A Functional Analysis of the Unclassified Pro2767Ser BRCA2 Variant Reveals Its Potential Pathogenicity that Acts by Hampering DNA Binding and Homology-Mediated DNA Repair

 ,
,  , ,
, ,  and
and 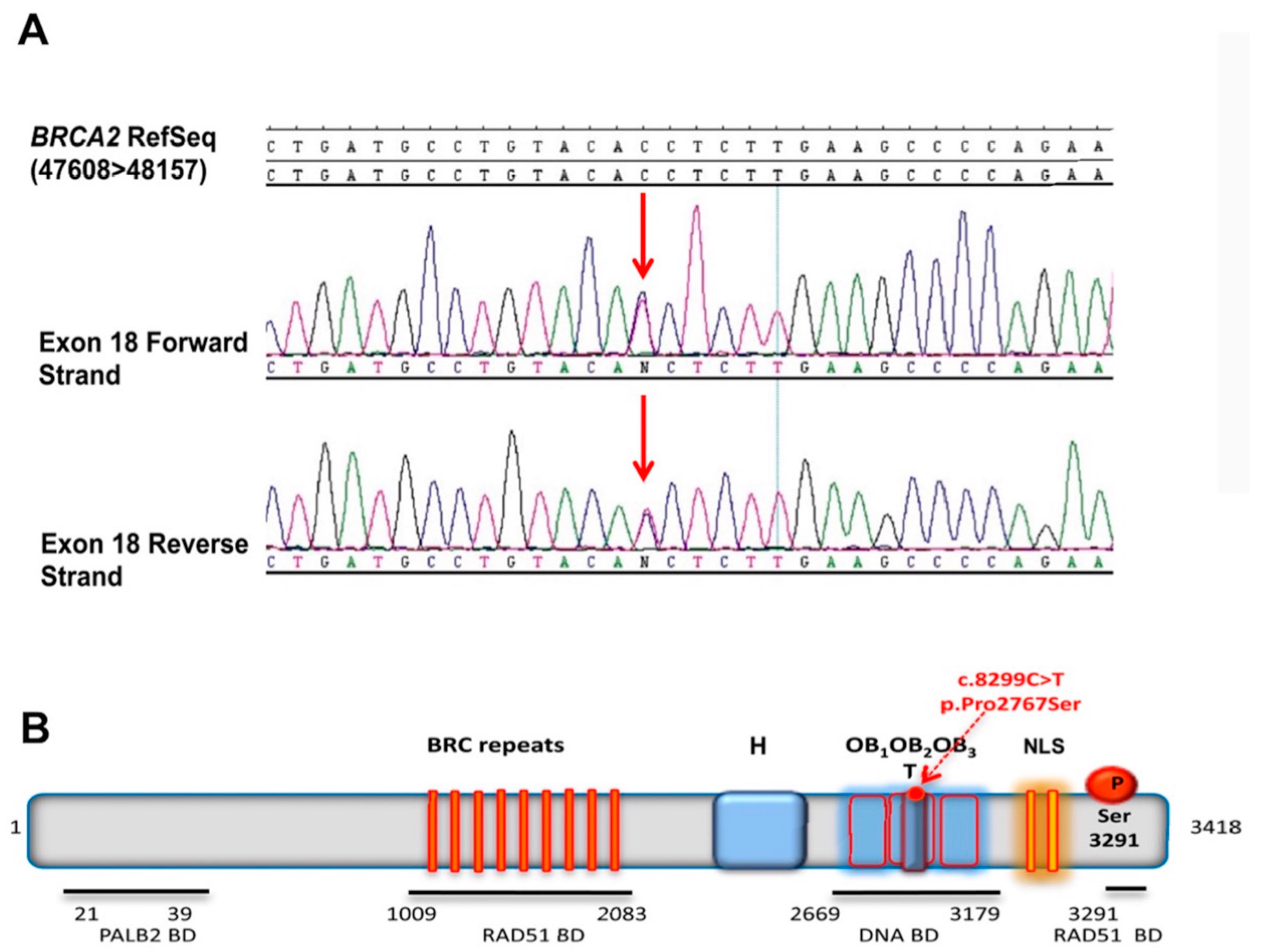{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The p.Pro2767Ser BRCA2 Variant Identification and Pathogenicity In Silico Predictions
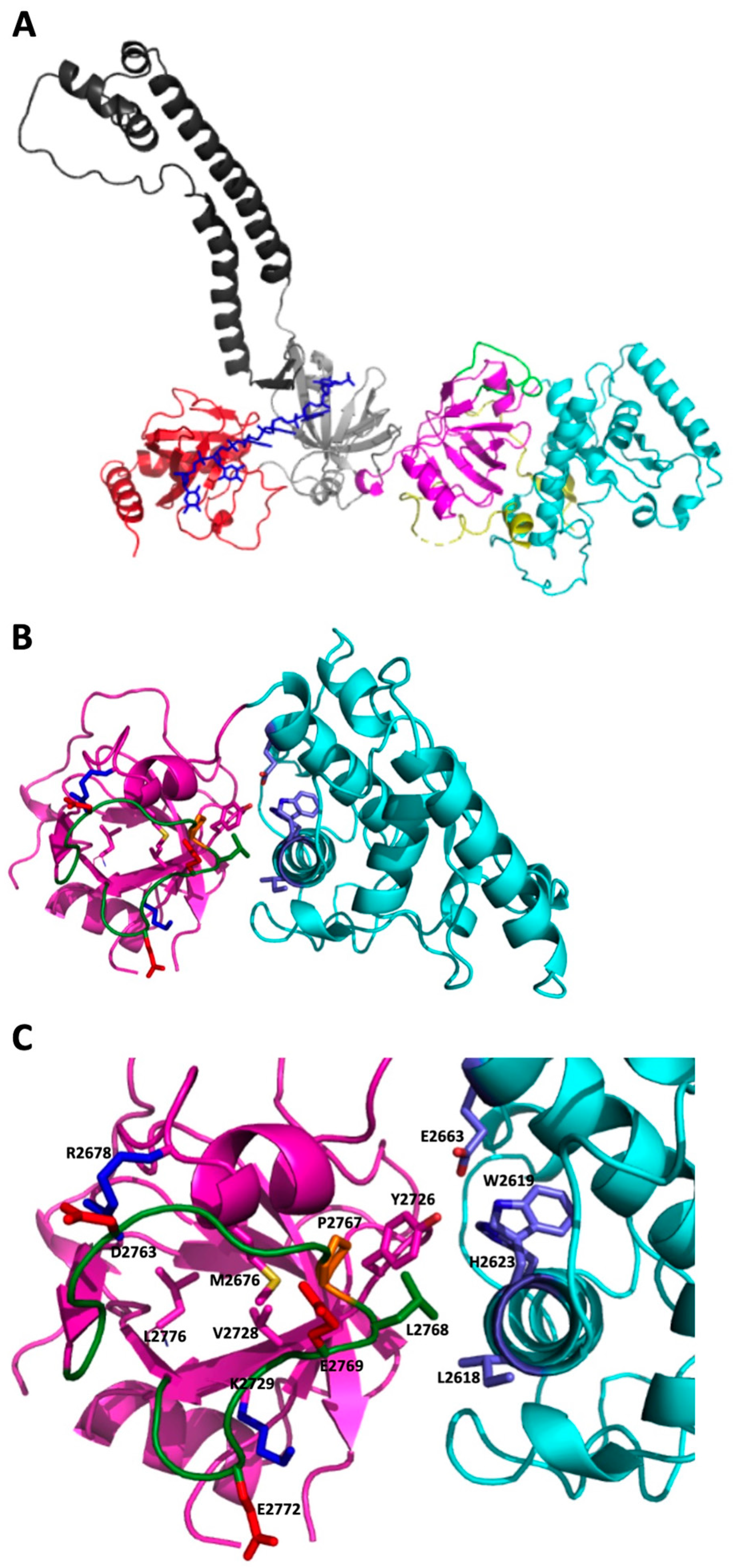
2.2. 3D Modeling Analysis of the Region Containing the Variant
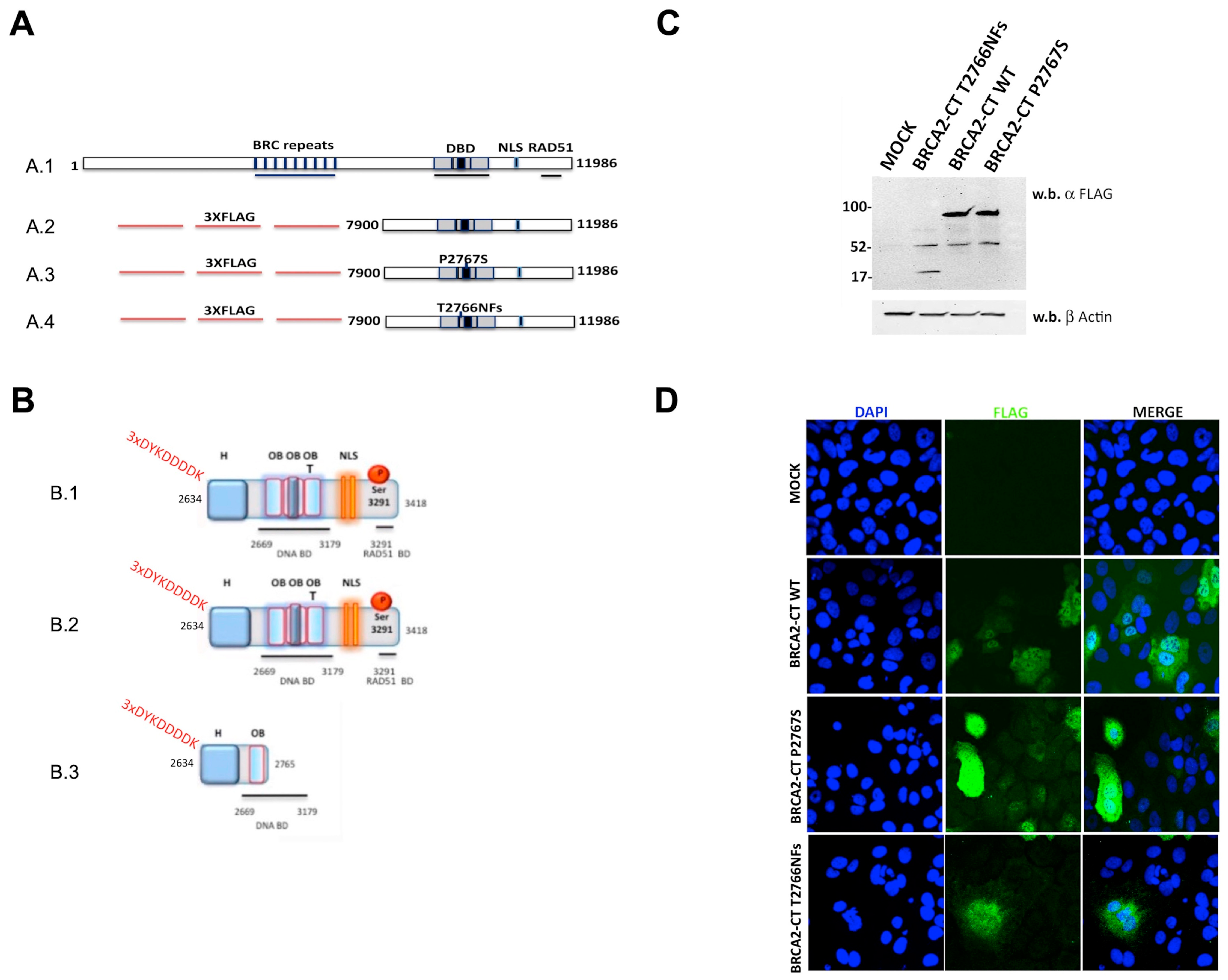
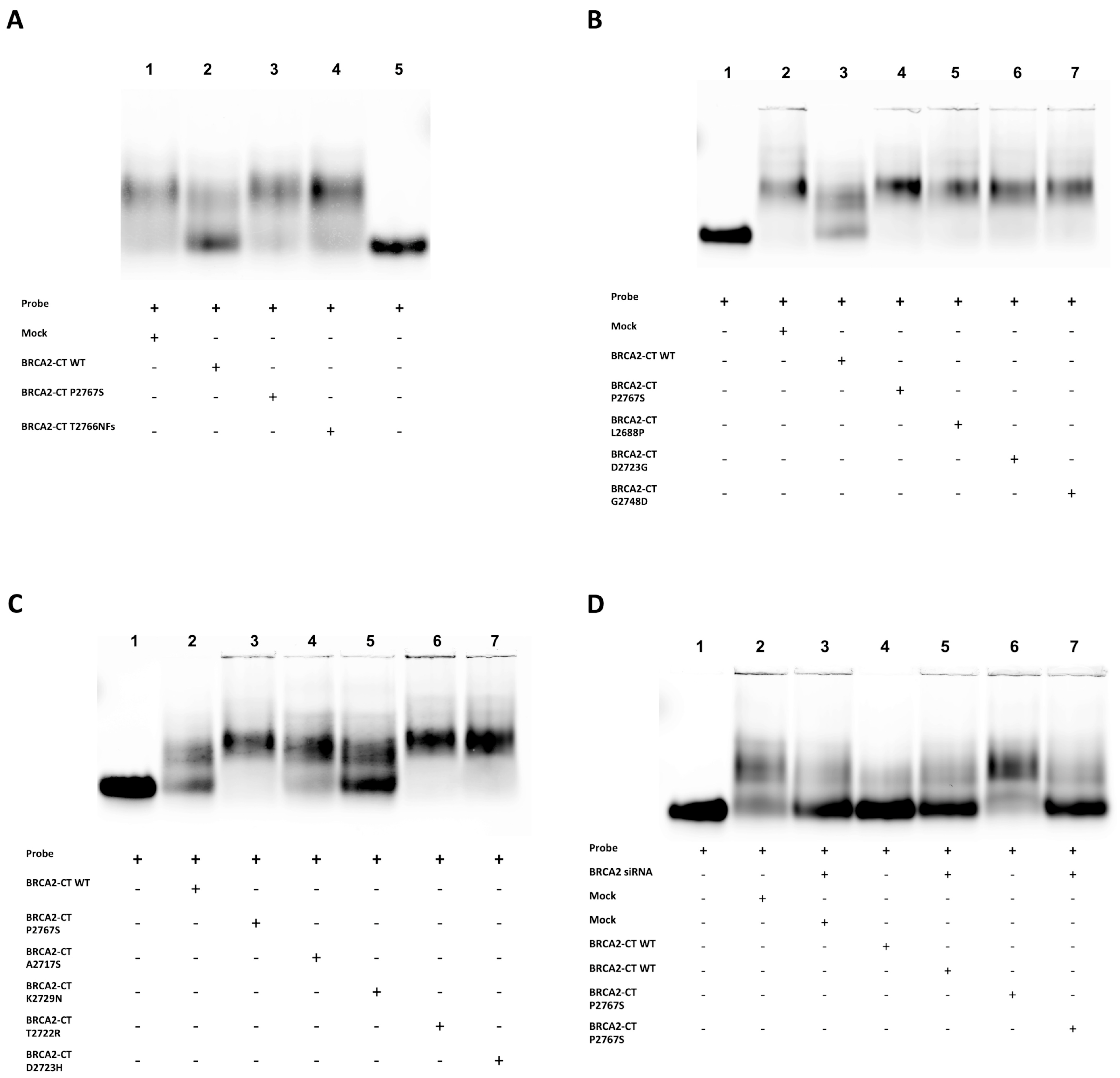
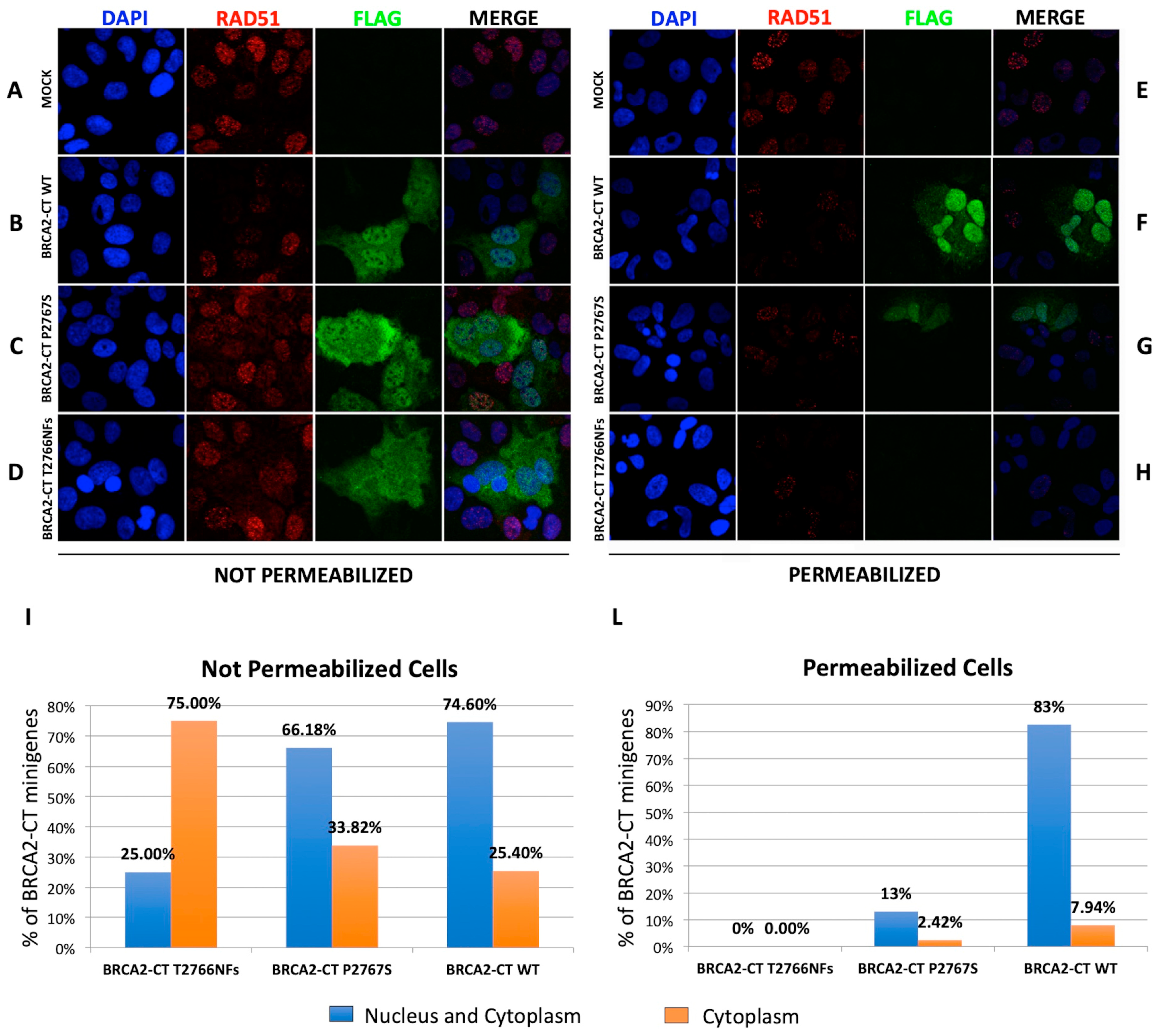
2.3. The p.Pro2767Ser Likely Pathogenic Variant Affects the Binding of BRCA2 to DNA
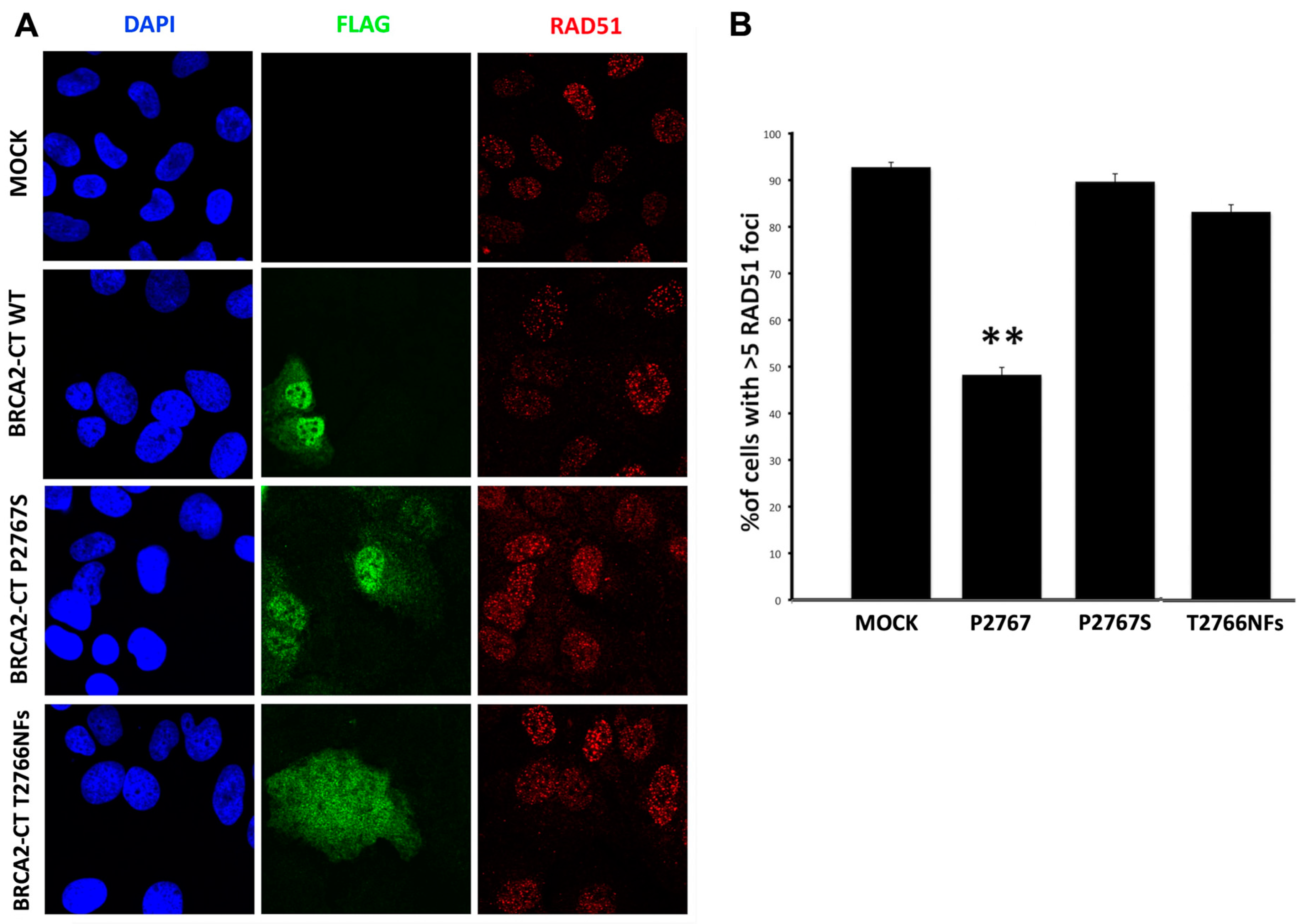
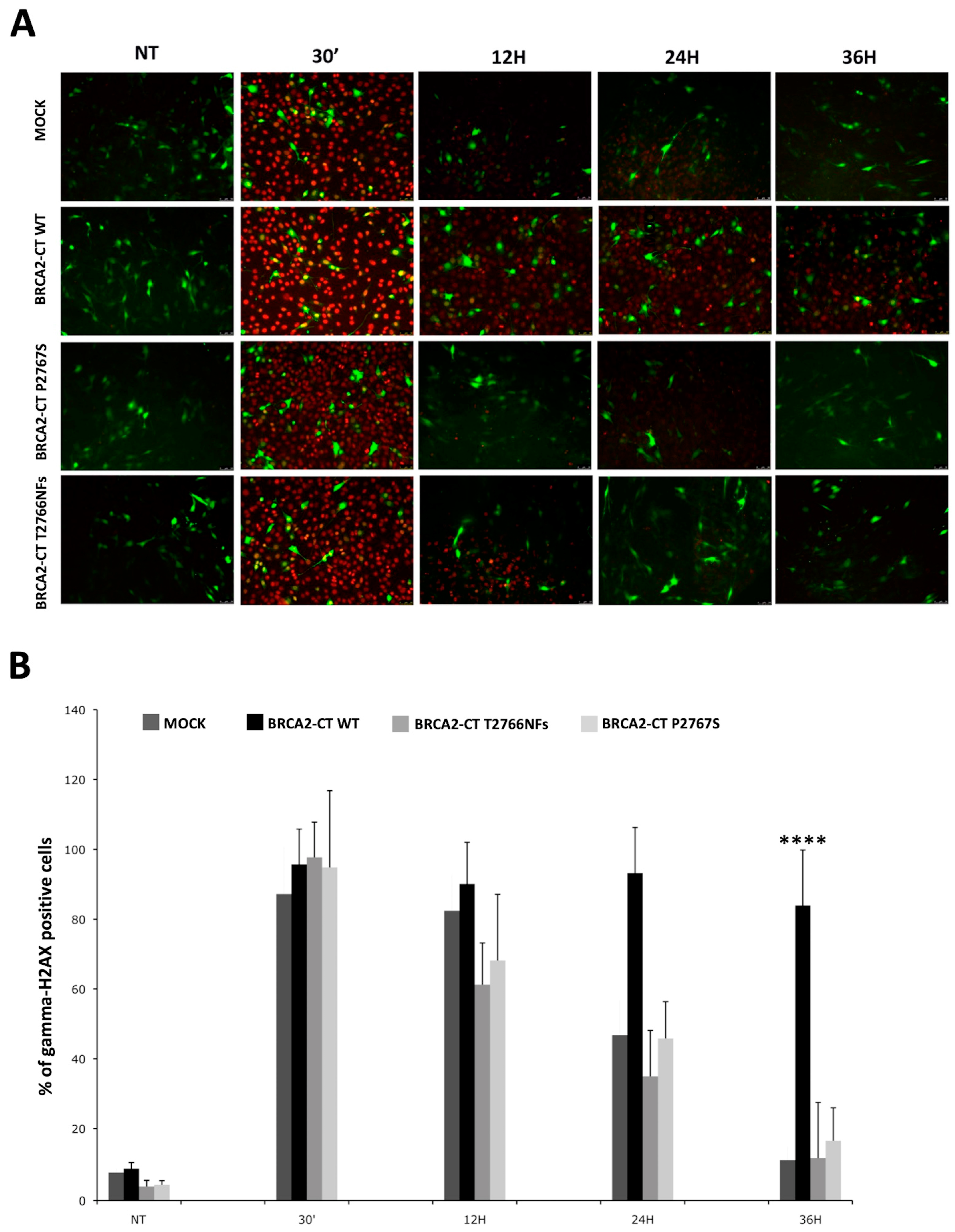
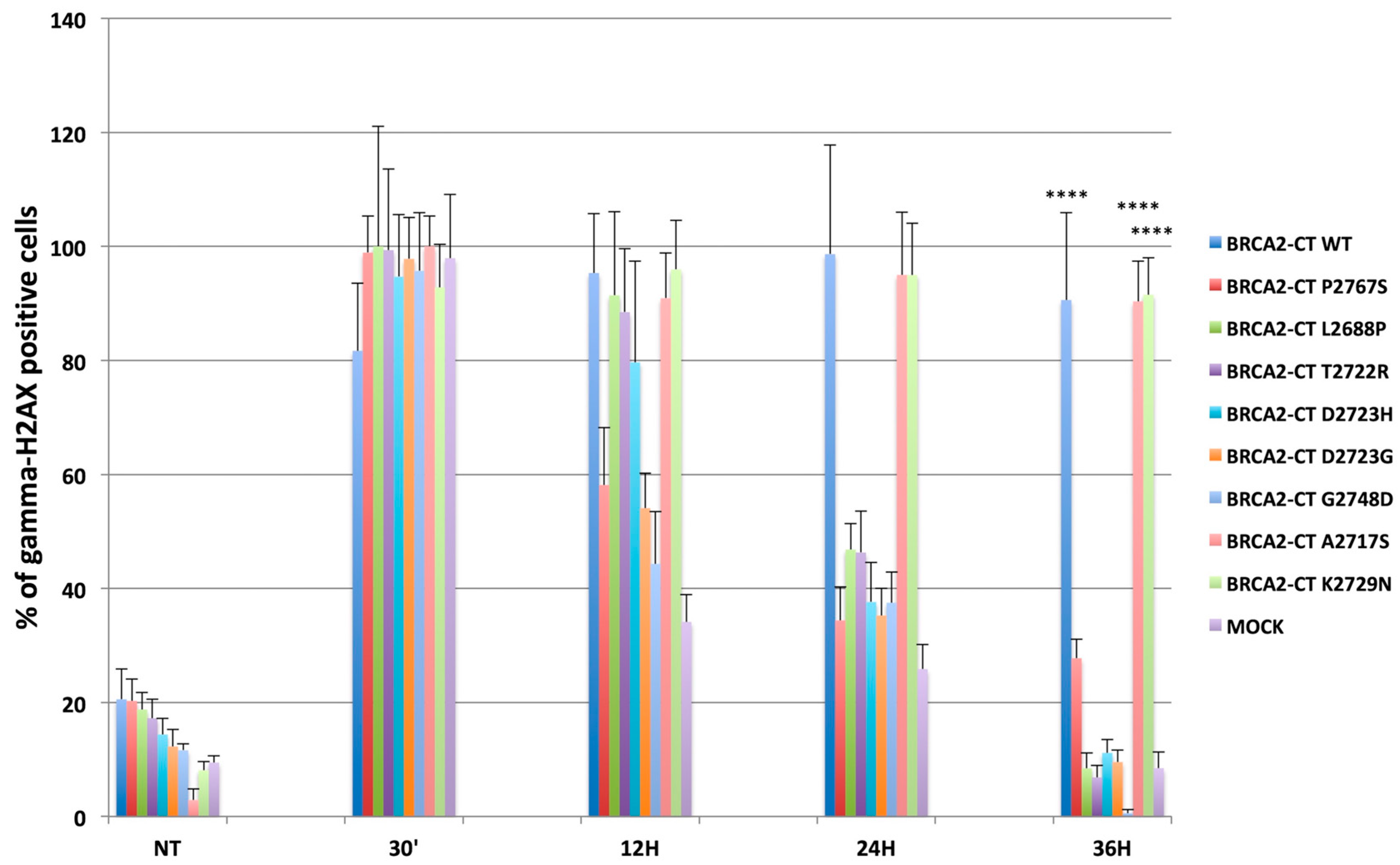
2.4. The p.Pro2767Ser Likley Pathogenic Variant Affects RAD51 Foci Formation and Repair of DNA Damage
3. Discussion
4. Materials and Methods
4.1. Patient and BRCA NGS Screening
4.2. Gene, Nomenclature and Databases
4.3. Bioinformatics and 3D Modeling
4.4. Cloning and Site-Directed Mutagenesis
4.5. Cell Cultures and Transfections
4.6. Protein Extracts and Western Blot Analysis
4.7. Cell Irradiation
4.8. In Vitro ssDNA Binding Assay
4.9. Endogenous BRCA2 Silencing
4.9.1. RT-PCR and qPCR
4.9.2. Western Blot Analysis
4.9.3. In Vitro ssDNA Binding Assay on Silenced and Scrambled Negative Controls Samples
4.10. RAD51 Immunofluorescence
4.11. DNA Damage Recovery Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Narod, S.A. Breast cancer in young women. Nat. Rev. Clin. Oncol. 2012, 9, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akram, M.; Iqbal, M.; Daniyal, M.; Khan, A.U. Awareness and current knowledge of breast cancer. Biol. Res. 2017, 50, 33. [Google Scholar] [CrossRef] [PubMed]
