Intracellular Hydrogen Peroxide Concentration: Relevance in Cancer Cell Susceptibility during Ascorbate Therapy. Free Radic. Biol. Med. 2018, 120, 356–367. [Google Scholar] [CrossRef]
- Ma, E.; Chen, P.; Wilkins, H.M.; Wang, T.; Swerdlow, R.H.; Chen, Q. Pharmacologic Ascorbate Induces Neuroblastoma Cell Death by Hydrogen Peroxide Mediated DNA Damage and Reduction in Cancer Cell Glycolysis. Free Radic. Biol. Med. 2017, 113, 36–47. [Google Scholar] [CrossRef]
- Kang, R.; Kroemer, G.; Tang, D. The Tumor Suppressor Protein P53 and the Ferroptosis Network. Free Radic. Biol. Med. 2018, 133, 162–168. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Glasauer, A.; Hoover, P.; Yang, S.; Blatt, H.; Mullen, A.R.; Getsios, S.; Gottardi, C.J.; DeBerardinis, R.J.; Lavker, R.M.; et al. Mitochondrial Reactive Oxygen Species Promote Epidermal Differentiation and Hair Follicle Development. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial Metabolism and ROS Generation Are Essential for Kras-Mediated Tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Chou, D.S.; Hsiao, G.; Lai, Y.A.; Tsai, Y.J.; Sheu, J.R. Baicalein Induces Proliferation Inhibition in B16F10 Melanoma Cells by Generating Reactive Oxygen Species via 12-Lipoxygenase. Free Radic. Biol. Med. 2009, 46, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Laplate, M.; Sabatini, D.M. MTOR Signaling in Growth Control and Disease. Cell 2012, 168, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Park, S.Y.; Kim, Y.M.; Lee, W.S.; Park, O.J. AMP Kinase/Cyclooxygenase-2 Pathway Regulates Proliferation and Apoptosis of Cancer Cells Treated with Quercetin. Exp. Mol. Med. 2009, 41, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Hall, M.N. Where Is MTOR and What Is It Doing There? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Yonezawa, K.; Weng, Q.P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino Acid Sufficiency and MTOR Regulate P70 S6 Kinase and EIF-4E BP1 through a Common Effector Mechanism. J. Biol. Chem. 1998, 273, 14484–14494. [Google Scholar] [CrossRef]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to MTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor Suppressor Complex with GAP Activity for the Rag GTPases That Signal Amino Acid Sufficiency to MTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef]
- Hardie, D.G.; Carling, D.; Gamblin, S.J. AMP-Activated Protein Kinase: Also Regulated by ADP? Trends Biochem. Sci. 2011, 36, 470–477. [Google Scholar] [CrossRef]
- Hardie, D.G. Sensing of Energy and Nutrients by AMP-Activated Protein Kinase. Am. J. Clin. Nutr. 2011, 93, 891S–896S. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.J.; Bardeesy, N.; Manning, B.D.; Lopez, L.; Kosmatka, M.; DePinho, R.A.; Cantley, L.C. The LKB1 Tumor Suppressor Negatively Regulates MTOR Signaling. Cancer Cell 2004, 6, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-Activated Protein Kinase Induces a P53-Dependent Metabolic Checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; De Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The Regulation of AMPK Β1, TSC2, and PTEN Expression by P53: Stress, Cell and Tissue Specificity, and the Role of These Gene Products in Modulating the IGF-1-AKT-MTOR Pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. P53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and MTOR Signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef]
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Dalla Pozza, E.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant P53 Proteins Counteract Autophagic Mechanism Sensitizing Cancer Cells to MTOR Inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function Mutant P53 Promotes Cell Growth and Cancer Cell Metabolism via Inhibition of AMPK Activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From Phenomenology to Molecular Understanding in Less than a Decade. Nat. Rev. Mol. Cell. Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 Induces Autophagy by Phosphorylating Beclin-1 and Activating VPS34 Lipid Kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Physiological Functions of Autophagy. Curr. Top Microbiol. Immunol. 