PTEN and Gynecological Cancers


Abstract
:1. Background
2. Overview
3. Ovarian Cancer
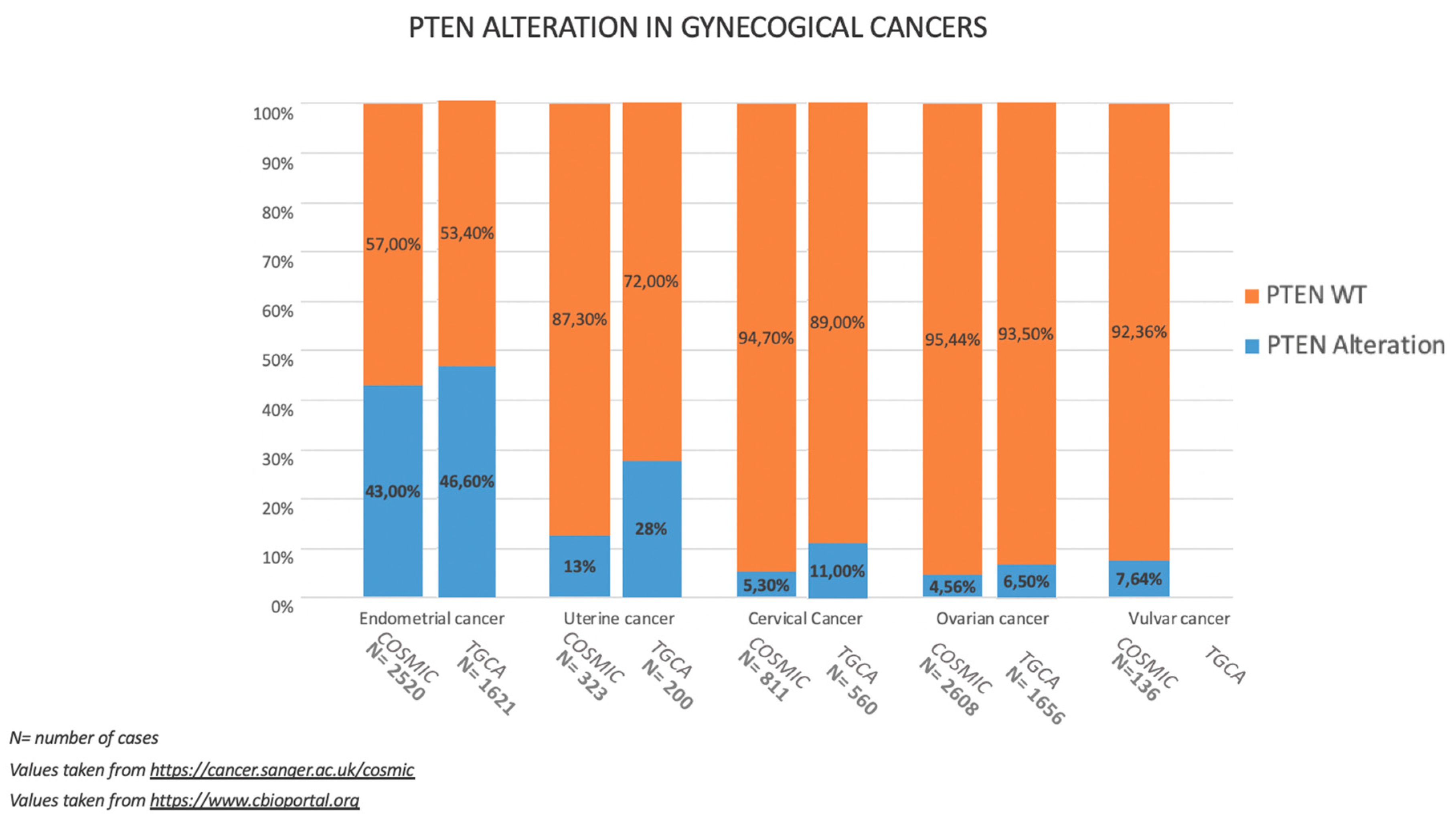
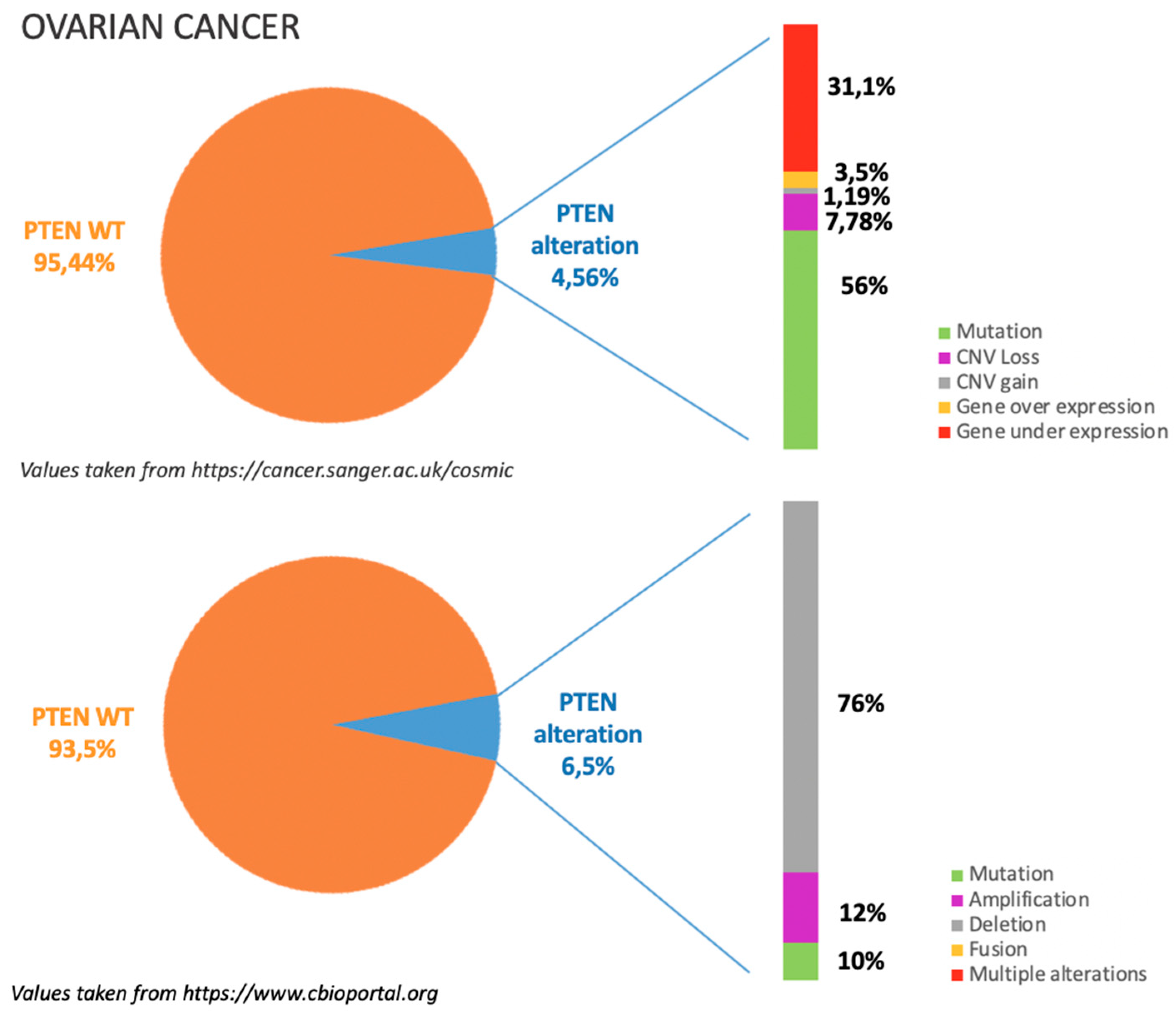
3.1. Incidence of PTEN Alteration
3.2. PTEN Pathways in OC Tumorigenesis
3.3. Clinical Implications
4. Endometrial Cancer
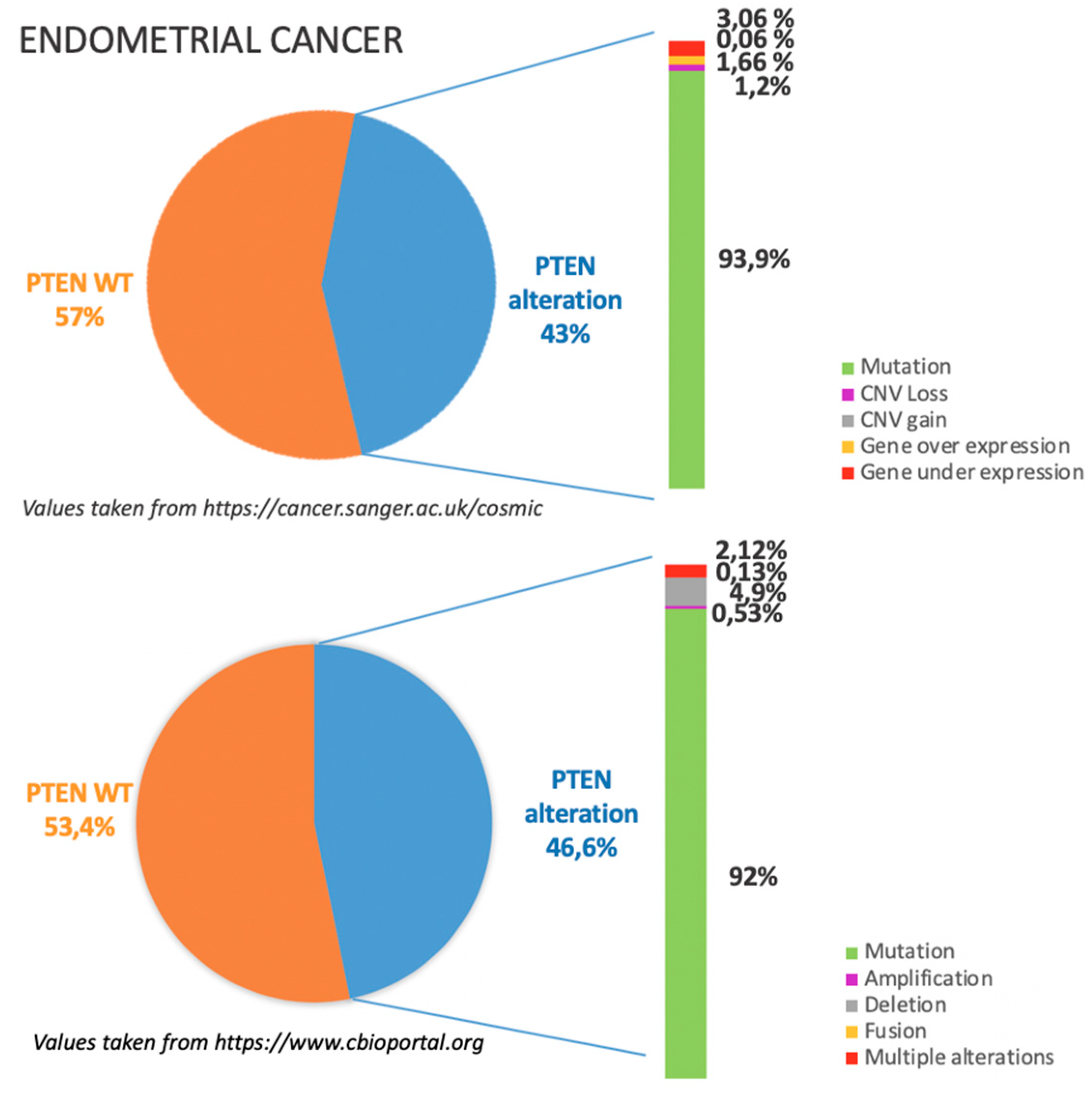
4.1. Incidence of PTEN Alteration
4.2. PTEN Pathways in EC Tumorigenesis
4.3. Clinical Implications
5. Cervical Cancer
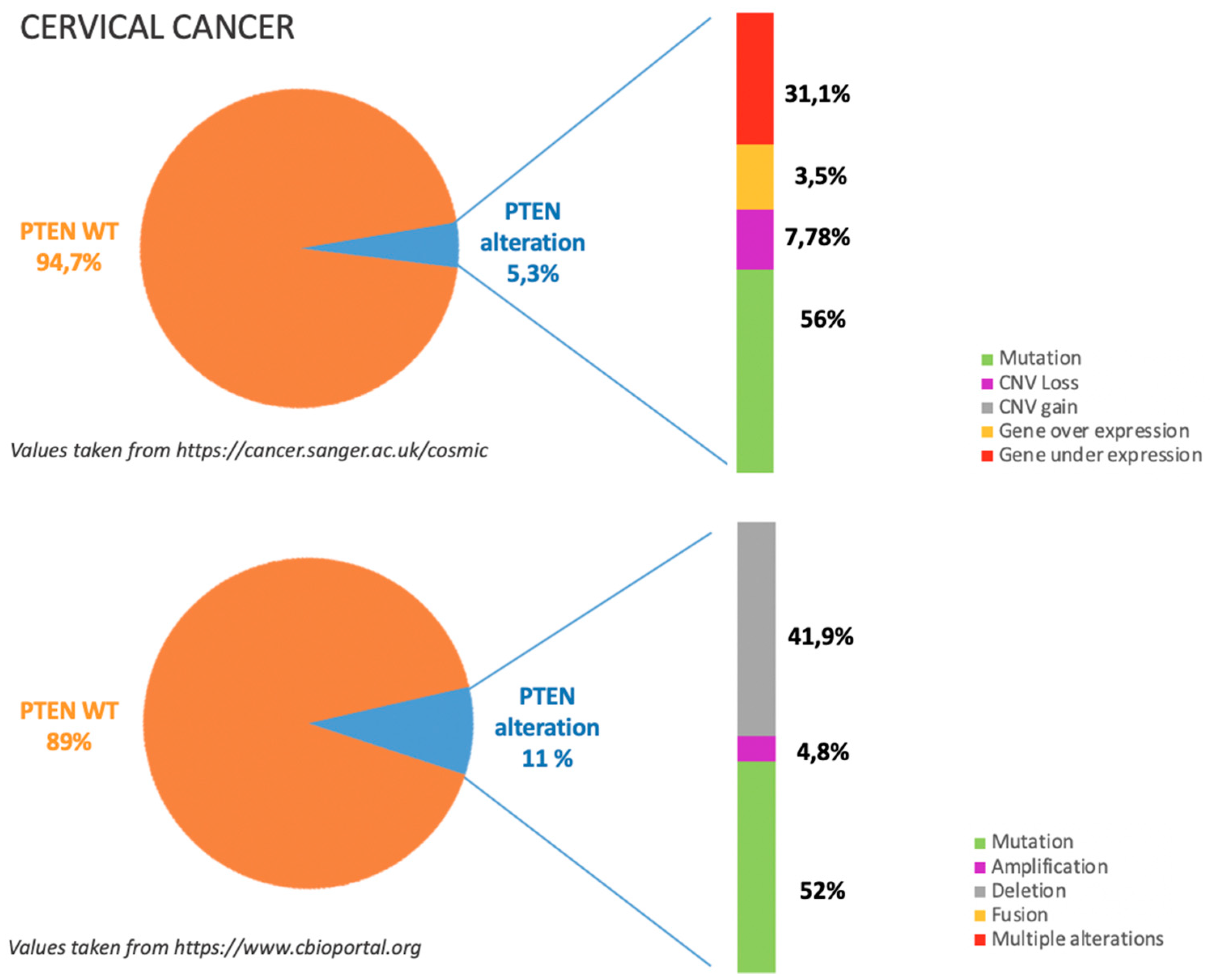
5.1. Incidence of PTEN Alteration