- Cobain, E.F.; Milliron, K.J.; Merajver, S.D. Updates on breast cancer genetics: Clinical implications of detecting syndromes of inherited increased susceptibility to breast cancer. Semin. Oncol. 2016, 43, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Engel, C.; Fischer, C. Breast cancer risks and risk prediction models. Breast Care (Basel) 2015, 10, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Nunziato, M.; Esposito, M.V.; Starnone, F.; Diroma, M.A.; Calabrese, A.; Del Monaco, V.; Buono, P.; Frasci, G.; Botti, G.; D’Aiuto, M.; et al. A multi-gene panel beyond BRCA1/BRCA2 to identify new breast cancer-predisposing mutations by a picodroplet PCR followed by a next-generation sequencing strategy: A pilot study. Anal. Chim. Acta 2018, 1046, 154–162. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Esposito, M.V.; Gilder, J.A.; Frisso, G.; Salvatore, F. Should a BRCA2 stop codon human variant, usually considered a polymorphism, be classified as a predisposing mutation? Cancer 2014, 120, 1594–1595. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.V.; Nunziato, M.; Starnone, F.; Telese, A.; Calabrese, A.; D’Aiuto, G.; Pucci, P.; D’Aiuto, M.; Baralle, F.; D’Argenio, V.; et al. A novel pathogenic BRCA1 splicing variant produces partial intron retention in the mature messenger RNA. Int. J. Mol. Sci. 2016, 17, 2145. [Google Scholar] [CrossRef]
- Toland, A.E.; Andreassen, P.R. DNA repair-related functional assays for the classification of BRCA1 and BRCA2 variants: A critical review and needs assessment. J. Med. Genet. 2017, 54, 721–731. [Google Scholar] [CrossRef]