2009, 335, 71–84. [Google Scholar] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.; Darley-Usmar, V.; Zhang, J. Cellular Metabolic and Autophagic Pathways: Traffic Control by Redox Signaling. Free Radic. Biol. Med. 2013, 63, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The Role of Autophagy in Cancer Development and Response to Therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual Role of Autophagy in Hallmarks of Cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-Induced Autophagy Promotes Tumor Cell Survival and Adaptation to Antiangiogenic Treatment in Glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef] [Green Version]
- White, E. Deconvoluting the Context-Dependent Role for Autophagy in Cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Chromosomal Instability Autophagy Suppresses Tumor Progression by Limiting Chromosomal Instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef]
- Lee, J.H.; Budanov, A.V.; Karin, M. Sestrins Orchestrate Cellular Metabolism to Attenuate Aging. Cell Metab. 2013, 18, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Budanov, A.V.; Shoshani, T.; Faerman, A.; Zelin, E.; Kamer, I.; Kalinski, H.; Gorodi, S.; Fishman, A.; Chajut, A.; Einat, P.; et al. Identification of a Novel Stress-Responsive Gene Hi95 Involved in Regulation of Cell Viability. Oncogene 2002, 21, 6017–6031. [Google Scholar] [CrossRef]
- Velasco-Miguel, S.; Buckbinder, L.; Jean, P.; Gelbert, L.; Talbott, R.; Laidlaw, J.; Seizinger, B.; Kley, N.; Laidlaw, J.; Seizinger, B.; et al. PA26, a Novel Target of the P53 Tumor Suppressor and Member of the GADD Family of DNA Damage and Growth Arrest Inducible Genes. Oncogene 1999, 18, 127–137. [Google Scholar] [CrossRef]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.L.; Karin, M.; Budanov, A.V. Sestrins Inhibit MTORC1 Kinase Activation through the GATOR Complex. Cell Rep. 2014, 9, 1281–1291. [Google Scholar] [CrossRef]
- Nogueira, V.; Park, Y.; Chen, C.C.; Xu, P.Z.; Chen, M.L.; Tonic, I.; Unterman, T.; Hay, N. Akt Determines Replicative Senescence and Oxidative or Oncogenic Premature Senescence and Sensitizes Cells to Oxidative Apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Kopnin, P.B.; Agapova, L.S.; Kopnin, B.P.; Chumakov, P.M. Repression of Sestrin Family Genes Contributes to Oncogenic Ras-Induced Reactive Oxygen Species upregulation and Genetic Instability. Cancer Res. 2007, 67, 4671–4678. [Google Scholar] [CrossRef]
- Ro, S.H.; Xue, X.; Ramakrishnan, S.K.; Cho, C.S.; Namkoong, S.; Jang, I.; Semple, I.A.; Ho, A.; Park, H.W.; Shah, Y.M.; et al. Tumor Suppressive Role of Sestrin2 during Colitis and Colon Carcinogenesis. eLife 2016, 5, e12204. [Google Scholar] [CrossRef]
- Dai, J.; Huang, Q.; Niu, K.; Wang, B.; Li, Y.; Dai, C.; Chen, Z.; Tao, K.; Dai, J. Sestrin 2 Confers Primary Resistance to Sorafenib by Simultaneously Activating AKT and AMPK in Hepatocellular Carcinoma. Cancer Med. 2018, 7, 5691–5703. [Google Scholar] [CrossRef]
- Kim, J.S.; Ro, S.H.; Kim, M.; Park, H.W.; Semple, I.A.; Park, H.; Cho, U.S.; Wang, W.; Guan, K.L.; Karin, M.; et al. Sestrin2 Inhibits MTORC1 through Modulation of GATOR Complexes. Sci. Rep. 2015, 5, 9502. [Google Scholar] [CrossRef]
- Saxton, R.A.; Knockenhauer, K.E.; Wolfson, R.L.; Chantranupong, L.; Pacold, M.E.; Wang, T.; Schwartz, T.U.; Sabatini, D.M. Structural Basis for Leucine Sensing by the Sestrin2-MTORC1 Pathway. Science 2016, 351, 53–58. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 Is a Leucine Sensor for the MTORC1 Pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins Activate Nrf2 by Promoting P62-Dependent Autophagic Degradation of Keap1 and Prevent Oxidative Liver Damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The Antioxidant Function of the P53 Tumor Suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef]
- Budanov, A.V.; Sablina, A.A.; Feinstein, E.; Koonin, E.V.; Chumakov, P.M. Regeneration of Peroxiredoxins by P53-Regulated Sestrins, Homologs of Bacterial AhpD. Science 2004, 304, 596–600. [Google Scholar] [CrossRef]
