Validation of Hepatocellular Carcinoma Experimental Models for TGF-β Promoting Tumor Progression

 ,
,  ,
, 
Abstract
:1. Introduction
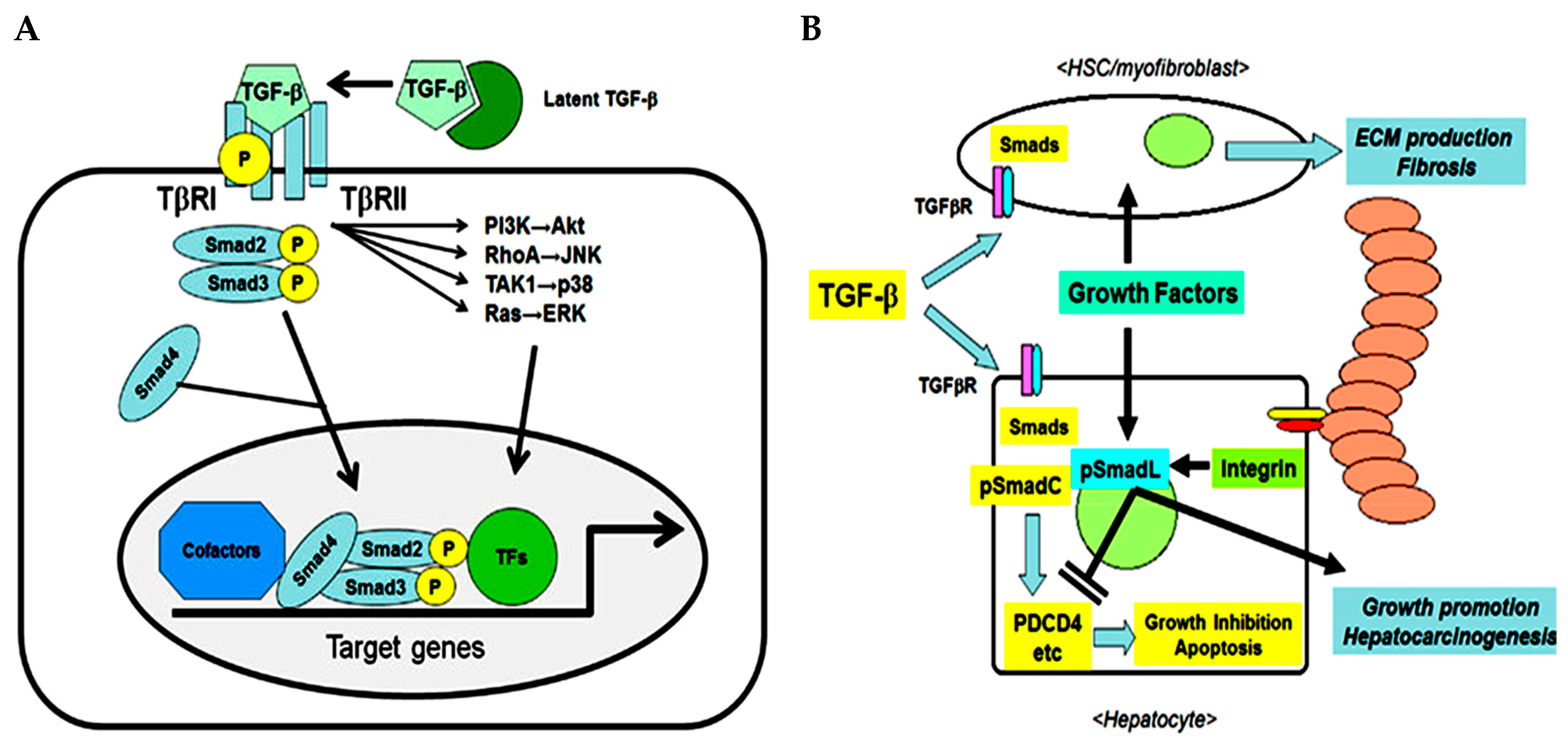
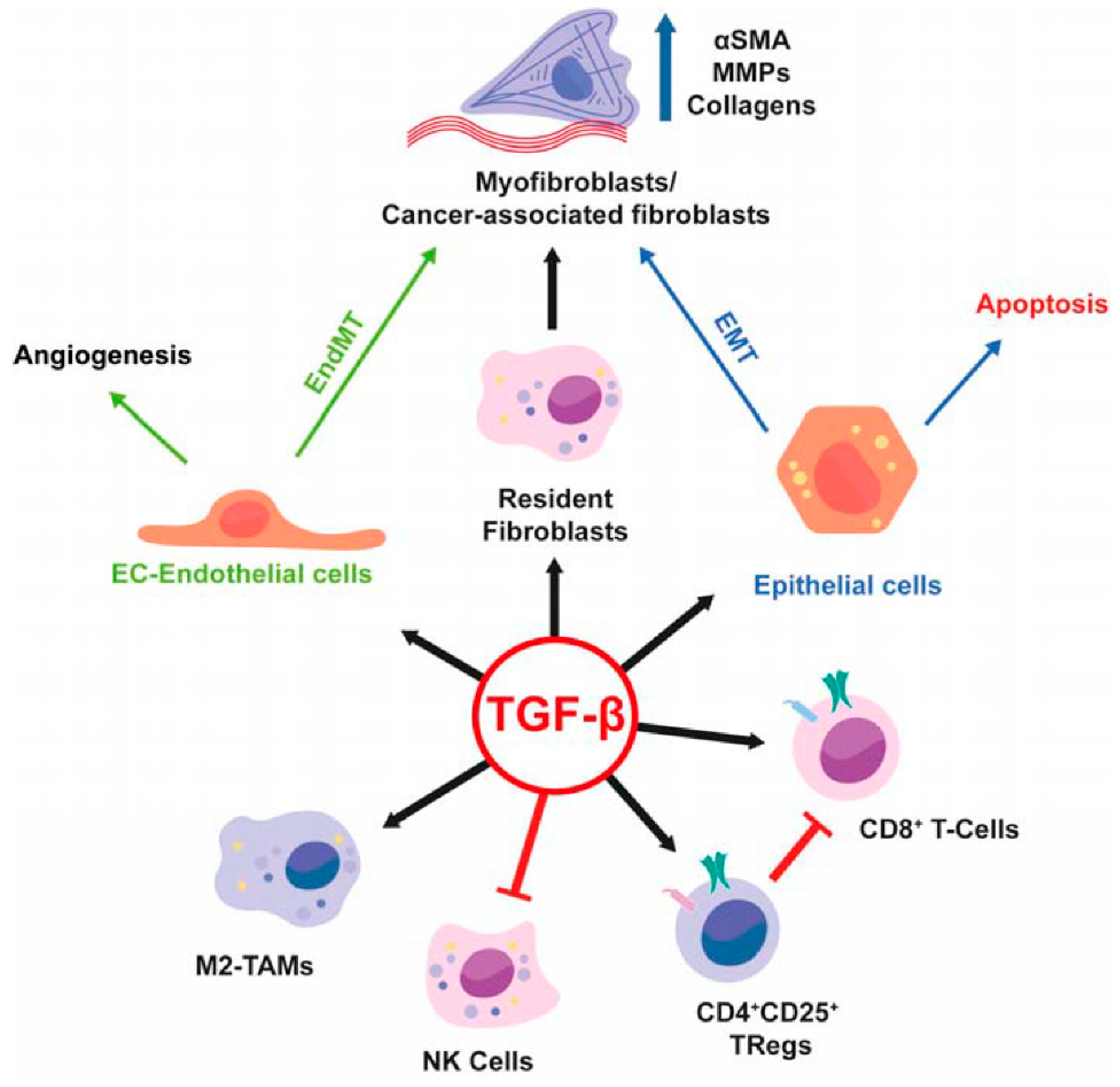