- Eccles, B.K.; Copson, E.; Maishman, T.; Abraham, J.E.; Eccles, D.M. Understanding of BRCA VUS genetic results by breast cancer specialists. BMC Cancer 2015, 15, 936. [Google Scholar] [CrossRef]
- Eccles, E.B.; Mitchell, G.; Monteiro, A.N.A.; Schmutzler, R.; Couch, F.J.; Spurdle, A.B.; Gómez-García, E.B.; Driessen, R.; Lindor, N.M.; Blok, M.J.; et al. BRCA1 and BRCA2 genetic testing-pitfalls and recommendations for managing variants of uncertain clinical significance. Ann. Oncol. 2015, 26, 2057–2065. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, H.; Mochizuki, H.; Inoue, M.; Hirotsu, Y.; Amemiya, K.; Sakamoto, I.; Nakagomi, S.; Kubota, T.; Omata, M. Combined annotation-dependent depletion score for BRCA1/2 variants in patients with breast and/or ovarian cancer. Cancer Sci. 2018, 109, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Haroun, I.; Graham, T.C.; Eisen, A.; Kiss, A.; Warner, E. Variants of unknown significance in BRCA testing impact on risk perception, worry, prevention and counseling. Ann. Oncol. 2013, 24 (Suppl. 8), viii69–viii74. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, F.C.; Van Overeem Hansen, T.; Sørensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Buechelmaier, E.S.; Powell, S.N. Rad51 Paralog Complexes BCDX2 and CX3 Act at Different Stages in the BRCA1-BRCA2-Dependent Homologous Recombination Pathway. Mol. Cell. Biol. 2013, 33, 387–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, L.; Yu, D.S.; Lo, T.; Anand, S.; Lee, M.Y.; Blundell, T.L.; Venkitaraman, A.R. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 2002, 420, 287–293. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu. Rev. Biochem. 2019, 88, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef] [Green Version]