- Woo, H.A.; Bae, S.H.; Park, S.; Rhee, S.G. Sestrin 2 Is Not a Reductase for Cysteine Sulfinic Acid of Peroxiredoxins. Antioxid. Redox Signal. 2009, 11, 739–745. [Google Scholar] [CrossRef]
- Lee, J.H.; Budanov, A.V.; Park, E.J.; Birse, R.; Kim, T.E.; Perkins, G.A.; Ocorr, K.; Ellisman, M.H.; Bodmer, R.; Bier, E.; et al. Sestrin as a Feedback Inhibitor of TOR That Prevents Age-Related Pathologies. Science 2010, 327, 1223–1228. [Google Scholar] [CrossRef] [Green Version]
- Tajan, M.; Hock, A.K.; Blagih, J.; Robertson, N.A.; Labuschagne, C.F.; Kruiswijk, F.; Humpton, T.J.; Adams, P.D.; Vousden, K.H. A Role for P53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab. 2018, 28, 721–736. [Google Scholar] [CrossRef]
- Chantranupong, L.; Wolfson, R.L.; Orozco, J.M.; Saxton, R.A.; Scaria, S.M.; Bar-Peled, L.; Spooner, E.; Isasa, M.; Gygi, S.P.; Sabatini, D.M. The Sestrins Interact with Gator2 to Negatively Regulate the Amino-Acid-Sensing Pathway Upstream of MTORC1. Cell Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef]
- Chen, X.; Ma, J.J.; Tan, M.; Yao, P.; Hu, Q.H.; Eriani, G.; Wang, E.D. Modular Pathways for Editing Non-Cognate Amino Acids by Human Cytoplasmic Leucyl-TRNA Synthetase. Nucleic Acids Res. 2011, 39, 235–247. [Google Scholar] [CrossRef]
- Xu, D.; Shimkus, K.L.; Lacko, H.A.; Kutzler, L.; Jefferson, L.S.; Kimball, S.R. Evidence for a Role for Sestrin1 in Mediating Leucine-Induced Activation of Mtorc1 in Skeletal Muscle. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E817–E828. [Google Scholar] [CrossRef]
- Ye, J.; Palm, W.; Peng, M.; King, B.; Lindsten, T.; Li, M.O.; Koumenis, C.; Thompson, C.B. GCN2 Sustains MTORC1 Suppression upon Amino Acid Deprivation by Inducing Sestrin2. Genes Dev. 2015, 29, 2331–2336. [Google Scholar] [CrossRef]
- Kimball, S.R.; Gordon, B.S.; Moyer, J.E.; Dennis, M.D.; Jefferson, L.S. Leucine Induced Dephosphorylation of Sestrin2 Promotes MTORC1 Activation. Cell. Signal. 2016, 28, 896–906. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Park, H.W.; Park, H.; Ro, S.H.; Jang, I.; Semple, I.A.; Kim, D.N.; Kim, M.; Nam, M.; Zhang, D.; Yin, L.; et al. Hepatoprotective Role of Sestrin2 against Chronic ER Stress. Nat. Commun. 2014, 5, 4233. [Google Scholar] [CrossRef]
- Hou, Y.S.; Guan, J.J.; Xu, H.D.; Wu, F.; Sheng, R.; Qin, Z.H. Sestrin2 Protects Dopaminergic Cells against Rotenone Toxicity through AMPK-Dependent Autophagy Activation. Mol. Cell. Biol. 2015, 35, 2740–2751. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Dirat, B.; Laurent, K.; Puissant, A.; Auberger, P.; Budanov, A.; Tanti, J.F.; Bost, F. Sestrin2 Integrates Akt and MTOR Signaling to Protect Cells against Energetic Stress-Induced Death. Cell Death Differ. 2013, 20, 611–619. [Google Scholar] [CrossRef]
- Hong-Brown, L.Q.; Brown, C.R.; Navaratnarajah, M.; Lang, C.H. Adamts1 Mediates Ethanol-Induced Alterations in Collagen and Elastin via a FoxO1-Sestrin3-AMPK Signaling Cascade in Myocytes. J. Cell. Biochem. 2015, 116, 91–101. [Google Scholar] [CrossRef]
- Sanli, T.; Linher-Melville, K.; Tsakiridis, T.; Singh, G. Sestrin2 Modulates AMPK Subunit Expression and Its Response to Ionizing Radiation in Breast Cancer Cells. PLoS ONE 2012, 7, e32035. [Google Scholar] [CrossRef]
- Tao, R.; Xiong, X.; Liangpunsakul, S.; Dong, X.C. Sestrin 3 Protein Enhances Hepatic Insulin Sensitivity by Direct Activation of the MTORC2-Akt Signaling. Diabetes 2015, 64, 1211–1223. [Google Scholar] [CrossRef]
- Kim, G.T.; Lee, S.H.; Kim, J.I.; Kim, Y.M. Quercetin Regulates the Sestrin 2-AMPK-P38 MAPK Signaling Pathway and Induces Apoptosis by Increasing the Generation of Intracellular ROS in a P53-Independent Manner. Int. J. Mol. Med. 2014, 33, 863–869. [Google Scholar] [CrossRef]
- Kim, G.T.; Lee, S.H.; Kim, Y.M. Quercetin Regulates Sestrin 2-AMPK-MTOR Signaling Pathway and Induces Apoptosis via Increased Intracellular ROS in HCT116 Colon Cancer Cells. J. Cancer Prev. 2013, 18, 264–270. [Google Scholar] [CrossRef]
- Khan, N.; Jajeh, F.; Khan, M.I.; Mukhtar, E.; Shabana, S.M.; Mukhtar, H. Sestrin-3 Modulation Is Essential for Therapeutic Efficacy of Cucurbitacin B in Lung Cancer Cells. Carcinogenesis 2017, 38, 184–195. [Google Scholar] [CrossRef]