1.1. Regulation Strategies of TGF-β Signaling in HCC Cells
1.2. Mouse as a Translational Model for TGF-β Action in HCC
1.3. Inhibition of TGF-β in Experimental Mouse Models
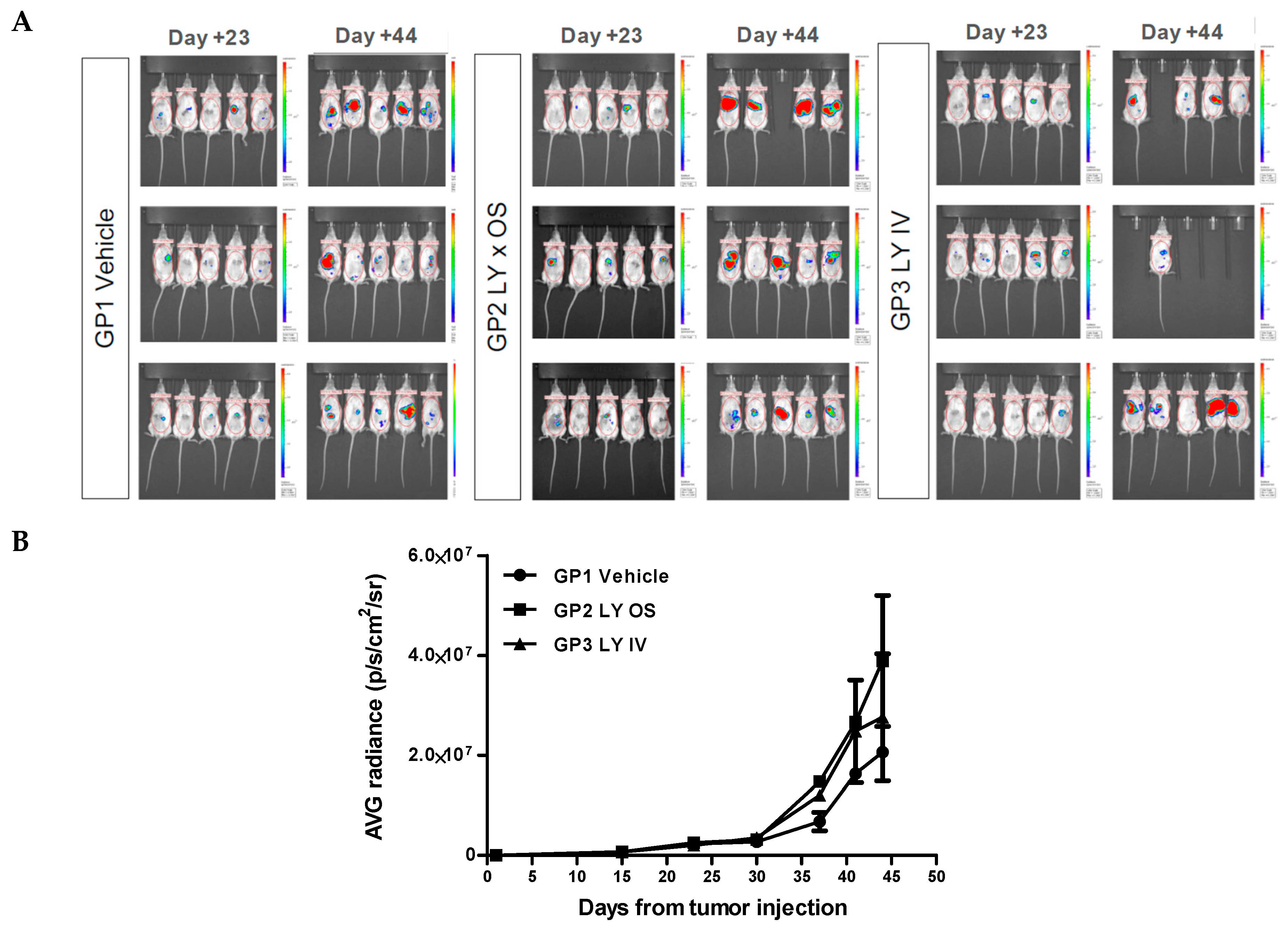
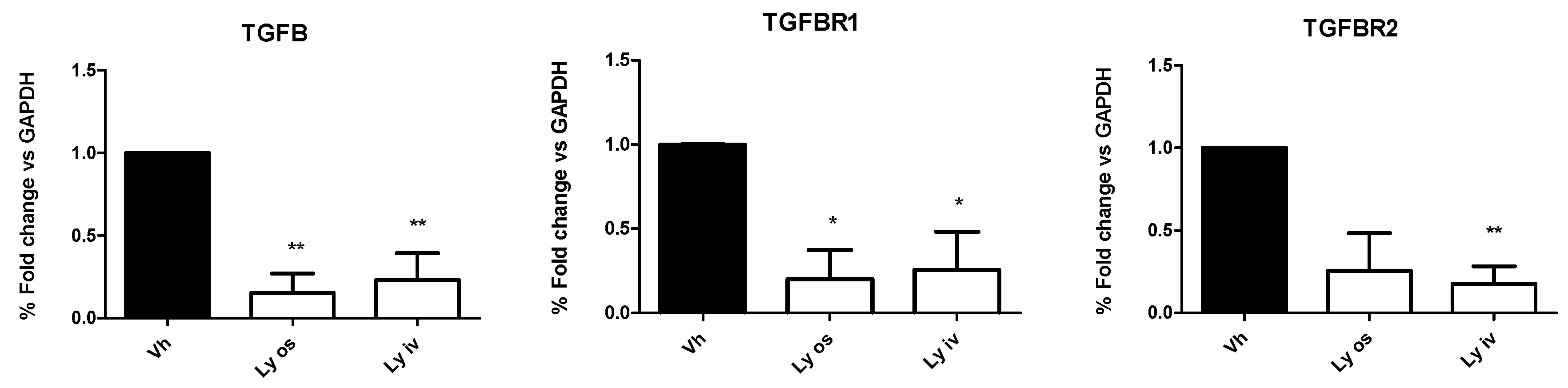