- Galkin, V.E.; Esashi, F.; Yu, X.; Yang, S.; West, S.C.; Egelman, E.H. BRCA2 BRC motifs bind RAD51-DNA filaments. Proc. Natl. Acad. Sci. USA 2005, 102, 8537–8542. [Google Scholar] [CrossRef] [PubMed]
- Jeyasekharan, A.D.; Liu, Y.; Hattori, H.; Pisupati, V.; Jonsdottir, A.B.; Rajendra, E.; Lee, M.; Sundaramoorthy, E.; Schlachter, S.; Kaminski, C.F.; et al. A cancer-associated BRCA2 mutation reveals masked nuclear export signals controlling localization. Nat. Struct. Mol. Biol. 2013, 20, 1191–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esashi, F.; Christ, N.; Cannon, J.; Liu, Y.; Hunt, T.; Jasin, M.; West, S.C. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005, 434, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Jeffrey, P.D.; Miller, J.; Kinnucan, E.; Sun, Y.; Thomä, N.H.; Zheng, N.; Chen, P.L.; Lee, W.H.; Pavletich, N.P. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 2002, 297, 1837–1848. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Shahid, T.; Soroka, J.; Kong, E.H.; Malivert, L.; McIlwraith, M.J.; Pape, T.; West, S.C.; Zhang, X. Structure and mechanism of action of the BRCA2 breast cancer tumor suppressor. Nat. Struct. Mol. Biol. 2014, 21, 962–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Vaithiyalingam, S.; San Filippo, J.; Maranon, D.G.; Jimenez-Sainz, J.; Fontenay, G.V.; Kwon, Y.; Leung, S.G.; Lu, L.; Jensen, R.B.; et al. Promotion of BRCA2-Dependent Homologous Recombination by DSS1 via RPA Targeting and DNA Mimicry. Mol. Cell 2015, 59, 176–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreira, A.; Kowalczykowski, S.C. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 10448–10453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esashi, F.; Galkin, V.E.; Yu, X.; Egelman, E.H.; West, S.C. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat. Struct. Mol. Biol. 2007, 14, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef]
- Thorslund, T.; McIlwraith, M.J.; Compton, S.A.; Lekomtsev, S.; Petronczki, M.; Griffith, J.D.; West, S.C. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Bertwistle, D.; Valentine, J.; Gabriel, A.; Swift, S.; Ross, G.; Griffin, C.; Thacker, J.; Ashworth, A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001, 20, 4704–4716. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Andreassen, P.R.; Andrea, A.D.D. Functional Interaction of Monoubiquitinated Functional Interaction of Monoubiquitinated FANCD2 and BRCA2/FANCD1 in Chromatin. Mol. Cell. Biol. 2004, 24, 5850–5862. [Google Scholar] [CrossRef] [PubMed]
- Guidugli, L.; Carreira, A.; Caputo, S.M.; Ehlen, A.; Galli, A.; Monteiro, A.N.A.; Neuhausen, S.L.; Hansen, T.V.O.; Couch, F.J.; Vreeswijk, M.P.G. Functional assays for analysis of variants of uncertain significance in BRCA2. Hum. Mutat. 2014, 35, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Davies, O.R.; Pellegrini, L. Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nat. Struct. Mol. Biol. 2007, 14, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Iacovoni, J.S.; Caron, P.; Lassadi, I.; Nicolas, E.; Massip, L.; Trouche, D.; Legube, G. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010, 29, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Minucci, A.; Lalle, M.; De Leo, R.; Mazzuccato, G.; Scambia, G.; Urbani, A.; Fagotti, A.; Concolino, P.; Capoluongo, E. Additional molecular and clinical evidence open the way to definitive IARC classification of the BRCA1 c.5332G > A (p.Asp1778Asn) variant. Clin. Biochem. 2019, 63, 54–58. [Google Scholar] [CrossRef]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.L.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Healey, S.; Devereau, A.; Hogervorst, F.B.L.; Monteiro, A.N.A.; Nathanson, K.L.; Radice, P.; Stoppa-Lyonnet, D.; Tavtigian, S.; Wappenschmidt, B.; et al. ENIGMA-evidence-based network for the interpretation of germline mutant alleles: An international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum. Mutat. 2012, 33, 2–7. [Google Scholar] [CrossRef]
- Easton, D.F.; Deffenbaugh, A.M.; Pruss, D.; Frye, C.; Wenstrup, R.J.; Allen-Brady, K.; Tavtigian, S.V.; Monteiro, A.N.A.; Iversen, E.S.; Couch, F.J.; et al. A Systematic Genetic Assessment of 1433 Sequence Variants of Unknown Clinical Significance in the BRCA1 and BRCA2 Breast Cancer–Predisposition Genes. Am. J. Hum. Genet. 2007, 81, 873–883. [Google Scholar] [CrossRef]
- Farrugia, D.J.; Agarwal, M.K.; Pankratz, V.S.; Deffenbaugh, A.M.; Pruss, D.; Frye, C.; Wadum, L.; Johnson, K.; Mentlick, J.; Tavtigian, S.V.; et al. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Res. 2008, 68, 3523–3531. [Google Scholar] [CrossRef] [PubMed]