- Jegal, K.H.; Ko, H.L.; Park, S.M.; Byun, S.H.; Kang, K.W.; Cho, I.J.; Kim, S.C. Eupatilin Induces Sestrin2-Dependent Autophagy to Prevent Oxidative Stress. Apoptosis 2016, 21, 642–656. [Google Scholar] [CrossRef]
- Liang, Y.; Zhu, J.; Huang, H.; Xiang, D.; Li, Y.; Zhang, D.; Li, J.; Wang, Y.; Jin, H.; Jiang, G.; et al. SESN2/Sestrin 2 Induction-Mediated Autophagy and Inhibitory Effect of Isorhapontigenin (ISO) on Human Bladder Cancers. Autophagy 2016, 12, 1229–1239. [Google Scholar] [CrossRef]
- Lin, L.; Liu, S.; Leu, J.; Chang, C. Arsenic Trioxide-Mediated Suppression of MiR-182-5p is Associated with Potent Anti-Oxidant Effects through upregulation of SESN2. Oncotarget 2018, 9, 16028–16042. [Google Scholar] [CrossRef]
- Jin, S.H.; Yang, J.H.; Shin, B.Y.; Seo, K.; Shin, S.M.; Cho, I.J.; Ki, S.H. Resveratrol Inhibits LXRα-Dependent Hepatic Lipogenesis through Novel Antioxidant Sestrin2 Gene Induction. Toxicol. Appl. Pharmacol. 2013, 271, 95–105. [Google Scholar] [CrossRef]
- Yan, M.; Vemu, B.; Veenstra, J.; Petiwala, S.M.; Johnson, J.J. Carnosol, A Dietary Diterpene from Rosemary (Rosmarinus Officinalis) Activates Nrf2 Leading to Sestrin 2 Induction in Colon Cells. Integr. Mol. Med. 2018, 5. [Google Scholar] [CrossRef]
- Yen, J.H.; Huang, S.T.; Huang, H.S.; Fong, Y.C.; Wu, Y.Y.; Chiang, J.H.; Su, Y.C. HGK-Sestrin 2 Signaling-Mediated Autophagy Contributes to Antitumor Efficacy of Tanshinone IIA in Human Osteosarcoma Cells. Cell Death Dis. 2018, 9, 1003. [Google Scholar] [CrossRef]
- Kosaka, T.; Hongo, H.; Miyazaki, Y.; Nishimoto, K.; Miyajima, A.; Oya, M.; Kosaka, T.; Hongo, H.; Miyazaki, Y.; Nishimoto, K.; et al. Reactive Oxygen Species Induction by Cabazitaxel through Inhibiting Sestrin-3 in Castration Resistant Prostate Cancer. Oncotarget 2017, 8, 87675–87683. [Google Scholar] [CrossRef]
- Brüning, A.; Rahmeh, M.; Friese, K. Nelfinavir and Bortezomib Inhibit MTOR Activity via ATF4-Mediated Sestrin-2 Regulation. Mol. Oncol. 2013, 7, 1012–1018. [Google Scholar] [CrossRef]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.H.; Yang, N.; Lin, Q.; Xia, D.; Shen, H.M. Histone Deacetylase Inhibitors Induce Autophagy through FOXO1-Dependent Pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef]
- Vakana, E.; Arslan, A.D.; Szilard, A.; Altman, J.K.; Platanias, L.C. Regulatory Effects of Sestrin 3 (SESN3) in BCR-ABL Expressing Cells. PLoS ONE 2013, 8, e78780. [Google Scholar] [CrossRef]
- Chen, J.H.; Zhang, P.; Chen, W.D.; Li, D.D.; Wu, X.Q.; Deng, R.; Jiao, L.; Li, X.; Ji, J.; Feng, G.K.; et al. ATM-Mediated PTEN Phosphorylation Promotes PTEN Nuclear Translocation and Autophagy in Response to DNA-Damaging Agents in Cancer Cells. Autophagy 2015, 11, 239–252. [Google Scholar] [CrossRef]
- Gonzalez, V.J.; Abbas-Aghababazadeh, F.; Fridley, B.L.; Ghansah, T.; Saligan, L.N. Expression of Sestrin Genes in Radiotherapy for Prostate Cancer and Its Association With Fatigue: A Proof-of-Concept Study. Biol. Res. Nurs. 2018, 20, 218–226. [Google Scholar] [CrossRef]
- Kumar, A.; Shaha, C. RBX1-Mediated Ubiquitination of SESN2 Promotes Cell Death upon Prolonged Mitochondrial Damage in SH-SY5Y Neuroblastoma Cells. Mol. Cell. Biochem. 2018, 446, 1–9. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, W.; Wang, Q.; Li, Q.; Wang, H.; Wang, J.; Teng, T.; Chen, M.; Ji, A.; Li, Y. New Drug Candidate Targeting the 4A1 Orphan Nuclear Receptor for Medullary Thyroid Cancer Therapy. Molecules 2018, 23, 565. [Google Scholar] [CrossRef]
- Piotrowska-Kempisty, H.; Rucinski, M.; Borys, S.; Kucinska, M.; Kaczmarek, M.; Zawierucha, P.; Wierzchowski, M.; Lazewski, D.; Murias, M.; Jodynis-Liebert, J. 3′-Hydroxy-3,4,5,4′-Tetramethoxystilbene, the Metabolite of Resveratrol Analogue DMU-212, Inhibits Ovarian Cancer Cell Growth in Vitro and in a Mice Xenograft Model. Sci. Rep. 2016, 6, 32627. [Google Scholar] [CrossRef]
- Zhao, B.; Shah, P.; Qiang, L.; He, T.C.; Budanov, A.; He, Y.Y. Distinct Role of Sesn2 in Response to UVB-Induced DNA Damage and UVA-Induced Oxidative Stress in Melanocytes. Photochem. Photobiol. 2017, 93, 375–381. [Google Scholar] [CrossRef]