5.2. PTEN Pathways in CC Tumorigenesis
5.3. Clinical Implications
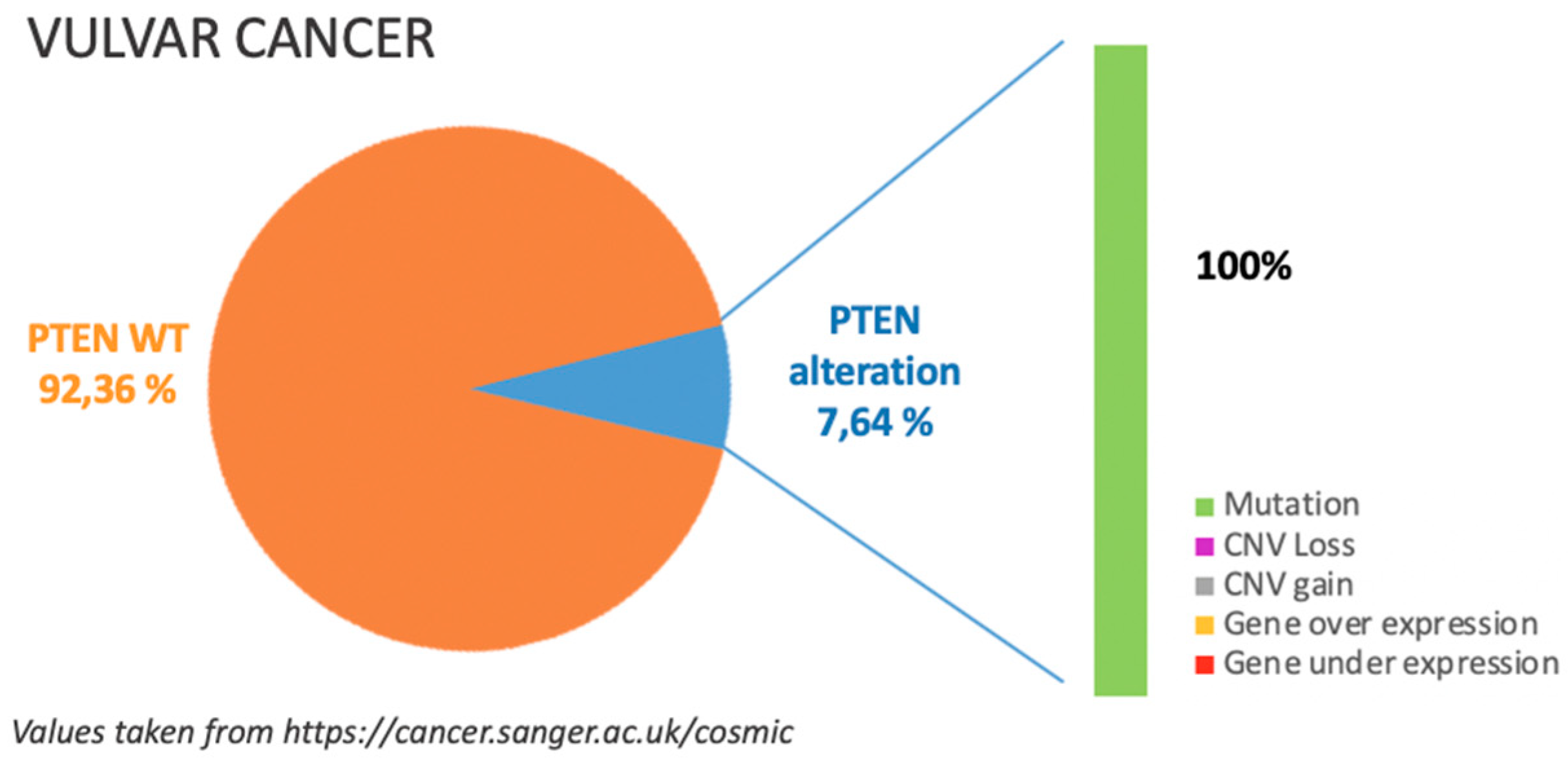
6. Vulvar Cancer
6.1. Incidence of PTEN Alteration
6.2. PTEN Pathways in VC Tumorigenesis
6.3. Clinical Implications
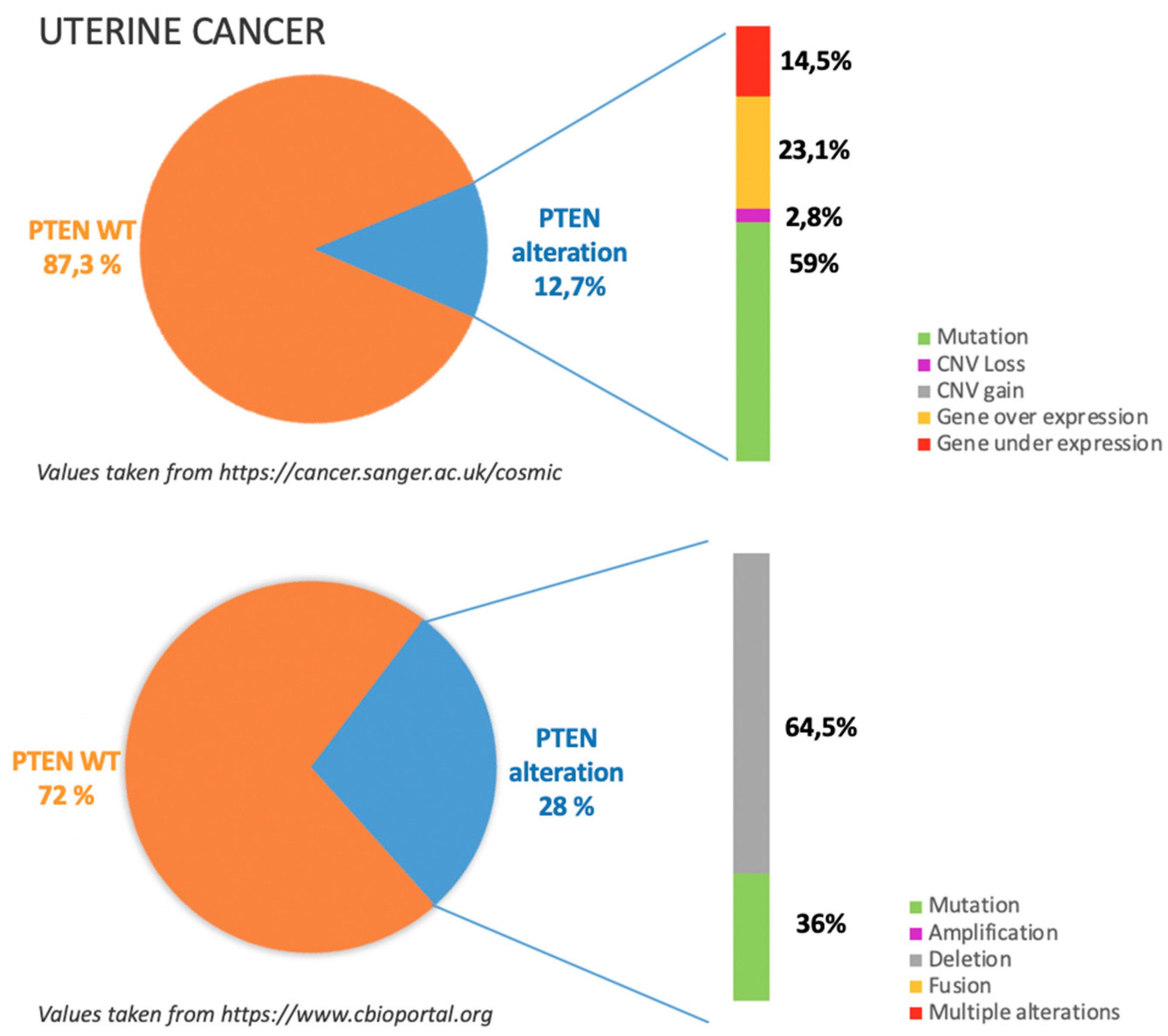
7. Uterine Cancers
7.1. Incidence of PTEN Alteration
7.2. PTEN Pathways in UC Tumorigenesis
7.3. Clinical Implications
8. Future Perspectives and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Di Cristofano, A.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten is essential for embryonic development and tumour suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef]
- Di Cristofano, A.; Pandolfi, P.P. The multiple roles of PTEN in tumor suppression. Cell 2000, 100, 387–390. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Goel, V.; Haluska, F.G. PTEN signaling pathways in melanoma. Oncogene 2003, 22, 3113–3122. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.S. Molecular carcinogenesis of endometrial cancer. Taiwan J. Obstet. Gynecol. 2007, 46, 26–32. [Google Scholar] [CrossRef]
- Bhyan, S.B.; Wee, Y.; Liu, Y.; Cummins, S.; Zhao, M. Integrative analysis of common genes and driver mutations implicated in hormone stimulation for four cancers in women. PeerJ 2019, 7, e6872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalogue of Somatic Mutations in Cancer. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 27 June 2019).
- CBioportal for Cancer Genonics. Available online: http://www.cbioportal.org (accessed on 27 June 2019).