- Goldgar, D.E.; Easton, D.F.; Deffenbaugh, A.M.; Monteiro, A.N.A.; Tavtigian, S.V.; Couch, F.J. Integrated Evaluation of DNA Sequence Variants of Unknown Clinical Significance: Application to BRCA1 and BRCA2. Am. J. Hum. Genet. 2004, 75, 535–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidugli, L.; Pankratz, V.S.; Singh, N.; Thompson, J.; Erding, C.A.; Engel, C.; Schmutzler, R.; Domchek, S.; Nathanson, K.; Radice, P.; et al. A classification model for BRCA2 DNA binding domain missense variants based on homology-directed repair activity. Cancer Res. 2013, 73, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Lindor, N.M.; Guidugli, L.; Wang, X.; Vallée, M.P.; Monteiro, A.N.A.; Tavtigian, S.; Goldgar, D.E.; Couch, F.J. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum. Mutat. 2012, 33, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Georgoulis, A.; Vorgias, C.E.; Chrousos, G.P.; Rogakou, E.P. Genome instability and γH2AX. Int. J. Mol. Sci. 2017, 18, 1979. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.B.; Pilarski, R.; Berry, M.; Buys, S.S.; Farmer, M.; Friedman, S.; Garber, J.E.; Kauff, N.D.; Khan, S.; Klein, C.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J. Natl. Compr. Cancer Netw. 2017, 15, 9–20. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Esposito, M.V.; Telese, A.; Precone, V.; Starnone, F.; Nunziato, M.; Cantiello, P.; Iorio, M.; Evangelista, E.; D’Aiuto, M.; et al. The molecular analysis of BRCA1 and BRCA2: Next-generation sequencing supersedes conventional approaches. Clin. Chim. Acta 2015, 446, 221–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 15, 5–6. [Google Scholar]
- Schrodinger. Schrodinger Software Release 2015-2 Induced Fit Docking; Schrodinger Press: Portland, OR, USA, 2015. [Google Scholar]