- Hua, X.; Xu, J.; Deng, X.; Xu, J.; Li, J.; Zhu, D.Q.; Zhu, J.; Jin, H.; Tian, Z.; Huang, H.; et al. New Compound ChlA-F Induces Autophagy-Dependent Anti-Cancer Effect via Upregulating Sestrin-2 in Human Bladder Cancer. Cancer Lett. 2018, 436, 38–51. [Google Scholar] [CrossRef]
- Cossarizza, A.; Gibellini, L.; Pinti, M.; Nasi, M.; Montagna, J.P.; De Biasi, S.; Roat, E.; Bertoncelli, L.; Cooper, E.L. Quercetin and Cancer Chemoprevention. Evid. Based Complement. Alternat. Med. 2011, 2011, 591356. [Google Scholar]
- Haritunians, T.; Gueller, S.; Zhang, L.; Badr, R.; Yin, D.; Xing, H.; Fung, M.C.; Koeffler, H.P. Cucurbitacin B Induces Differentiation, Cell Cycle Arrest, and Actin Cytoskeletal Alterations in Myeloid Leukemia Cells. Leuk. Res. 2008, 32, 1366–1373. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, H.; Sun, C.; Shan, X.; Yang, X.; Li-Ling, J.; Deng, Y. Targeted Constitutive Activation of Signal Transducer and Activator of Transcription 3 in Human Hepatocellular Carcinoma Cells by Cucurbitacin B. Cancer Chemother. Pharmacol. 2009, 63, 635–642. [Google Scholar] [CrossRef]
- Thoennissen, N.H.; Iwanski, G.B.; Doan, N.B.; Okamoto, R.; Lin, P.; Abbassi, S.; Jee, H.S.; Yin, D.; Toh, M.; Wei, D.X.; et al. Cucurbitacin B Induces Apoptosis by Inhibition of the JAK/STAT Pathway and Potentiates Antiproliferative Effects of Gemcitabine on Pancreatic Cancer Cells. Cancer Res. 2009, 69, 5876–5884. [Google Scholar] [CrossRef]
- Iwanski, G.B.; Lee, D.H.; En-Gal, S.; Doan, N.B.; Castor, B.; Vogt, M.; Toh, M.; Bokemeyer, C.; Said, J.W.; Thoennissen, N.H.; et al. Cucurbitacin B, a Novel in Vivo Potentiator of Gemcitabine with Low Toxicity in the Treatment of Pancreatic Cancer. Br. J. Pharmacol. 2010, 160, 998–1007. [Google Scholar] [CrossRef]
- Cai, M.; Phan, P.T.T.; Hong, J.G.; Kim, D.H.; Kim, J.M.; Park, S.J.; Liu, X.; Han, J.E.; Park, H.; Choi, J.W.; et al. The Neuroprotective Effect of Eupatilin against Ischemia/Reperfusion-Induced Delayed Neuronal Damage in Mice. Eur. J. Pharmacol. 2012, 689, 104–110. [Google Scholar] [CrossRef]
- Lee, S.; Lee, M.; Kim, S.H. Eupatilin Inhibits H2O2-Induced Apoptotic Cell Death through Inhibition of Mitogen-Activated Protein Kinases and Nuclear Factor-ΚB. Food Chem. Toxicol. 2008, 46, 2865–2870. [Google Scholar] [CrossRef]
- Jeong, J.H.; Moon, S.J.; Jhun, J.Y.; Yang, E.J.; Cho, M.L.; Min, J.K. Eupatilin Exerts Antinociceptive and Chondroprotective Properties in a Rat Model of Osteoarthritis by Downregulating Oxidative Damage and Catabolic Activity in Chondrocytes. PLoS ONE 2015, 10, e0130882. [Google Scholar] [CrossRef]
- Min, S.W.; Kim, N.J.; Baek, N.I.; Kim, D.H. Inhibitory Effect of Eupatilin and Jaceosidin Isolated from Artemisia Princeps on Carrageenan-Induced Inflammation in Mice. J. Ethnopharmacol. 2009, 125, 497–500. [Google Scholar] [CrossRef]
- Son, J.E.; Lee, E.; Seo, S.G.; Lee, J.; Kim, J.E.; Kim, J.; Lee, K.W.; Lee, H.J. Eupatilin, a Major Flavonoid of Artemisia, Attenuates Aortic Smooth Muscle Cell Proliferation and Migration by Inhibiting PI3K, MKK3/6, and MKK4 Activities. Planta Med. 2013, 79, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Cheong, J.H.; Hong, S.Y.; Zheng, Y.; Noh, S.H. Eupatilin Inhibits Gastric Cancer Cell Growth by Blocking STAT3-Mediated VEGF Expression. J. Gastric Cancer 2011, 11, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.D.; Chen, G.Q.; Wang, Z.G.; Wang, Z.Y.; Chen, S.J.; Chen, Z. Arsenic Trioxide, a Therapeutic Agent for APL. Oncogene 2001, 20, 7146–7153. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, Z.; Lallemand-Breitenbach, V.; De Thé, H. How Acute Promyclocytic Leukaemia Revived Arsenic. Nat. Rev. Cancer 2002, 2, 705–713. [Google Scholar] [CrossRef]
- Zhang, T.; Cao, E.; Li, J.; Ma, W.; Qin, J. Induction of Apoptosis and Inhibition of Human Gastric Cancer MGC-803 Cell Growth by Arsenic Trioxide. Eur. J. Cancer 1999, 35, 1258–1263. [Google Scholar] [CrossRef]
- Akao, Y.; Nakagawa, Y.; Akiyama, K. Arsenic Trioxide Induces Apoptosis in Neuroblastoma Cell Lines through the Activation of Caspase 3 in Vitro. FEBS Lett. 1999, 455, 59–62. [Google Scholar] [CrossRef]