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.E.M. The origin and pathogenesis of epithelial ovarian cancer: A proposed unifying theory. Am. J. Surg. Pathol. 2010, 34, 433–443. [Google Scholar] [CrossRef]
- Murakami, R.; Matsumura, N.; Brown, J.B.; Higasa, K.; Tsutsumi, T.; Kamada, M.; Abou-Taleb, H.; Hosoe, Y.; Kitamura, S.; Yamaguchi, K.; et al. Exome Sequencing Landscape Analysis in Ovarian Clear Cell Carcinoma Shed Light on Key Chromosomal Regions and Mutation Gene Networks. Am. J. Pathol. 2017, 187, 2246–2258. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Tinker, A.; Clarke, B.; Ghatage, P.; Welch, S.; Weberpals, J.I.; Dhani, N.C.; Butler, M.O.; Tonkin, K.; Tan, Q.; et al. A Clinical and Molecular Phase II Trial of Oral ENMD-2076 in Ovarian Clear Cell Carcinoma (OCCC): A Study of the Princess Margaret Phase II Consortium. Clin. Cancer Res. 2018, 24, 6168–6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, M.; Nakayama, K.; Nakamura, K.; Ono, R.; Sanuki, K.; Yamashita, H.; Ishibashi, T.; Minamoto, T.; Iida, K.; Razia, S.; et al. Affinity-purified DNA-based mutation profiles of endometriosis-related ovarian neoplasms in Japanese patients. Oncotarget 2018, 9, 14754–14763. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Wang, H.J.; Xu, P.; Chen, J.; Pan, H.Y.; Liu, Y. [Expressions of Livin and PTEN in Cancerous Tissues of Ovary Endometriosis]. Sichuan Da Xue Xue Bao Yi Xue Ban 2016, 47, 512–515. (In Chinese) [Google Scholar] [PubMed]
- Smith, I.N.; Briggs, J.M. Structural mutation analysis of PTEN and its genotype-phenotype correlations in endometriosis and cancer. Proteins 2016, 84, 1625–1643. [Google Scholar] [CrossRef] [Green Version]
- Worley, M.J., Jr.; Liu, S.; Hua, Y.; Kwok, J.S.; Samuel, A.; Hou, L.; Shoni, M.; Lu, S.; Sandberg, E.M.; Keryan, A.; et al. Molecular changes in endometriosis-associated ovarian clear cell carcinoma. Eur. J. Cancer 2015, 51, 1831–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, F.C.; Santiago, I.; Trinh, A.; Xian, J.; Guo, A.; Sayal, K.; Jimenez-Linan, M.; Deen, S.; Driver, K.; Mack, M.; et al. Combined image and genomic analysis of high-grade serous ovarian cancer reveals PTEN loss as a common driver event and prognostic classifier. Genome Biol. 2014, 15, 526. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, A.J.; Schultz, N.; Westfal, M.L.; Sakr, R.A.; Giri, D.D.; Scarperi, S.; Janakiraman, M.; Olvera, N.; Stevens, E.V.; She, Q.B.; et al. Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discov. 2014, 2, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Willner, J.; Wurz, K.; Allison, K.H.; Galic, V.; Garcia, R.L.; Goff, B.A.; Swisher, E.M. Alternate molecular genetic pathways in ovarian carcinomas of common histological types. Hum. Pathol. 2007, 38, 607–613. [Google Scholar] [CrossRef]
- Obata, K.; Morland, S.J.; Watson, R.H.; Hitchcock, A.; Chenevix-Trench, G.; Thomas, E.J.; Campbell, I.G. Frequent PTEN/MMAC mutations in endometrioid but not serous or mucinous epithelial ovarian tumors. Cancer Res. 1998, 58, 2095–2097. [Google Scholar] [PubMed]
- Singer, G.; Oldt, R., III; Cohen, Y.; Wang, B.G.; Sidransky, D.; Kurman, R.J.; Shih, I.M. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J. Natl. Cancer Inst. 2003, 95, 484–486. [Google Scholar] [CrossRef] [PubMed]
- Dinulescu, D.M.; Ince, T.A.; Quade, B.J.; Shafer, S.A.; Crowley, D.; Jacks, T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat. Med. 2005, 11, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Hendrix-Lucas, N.; Kuick, R.; Zhai, Y.; Schwartz, D.R.; Akyol, A.; Hanash, S.; Misek, D.E.; Katabuchi, H.; Williams, B.O.; et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell 2007, 11, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Baust, C.A.; Kuhn, E.; Valle, B.L.; Shih Ie, M.; Kurman, R.J.; Wang, T.L.; Amano, T.; Ko, M.S.; Miyoshi, I.; Araki, Y.; et al. A genetically engineered ovarian cancer mouse model based on fallopian tube transformation mimics human high-grade serous carcinoma development. J. Pathol. 2014, 233, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Endsley, M.P.; Moyle-Heyrman, G.; Karthikeyan, S.; Lantvit, D.D.; Davis, D.A.; Wei, J.J.; Burdette, J.E. Spontaneous transformation of murine oviductal epithelial cells: A model system to investigate the onset of fallopian-derived tumors. Front. Oncol. 2015, 5, 154. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Czarnecki, A.A.; Dean, M.; Modi, D.A.; Lantvit, D.D.; Hardy, L.; Baligod, S.; Davis, D.A.; Wei, J.J.; Burdette, J.E. PTEN loss in the fallopian tube induces hyperplasia and ovarian tumor formation. Oncogene 2018, 37, 1976–1990. [Google Scholar] [CrossRef] [PubMed]
- Coscia, F.; Watters, K.M.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef] [PubMed]
- Roh, M.H.; Yassin, Y.; Miron, A.; Mehra, K.K.; Mehrad, M.; Monte, N.M.; Mutter, G.L.; Nucci, M.R.; Ning, G.; Mckeon, F.D.; et al. High-grade fimbrial-ovarian carcinomas are unified by alteredp53, PTEN and PAX2 expression. Mod. Pathol. 2010, 23, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Chivukula, M.; Edwards, R.; Nikiforova, M.; Mantha, G.; McManus, K.; Carter, G. Loss of heterozygosity (LOH) and immunohistochemical ana-lysis (IHC) of a subset of primary fallopian tube carcinomas (PFTC) arising in the background of tubal intraepithelial carcinoma ‘TIC’ from primary perito-neal serous carcinomas with/without associated tubal intraepithelial carcinoma ‘TIC’. Mod. Pathol. 2010, 23, 237A. [Google Scholar]
- Dean, M.; Jin, V.; Bergsten, T.M.; Austin, J.R.; Lantvit, D.D.; Russo, A.; Burdette, J.E. Loss of PTEN in Fallopian Tube Epithelium Results in Multicellular Tumor Spheroid Formation and Metastasis to the Ovary. Cancers 2019, 11, 884. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, T.; Brozick, J.; Babalola, K.; Budiu, R.; Tseng, G.; Vlad, A.M. Effects of Kras activation and Pten deletion alone or in combination on MUC1 biology and epithelial-to-mesenchymal transition in ovarian cancer. Oncogene 2016, 35, 5010–5020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalin, A.; Merideth, M.A.; Regier, D.S.; Blumenthal, G.M.; Dennis, P.A.; Stratton, P. Management of reproductive health in Cowden syndrome complicated by endometrial polyps and breast cancer. Obstet. Gynecol. 2013, 121 Pt 2 (Suppl. 1), 461–464. [Google Scholar] [CrossRef]
- Bakkar, R.M.; Xie, S.S.; Urbauer, D.L.; Djordjevic, B.; Vu, K.; Broaddus, R.R. Intact PTEN Expression by Immunohistochemistry is Associated with Decreased Survival in Advanced Stage Ovarian/Primary Peritoneal High-grade Serous Carcinoma. Int. J. Gynecol. Pathol. 2015, 34, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Li, H.L.; Liu, L.; Cheng, J.X. Expression levels of PTEN, HIF-1α, and VEGF as prognostic factors in ovarian cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2596–2603. [Google Scholar]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Privately and Publicly Funded Clinical Studies Conducted around the World. Available online: www. clinicaltrials.gov (accessed on 27 June 2019).