- Wang, S.C.; Shao, R.; Pao, A.Y.; Zhang, S.; Hung, M.C.; Su, L.K. Inhibition of cancer cell growth by BRCA2. Cancer Res. 2002, 62, 1311–1314. [Google Scholar]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, M.V.; Minopoli, G.; Esposito, L.; D’Argenio, V.; Di Maggio, F.; Sasso, E.; D’Aiuto, M.; Zambrano, N.; Salvatore, F. A Functional Analysis of the Unclassified Pro2767Ser BRCA2 Variant Reveals Its Potential Pathogenicity that Acts by Hampering DNA Binding and Homology-Mediated DNA Repair. Cancers 2019, 11, 1454. https://doi.org/10.3390/cancers11101454
Esposito MV, Minopoli G, Esposito L, D’Argenio V, Di Maggio F, Sasso E, D’Aiuto M, Zambrano N, Salvatore F. A Functional Analysis of the Unclassified Pro2767Ser BRCA2 Variant Reveals Its Potential Pathogenicity that Acts by Hampering DNA Binding and Homology-Mediated DNA Repair. Cancers. 2019; 11(10):1454. https://doi.org/10.3390/cancers11101454
Chicago/Turabian StyleEsposito, Maria Valeria, Giuseppina Minopoli, Luciana Esposito, Valeria D’Argenio, Federica Di Maggio, Emanuele Sasso, Massimiliano D’Aiuto, Nicola Zambrano, and Francesco Salvatore. 2019. "A Functional Analysis of the Unclassified Pro2767Ser BRCA2 Variant Reveals Its Potential Pathogenicity that Acts by Hampering DNA Binding and Homology-Mediated DNA Repair" Cancers 11, no. 10: 1454. https://doi.org/10.3390/cancers11101454
APA StyleEsposito, M. V., Minopoli, G., Esposito, L., D’Argenio, V., Di Maggio, F., Sasso, E., D’Aiuto, M., Zambrano, N., & Salvatore, F. (2019). A Functional Analysis of the Unclassified Pro2767Ser BRCA2 Variant Reveals Its Potential Pathogenicity that Acts by Hampering DNA Binding and Homology-Mediated DNA Repair. Cancers, 11(10), 1454. https://doi.org/10.3390/cancers11101454