- Seol, J.G.; Park, W.H.; Kim, E.S.; Jung, C.W.; Hyun, J.M.; Kim, B.K.; Lee, Y.Y. Effect of Arsenic Trioxide on Cell Cycle Arrest in Head and Neck Cancer Cell Line PCI-1. Biochem. Biophys. Res. Commun. 1999, 265, 400–404. [Google Scholar] [CrossRef]
- Chan, J.Y.W.; Siu, K.P.Y.; Fung, K.P. Effect of Arsenic Trioxide on Multidrug Resistant Hepatocellular Carcinoma Cells. Cancer Lett. 2006, 236, 250–258. [Google Scholar] [CrossRef]
- Leung, L.L.; Lam, S.K.; Li, Y.Y.; Ho, J.C.M. Tumor Growth-Suppressive Effect of Arsenic Trioxide in Squamous Cell Lung Carcinoma. Oncol. Lett. 2017, 14, 3748–3754. [Google Scholar] [CrossRef]
- Kumar, S.; Yedjou, C.G.; Tchounwou, P.B. Arsenic Trioxide Induces Oxidative Stress, DNA Damage, and Mitochondrial Pathway of Apoptosis in Human Leukemia (HL-60) Cells. J. Exp. Clin. Cancer Res. 2014, 33, 42. [Google Scholar] [CrossRef]
- Rossman, T.G. Mechanism of Arsenic Carcinogenesis: An Integrated Approach. Mutat. Res. 2003, 533, 37–65. [Google Scholar] [CrossRef]
- Morales, A.A.; Gutman, D.; Cejas, P.J.; Lee, K.P.; Boise, L.H. Reactive Oxygen Species Are Not Required for an Arsenic Trioxide-Induced Antioxidant Response or Apoptosis. J. Biol. Chem. 2009, 284, 12886–12895. [Google Scholar] [CrossRef]
- Joseph, S.B.; Laffitte, B.A.; Patel, P.H.; Watson, M.A.; Matsukuma, K.E.; Walczak, R.; Collins, J.L.; Osborne, T.F.; Tontonoz, P. Direct and Indirect Mechanisms for Regulation of Fatty Acid Synthase Gene Expression by Liver X Receptors. J. Biol. Chem. 2002, 277, 11019–11025. [Google Scholar] [CrossRef] [Green Version]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.M.A.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of Mouse Sterol Regulatory Element-Binding Protein-1c Gene (SREBP-1c) by Oxysterol Receptors, LXRα and LXRβ. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef]
- Steffensen, K.R.; Gustafsson, J.A. Putative Metabolic Effects of the Liver X Receptor (LXR). Diabetes 2004, 53, S36–S42. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP Maintains Lipid Biosynthesis and Viability of Cancer Cells under Lipid- and Oxygen-Deprived Conditions and Defines a Gene Signature Associated with Poor Survival in Glioblastoma Multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef]
- Young, R.M.; Ackerman, D.; Quinn, Z.L.; Mancuso, A.; Gruber, M.; Liu, L.; Giannoukos, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; et al. Dysregulated MTORC1 Renders Cells Critically Dependent on Desaturated Lipids for Survival under Tumor-Like Stress. Genes Dev. 2013, 27, 1115–1131. [Google Scholar] [CrossRef]
- Pandey, K.B.; Rizvi, S.I. Plant Polyphenols as Dietary Antioxidants in Human Health and Disease. Oxid. Med. Cell. Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Baur, J.A.; Sinclair, D.A. Therapeutic Potential of Resveratrol: The in Vivo Evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef]
- Tennen, R.I.; Michishita-Kioi, E.; Chua, K.F. Finding a Target for Resveratrol. Cell 2012, 148, 387–389. [Google Scholar] [CrossRef] [Green Version]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; MacHiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; MacKenzie, M.J.; Shen, L.; et al. Prednisone plus Cabazitaxel or Mitoxantrone for Metastatic Castration-Resistant Prostate Cancer Progressing after Docetaxel Treatment: A Randomised Open-Label Trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Mita, A.C.; Denis, L.J.; Rowinsky, E.K.; De Bono, J.S.; Goetz, A.D.; Ochoa, L.; Forouzesh, B.; Beeram, M.; Patnaik, A.; Molpus, K.; et al. Phase I and Pharmacokinetic Study of XRP6258 (RPR 116258A), a Novel Taxane, Administered as a 1-Hour Infusion Every 3 Weeks in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2009, 15, 723–730. [Google Scholar] [CrossRef] [Green Version]
- Pivot, X.; Koralewski, P.; Hidalgo, J.L.; Chan, A.; Gonçalves, A.; Schwartsmann, G.; Assadourian, S.; Lotz, J.P. A Multicenter Phase II Study of XRP6258 Administered as a 1-h i.v. Infusion Every 3 Weeks in Taxane-Resistant Metastatic Breast Cancer Patients. Ann. Oncol. 2008, 19, 1547–1552. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, Y.; Liao, W.; Yang, J.; Wu, L.; Zheng, Z.; Yu, Y.; Zhou, W.; Li, L.; Feng, J.; et al. Acetylation of FoxO1 Activates Bim Expression to Induce Apoptosis in Response to Histone Deacetylase Inhibitor Depsipeptide Treatment. Neoplasia 2009, 11, 313–324. [Google Scholar] [CrossRef]