- Hauke, J.; Hahnen, E.; Schneider, S.; Reuss, A.; Richters, L.; Kommoss, S.; Heimbach, A.; Marmé, F.; Schmidt, S.; Prieske, K.; et al. Deleterious somatic variants in 473 consecutive individuals with ovarian cancer: Results of the observational AGO-TR1 study (NCT02222883). J. Med. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Freixinos, V.; Ruiz-Pace, F.; Fariñas-Madrid, L.; Garrido-Castro, A.C.; Villacampa, G.; Nuciforo, P.; Vivancos, A.; Dienstmann, R.; Oaknin, A. Genomic heterogeneity and efficacy of PI3K pathway inhibitors in patients with gynaecological cancer. ESMO Open 2019, 4, e000444. [Google Scholar] [CrossRef] [Green Version]
- ACOG. ACOG practice bulletin, clinical management guidelines for obstetrician-gynecologists, number 65, August 2005: Management of endometrial cancer. Obstet. Gynecol. 2005, 106, 413–425. [Google Scholar]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [Green Version]
- Cheung, L.W.; Hennessy, B.T.; Li, J.; Yu, S.; Myers, A.P.; Djordjevic, B.; Lu, Y.; Stemke-Hale, K.; Dyer, M.D.; Zhang, F.; et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011, 1, 170–185. [Google Scholar] [CrossRef]
- Levine, R.L.; Cargile, C.B.; Blazes, M.S.; van Rees, B.; Kurman, R.J.; Ellenson, L.H. PTEN mutations and microsatellite instability in complex atypical hyperplasia, a precursor lesion to uterine endometrioid carcinoma. Cancer Res. 1998, 58, 3254–3258. [Google Scholar] [PubMed]
- Wang, H.; Douglas, W.; Lia, M.; Edelmann, W.; Kucherlapati, R.; Podsypanina, K.; Parsons, R.; Ellenson, L.H. DNA mismatch repair deficiency accelerates endometrial tumorigenesis in Pten heterozygous mice. Am. J. Pathol. 2002, 160, 1481–1486. [Google Scholar] [CrossRef]
- Stambolic, V.; Tsao, M.S.; Macpherson, D.; Suzuki, A.; Chapman, W.B.; Mak, T.W. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten +/− mice. Cancer Res. 2000, 60, 3605–3611. [Google Scholar] [PubMed]
- Mutter, G.L.; Ince, T.A.; Baak, J.P.; Kust, G.A.; Zhou, X.P.; Eng, C. Molecular identification of latent precancers in histologically normal endometrium. Cancer Res. 2001, 61, 4311–4314. [Google Scholar] [PubMed]
- Raffone, A.; Travaglino, A.; Saccone, G.; Viggiani, M.; Giampaolino, P.; Insabato, L.; Mollo, A.; De Placido, G.; Zullo, F. PTEN expression in endometrial hyperplasia and risk of cancer: A systematic review and meta-analysis. Arch. Gynecol. Obstet. 2019, 299, 1511–1524. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Creutzberg, C.; Amant, F.; Bosse, T.; González-Martín, A.; Ledermann, J.; Marth, C.; Nout, R.; Querleu, D.; Mirza, M.R.; et al. ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: Diag-nosis, treatment and follow-up. Ann. Oncol. 2016, 27, 16–41. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, R. PTEN Hamartoma Tumor Syndrome: A Clinical Overview. Cancers 2019, 11, 886. [Google Scholar] [CrossRef] [PubMed]
- Starink, T.M.; van der Veen, J.P.; Arwert, F.; de Waal, L.P.; de Lange, G.G.; Gille, J.J.; Eriksson, A.W. The Cowden syndrome: A clinical and genetic study in 21 patients. Clin. Genet. 1986, 29, 222–233. [Google Scholar] [CrossRef]
- NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines). Genetic/Familial High-risk Assessment: Breast and Ovarian. Version 2. 2019. Available online: www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf (accessed on 10 September 2018).
- Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Services; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope; Buck Institute for Research on Aging; Canada’s Michael Smith Genome Sciences Centre; Harvard Medical School; Helen F. Graham Cancer Center & Research Institute at Christiana Care Health Services; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef]
- Cheung, T.H.; Lo, K.W.; Yim, S.F.; Chan, L.K.; Heung, M.S.; Chan, C.S.; Cheung, A.Y.; Chung, T.K.; Wong, Y.F. Epigenetic and genetic alternation of PTEN in cervical neoplasm. Gynecol. Oncol. 2004, 93, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, M.M.; Alam, M.S.; Mehdi, S.J.; Ali, A.; Batra, S. Allelic loss of 10q23.3, the PTEN gene locus in cervical carcinoma from Northern Indian population. Pathol. Oncol. Res. 2012, 18, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Li, J.; Jiang, W.; Shen, X.; Yang, W.; Wu, X.; Yang, H. Comprehensive analysis of targetable oncogenic mutations in chinese cervical cancers. Oncotarget 2015, 6, 4968–4975. [Google Scholar] [CrossRef] [PubMed]
- Minaguchi, T.; Yoshikawa, H.; Nakagawa, S.; Yasugi, T.; Yano, T.; Iwase, H.; Mizutani, K.; Shiromizu, K.; Ohmi, K.; Watanabe, Y.; et al. Association of PTEN mutation with HPV-negative adenocarcinoma of the uterine cervix. Cancer Lett. 2004, 210, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Choi, Y.D.; Lee, J.H.; Nam, J.H.; Choi, C.; Lee, M.C.; Park, C.S.; Kim, H.S.; Min, K.W. Expression of PTEN in the progression of cervical neoplasia and its relation to tumor behavior and angiogenesis in invasive squamous cell carcinoma. J. Surg. Oncol. 2006, 93, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Ulloa, E.; Lizano, M.; Avilés-Salas, A.; Alfaro-Moreno, E.; Contreras-Paredes, A. Abnormal distribution of hDlg and PTEN in premalignant lesions and invasive cervical cancer. Gynecol. Oncol. 2011, 122, 663–668. [Google Scholar] [CrossRef]
- Huang, M.; Li, W.C.; Gao, D.L.; Wang, Y.P.; Gu, Y.L. Mutation and protein expression of PTEN gene in cervical adenocarcinoma and glandular intraepithelial neoplasia. Zhonghua Bing Li Xue Za Zhi 2009, 38, 397–401. (In Chinese) [Google Scholar] [PubMed]
- Lee, M.S.; Jeong, M.H.; Lee, H.W.; Han, H.J.; Ko, A.; Hewitt, S.M.; Kim, J.H.; Chun, K.H.; Chung, J.Y.; Lee, C.; et al. PI3K/AKT activation induces PTEN ubiquitination and destabilization accelerating tumourigenesis. Nat. Commun. 2015, 6, 7769. [Google Scholar] [CrossRef]
- Qi, Q.; Ling, Y.; Zhu, M.; Zhou, L.; Wan, M.; Bao, Y.; Liu, Y. Promoter region methylation and loss of protein expression of PTEN and significance in cervical cancer. BioMed. Rep. 2014, 2, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Harima, Y.; Sawada, S.; Nagata, K.; Sougawa, M.; Ostapenko, V.; Ohnishi, T. Mutation of the PTEN gene in advanced cervical cancer correlated with tumor progression and poor outcome after radiotherapy. Int. J. Oncol. 2001, 18, 493–497. [Google Scholar] [CrossRef]
- Bosch, F.X.; Lorincz, A.; Muñoz, N.; Meijer CJShah, K.V. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 2002, 55, 244–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Jiang, Y.; Lu, X.; Zhao, H.; Chen, C.; Wang, Y.; Hu, W.; Zhu, Y.; Yan, H.; Yan, F. Genomic characterization of cervical cancer based on human papillomavirus status. Gynecol. Oncol. 2019, 152, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.M.; Liu, X.; Wheler, J.; Naing, A.; Hong, D.; Coleman, R.L.; Tsimberidou, A.; Janku, F.; Zinner, R.; Lu KKurzrock, R.; et al. Targeted PI3K/AKT/mTOR therapy for metastatic carcinomas of the cervix: A phase I clinical experience. Oncotarget 2014, 5, 11168–11179. [Google Scholar] [CrossRef] [PubMed]
- Grigsby, P.; Elhammali, A.; Ruiz, F.; Markovina, S.; McLellan, M.D.; Miller, C.A.; Chundury, A.; Ta, N.L.; Rashmi, R.; Pfeifer, J.D.; et al. Clinical outcomes and differential effects of PI3K pathway mutation in obese versus non-obese patients with cervical cancer. Oncotarget 2017, 9, 4061–4073. [Google Scholar] [CrossRef] [PubMed]
- Weberpals, J.I.; Lo, B.; Duciaume, M.M.; Spaans, J.N.; Clancy, A.A.; Dimitroulakos, J.; Goss, G.D.; Sekhon, H.S. Vulvar Squamous Cell Carcinoma (VSCC) as Two Diseases: HPV Status Identifies Distinct Mutational Profiles Including Oncogenic Fibroblast Growth Factor Receptor 3. Clin. Cancer Res. 2017, 23, 4501–4510. [Google Scholar] [CrossRef] [Green Version]
- Zięba, S.; Kowalik, A.; Zalewski, K.; Rusetska, N.; Goryca, K.; Piaścik, A.; Misiek, M.; Bakuła-Zalewska, E.; Kopczyński, J.; Kowalski, K.; et al. Somatic mutation profiling of vulvar cancer: Exploring therapeutic targets. Gynecol. Oncol. 2018, 150, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Palisoul, M.L.; Mullen, M.M.; Feldman, R.; Thaker, P.H. Identification of molecular targets in vulvar cancers. Gynecol. Oncol. 2017, 146, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, J.; Ling, M.T.; Zhao, L.; Zhao, K.N. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol. Cancer 2015, 14, 87. [Google Scholar] [CrossRef]
- Koncar, R.F.; Feldman, R.; Bahassi, E.M.; Hashemi Sadraei, N. Comparative molecular profiling of HPV-induced squamous cell carcinoma. Cancer Med. 2017, 6, 1673–1685. [Google Scholar] [CrossRef]
- Movva, S.; Wen, W.; Chen, W.; Millis, S.Z.; Gatalica, Z.; Reddy, S.; von Mehren, M.; Van Tine, B.A. Multi-platform profiling of over 2000 sarcomas: Identification of biomarkers and novel therapeutic targets. Oncotarget 2015, 6, 12234–12247. [Google Scholar] [CrossRef] [Green Version]