- Emanuele, S.; Lauricella, M.; Tesoriere, G. Histone Deacetylase Inhibitors: Apoptotic Effects and Clinical Implications (Review). Int. J. Oncol. 2008, 33, 637–646. [Google Scholar] [CrossRef]
- Dalla Pozza, E.; Manfredi, M.; Brandi, J.; Buzzi, A.; Conte, E.; Pacchiana, R.; Cecconi, D.; Marengo, E.; Donadelli, M. Trichostatin A Alters Cytoskeleton and Energy Metabolism of Pancreatic Adenocarcinoma Cells: An in Depth Proteomic Study. J. Cell. Biochem. 2018, 119, 2696–2707. [Google Scholar] [CrossRef]
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy Potentiates the Anti-Cancer Effects of the Histone Deacetylase Inhibitors in Hepatocellular Carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of Autophagy in Histone Deacetylase Inhibitor-Induced Apoptotic and Nonapoptotic Cell Death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.G. Cytosolic FoxO1 Is Essential for the Induction of Autophagy and Tumor Suppressor Activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef]
- Xiao, D.; Bommareddy, A.; Kim, S.H.; Sehrawat, A.; Hahm, E.R.; Singh, S.V. Benzyl Isothiocyanate Causes FoxO1-Mediated Autophagic Death in Human Breast Cancer Cells. PLoS ONE 2012, 7, e32597. [Google Scholar] [CrossRef]
- Han, J.; Pan, X.Y.; Xu, Y.; Xiao, Y.; An, Y.; Tie, L.; Pan, Y.; Li, X.J. Curcumin Induces Autophagy to Protect Vascular Endothelial Cell Survival from Oxidative Stress Damage. Autophagy 2012, 8, 812–825. [Google Scholar] [CrossRef]
- Ambrosio, S.; Saccà, C.D.; Amente, S.; Paladino, S.; Lania, L.; Majello, B. Lysine-Specific Demethylase LSD1 Regulates Autophagy in Neuroblastoma through SESN2-Dependent Pathway. Oncogene 2017, 36, 6701–6711. [Google Scholar] [CrossRef]
- Zhuo, X.; Wu, Y.; Yang, Y.; Gao, L.; Qiao, X.; Chen, T. Knockdown of LSD1 Meliorates Ox-LDL-Stimulated NLRP3 Activation and Inflammation by Promoting Autophagy via SESN2-Mesiated PI3K/Akt/MTOR Signaling Pathway. Life Sci. 2019, 233, 116696. [Google Scholar] [CrossRef]
- Oricchio, E.; Katanayeva, N.; Donaldson, M.C.; Sungalee, S.; Joyce, P.P.; Béguelin, W.; Battistello, E.; Sanghvi, V.R.; Jiang, M.; Jiang, Y.; et al. Genetic and Epigenetic Inactivation of SESTRIN1 Controls MTORC1 and Response to EZH2 Inhibition in Follicular Lymphoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Donaldson, M.C.; Katanayeva, N.; Oricchio, E. Sestrin1, A Tumor Suppressor That Can Be Rescued. Mol. Cell. Oncol. 2017, 4, 1–2. [Google Scholar] [CrossRef]
- Druker, B.J. Translation of the Philadelphia Chromosome into Therapy for CML. Blood 2008, 112, 4808–4817. [Google Scholar] [CrossRef]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.N.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-Year Follow-up of Patients Receiving Imatinib for Chronic Myeloid Leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef]
- Kim, S.K.; Su, L.K.; Oh, Y.; Kemp, B.L.; Hong, W.K.; Mao, L. Alterations of PTEN/MMAC1, a Candidate Tumor Suppressor Gene, and Its Homologue, PTH2, in Small Cell Lung Cancer Cell Lines. Oncogene 1998, 16, 89–93. [Google Scholar] [CrossRef]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Cordani, M.; Sánchez-Álvarez, M.; Strippoli, R.; Bazhin, A.V.; Donadelli, M. Sestrins at the Interface of ROS Control and Autophagy Regulation in Health and Disease. Oxid. Med. Cell. Longev. 2019, 2019, 1283075. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.; Ye, Y.; Xie, L.; Li, W. Oxidative Stress and Liver Cancer: Etiology and Therapeutic Targets. Oxid. Med. Cell. Longev. 2016, 2016, 7891574. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Xu, Y.; Isenberg, J.S.; Bachowski, S.; Kolaja, K.L.; Jiang, J.; Stevenson, D.E.; Walborg, E.F. The Role of Oxidative Stress in Chemical Carcinogenesis. Environ. Health Perspect. 1998, 106, 289–295. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Entry | Therapeutic Approach | Cell Lines | Type of Cancer | Effect on SESNs | Molecular Mechanisms | Biological Effect | Refs |
|---|---|---|---|---|---|---|---|
| 1 | Quercetin | HCT116, HT-29 | Colon cancer | SESN2 ↑ | mTOR ↓, AMPK/p38 ↑ | ROS, apoptosis, cell death | [80,81] |
| 2 | Cucurbitacin B | A549, H1792, H1650, H1975 | Lung cancer | SESN3 ↑ | PI3K/mTOR ↓, STAT-3 ↓ AMPKα ↑ | Anti-proliferative, apoptosis | [82] |
| 3 | Eupatilin | HepG2 | Liver cancer | SESN2 ↑ | Autophagy and antioxidant genes ↑ | Protective autophagy, reduction ROS, hepatoprotection | [83] |
| 4 | Isorhapontigenin | UMUC3, T24T, HeLa | Bladder, cervix cancer | SESN2 ↑ | MAPK8-Jun | Autophagy, inhibition of cell growth | [84] |
| 5 | Arsenic trioxide | U87MG, patients derived glioma cells, A549, H1299 | Glioma, lung cancer | SESN2 ↑ | miR-182-5p ↓ | Antioxidant response, increased patient survival | [85] |
| 6 | Resveratrol | HepG2 | Liver cancer | SESN2 ↑ | LXRα-SREBP-1c ↓ | Inhibition of hepatic lipogenesis | [86] |
| 7 | Carnosol | HCT116, SW480 | Colon cancer | SESN2 ↑ | PERK/Nrf2/SESN2↑ | Reduction of cell viability, apoptosis, | [87] |
| 8 | Tanshinone IIA | 43B, MG63 | Osteosarcoma | SESN2↑ | MAP4K4 SAPK/JNK1/Jun kinase↑, Jun recruitment to AP-1-binding site in the SESN2 promoter region, PI3K/Akt ↓ | Anchorage-independent growth inhibition; osteosarcoma progression; mitochondrial dysfunction, Autophagy induction | [88] |
| 9 | Cabazitaxel | C4-2AT6 | Prostate cancer | SESN3 ↓ | Cleaved-PARP ↑ | ROS, citotoxicity | [89] |
| 10 | Bortezomib, Nelfinavir | MDA-MB-453, OVCAR3, HeLa | Breast, ovarian, cervix cancer | SESN2 ↑ | ATF4, ATF3, CHOP ↑ mTOR ↓ | Autophagy, ER stress, Proteasome inhibition | [90] |
| 11 | Suberoylanilide hydroxamic acid, trichostatin A, depsipeptide | HCT116, HepG2 | Lung, liver cancer | SESN3 ↑ | FOXO1 ↑, mTOR ↓ | Protective autophagy | [91] |
| 12 | Tyrosine kinase inhibitors | BV173, BV173R, Ba/F3 p210T315I, U937, KT-1 | Leukemia | SESN3 ↑ | mTORC1 ↓ | Antileukemic response | [92] |
| 13 | Topotecan | A549, HeLa | Lung, cervix cancer | SESN2 ↑ | PTEN nuclear translocation, p-Jun-SESN2-AMPK ↑ | Autophagy | [93] |
| 14 | External beam radiation therapy | Prostate cancer | SESN3 ↓ | AMPK-mTORC1↓ | Mitophagy, Oxidative Stress, Fatigue intensification during EBRT. | [94] | |
| 15 | Carbonyl cyanide m-chlorophenyl hydrazine (CCCP) | SH-SY5Y | Neuroblastoma | SESN2 ↑ (early time) SESN2 ↓ (prolonged exposure) | RBX1 mediated ubiquitination | Protection from mitochondrial damage | [95] |
| 16 | 2-imino-6-methoxy-2H-chromene-3-carbothioamide (IMCA) | TT | Thyroid cancer | SESN1 ↑, SESN2 ↑ | AMPK ↑ mTORC1 ↓ | Inhibition of cell proliferation, Apoptosis induction | [96] |
| 17 | 3,4,5,4′-tetramethoxystilbene (DMU-212) | A-2780, SKOV-3 | Ovarian cancer | SESNs ↑ | P53 signaling ↑ | Apoptosis induction, Inhibition of cell proliferation, reduction of tumor growth in vivo | [97] |
| 18 | Ultraviolet radiations (UVA, UVB) | NHEM, iMC23 | Melanoma | SESN2 ↑ | P53 and AKT3 pathway | Inhibition of UVB-induced DNA damage repair; Promotion of UVA-induced ROS generation | [98] |
| 19 | ChlA-F | RT4, T24T, UMUC3 | Bladder cancer | SESN2 ↑ | Autophagy signaling ↑ | Anchorage-independent growth inhibition | [99] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Álvarez, M.; Strippoli, R.; Donadelli, M.; Bazhin, A.V.; Cordani, M. Sestrins as a Therapeutic Bridge between ROS and Autophagy in Cancer. Cancers 2019, 11, 1415. https://doi.org/10.3390/cancers11101415
Sánchez-Álvarez M, Strippoli R, Donadelli M, Bazhin AV, Cordani M. Sestrins as a Therapeutic Bridge between ROS and Autophagy in Cancer. Cancers. 2019; 11(10):1415. https://doi.org/10.3390/cancers11101415
Chicago/Turabian StyleSánchez-Álvarez, Miguel, Raffaele Strippoli, Massimo Donadelli, Alexandr V. Bazhin, and Marco Cordani. 2019. "Sestrins as a Therapeutic Bridge between ROS and Autophagy in Cancer" Cancers 11, no. 10: 1415. https://doi.org/10.3390/cancers11101415
APA StyleSánchez-Álvarez, M., Strippoli, R., Donadelli, M., Bazhin, A. V., & Cordani, M. (2019). Sestrins as a Therapeutic Bridge between ROS and Autophagy in Cancer. Cancers, 11(10), 1415. https://doi.org/10.3390/cancers11101415