- Setsu, N.; Yamamoto, H.; Kohashi, K.; Endo, M.; Matsuda, S.; Yokoyama, R.; Nishiyama, K.; Iwamoto, Y.; Dobashi, Y.; Oda, Y. The Akt/mammalian target of rapamycin pathway is activated and associated with adverse prognosis in soft tissue leiomyosarcomas. Cancer 2012, 118, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherniack, A.D.; Shen, H.; Walter, V.; Stewart, C.; Murray, B.A.; Bowlby, R.; Hu, X.; Ling, S.; Soslow, R.A.; Broaddus, R.R.; et al. Integrated Molecular Characterization of Uterine Carcinosarcoma. Cancer Cell. 2017, 31, 411–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuppens, T.; Annibali, D.; Coosemans, A.; Trovik, J.; Ter Haar, N.; Colas, E.; Garcia-Jimenez, A.; Van de Vijver, K.; Kruitwagen, R.P.; Brinkhuis, M.; et al. Potential Targets’ Analysis Reveals Dual PI3K/mTOR Pathway Inhibition as a Promising Therapeutic Strategy for Uterine Leiomyosarcomas-an ENITEC Group Initiative. Clin. Cancer Res. 2017, 23, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Cuppens, T.; Moisse, M.; Depreeuw, J.; Annibali, D.; Colas, E.; Gil-Moreno, A.; Huvila, J.; Carpén, O.; Zikán, M.; Matias-Guiu, X.; et al. Integrated genome analysis of uterine leiomyosarcoma to identify novel driver genes and targetable pathways. Int. J. Cancer 2018, 142, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Oda, Y.; Saito, T.; Takahira, T.; Yamamoto, H.; Tamiya, S.; Iwamoto, Y.; Tsuneyoshi, M. Genetic and epigenetic alterations of the PTEN gene in soft tissue sarcomas. Hum. Pathol. 2005, 36, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN is associated with re-sistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef]
- Shrestha, S.; Yang, K.; Guy, C.; Vogel, P.; Neale, G.; Chi, H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat. Immunol. 2015, 16, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Xi, Z.; Jing, L.; Le-Ni, K.; Zhu, L.; Ze-Wen, D.; Hui, Y.; Ming-Rong, X.; Guang-Dong, L. Evaluation of PTEN and CD4+FOXP3+ T cell expressions as diagnostic and predictive factors in endometrial cancer: A case control study. Medicine (Baltim.) 2019, 98, e16345. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Rabadan RImmune and genomic correlates of response to anti-PD 1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Mansour, W.Y.; Tennstedt, P.; Volquardsen, J.; Oing, C.; Kluth, M.; Hube-Magg, C.; Borgmann, K.; Simon, R.; Petersen, C.; Dikomey, E.; et al. Loss of PTEN-assisted G2/M checkpoint impedes homologous recombination repair and enhances radio-curability and PARP inhibitor treatment response in prostate cancer. Sci. Rep. 2018, 8, 3947. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histology | Average % | N of Samples Tested | Details (Source: Number of Altered Samples/Total of Samples) |
|---|---|---|---|
| Malignant germ cell tumours (GCTs) | 1.75 | 60 | COSMIC dataset: 1/60 * |
| Sex cord-stromal tumour (SCSTs) | NA | 36 | COSMIC dataset ** |
| Endometrioid | 17.81 | 219 | COSMIC dataset: 39/219 |
| Clear cell | 3.77 | 159 | COSMIC dataset: 6/159 |
| Mucinous | 4.31 | 116 | COSMIC dataset: 5/116 |
| Low grade serous | NA | NA | COSMIC dataset |
| High grade serous carcinomas | 5.4% | 3667 | COSMIC dataset: 92/2011 TGCA dataset: 108/1656 |
| Undifferentiated carcinomas | NA | 9 | COSMIC dataset |
| Carcinosarcomas | 6.98 | 43 | COSMIC dataset: 3/43 |
| Histology | Average % | N of Samples Tested | Details (Source: Number of Altered Samples/Total of Samples) |
|---|---|---|---|
| Endometrioid | 52.6 | 3319 | COSMIC dataset: 1038/2092 TGCA dataset: 708/1227 |
| Serous | 8 | 467 | COSMIC dataset: 10/144 TGCA dataset: 28/323 |
| Clear cell | 7.14 | 168 | COSMIC dataset: 10/139 TGCA dataset: 2/29 |
| Mixed | 27 | 159 | COSMIC dataset: 28/123 * TGCA dataset: 15/36 |
| Dedifferentiated | 36.36 | 22 | COSMIC dataset: 8/22 |
| Histology | Average % | N of Samples Tested | Details (Source: Number of Altered Samples/Total of Samples) |
|---|---|---|---|
| Squamous | 6.7 | 1125 | COSMIC dataset: 19/628 TGCA dataset: 57/497 |
| Adenocarcinoma | 6.25 | 112 | COSMIC dataset: 5/81 TGCA dataset: 2/31 |
| Endocervical | 7.07 | 113 | COSMIC dataset: 3/60 TGCA dataset: 5/53 |
| Small cell | 16.67 | 18 | COSMIC dataset: 3/18 |
| Endometrioid | 22.22 | 9 | COSMIC dataset: 2/9 |
| Mixed adenosquamous | 5.5 | 18 | COSMIC dataset: 1/15 TGCA dataset:0/3 |
| Histology | Average % | N of Samples Tested | Details (Source: Number of Alterated Samples/Total of Samples) |
|---|---|---|---|
| Squamous | 5.88 | 136 | COSMIC dataset: 8/136 |
| Histology | Average % | N of Samples Tested | Details (Source: Number of Altered Samples/Total of Samples) |
|---|---|---|---|
| Leiomyosarcoma | 70.37 | 27 | TGCA dataset: 19/27 |
| Adenosarcoma | 18.18 | 11 | COSMIC dataset: 2/11 |
| Carcinosarcoma/MMMT | 15.4 | 485 | COSMIC dataset: 39/312 TGCA dataset: 36/173 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nero, C.; Ciccarone, F.; Pietragalla, A.; Scambia, G. PTEN and Gynecological Cancers. Cancers 2019, 11, 1458. https://doi.org/10.3390/cancers11101458
Nero C, Ciccarone F, Pietragalla A, Scambia G. PTEN and Gynecological Cancers. Cancers. 2019; 11(10):1458. https://doi.org/10.3390/cancers11101458
Chicago/Turabian StyleNero, Camilla, Francesca Ciccarone, Antonella Pietragalla, and Giovanni Scambia. 2019. "PTEN and Gynecological Cancers" Cancers 11, no. 10: 1458. https://doi.org/10.3390/cancers11101458
APA StyleNero, C., Ciccarone, F., Pietragalla, A., & Scambia, G. (2019). PTEN and Gynecological Cancers. Cancers, 11(10), 1458. https://doi.org/10.3390/cancers11101458