1.4. Efficacy Study of Galunisertib in Orthotopic Mouse Model
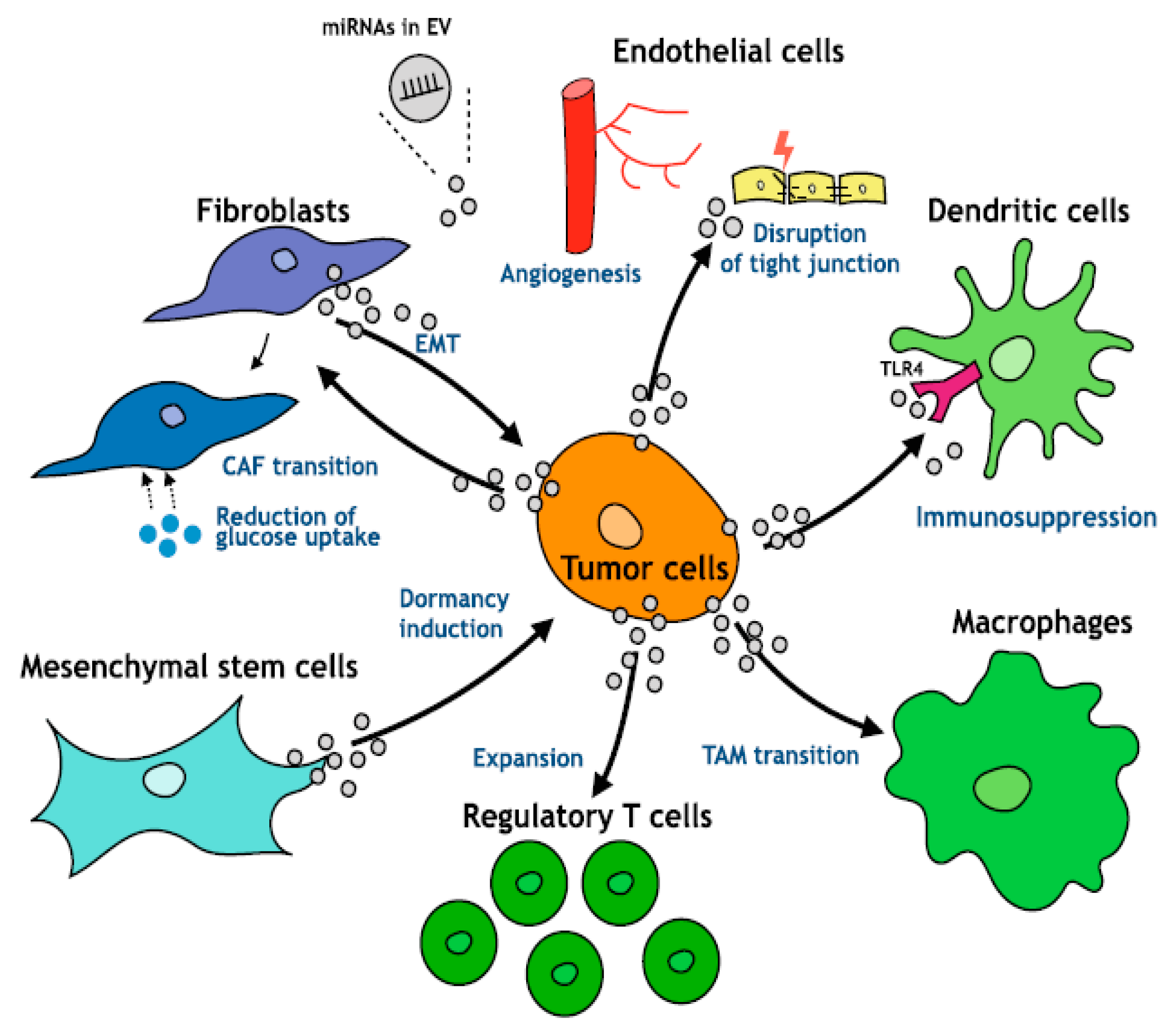
1.5. Exosomes-Moderator of Cross-Talk between Tumor Cells and Microenvironment of HCC
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAFs | cancer-associated fibroblasts |
| DCs | dendritic cells |
| DEN | N-nitrosodiethylamine |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| ELF | embryonic liver fodrin |
| EMT | epithelial-mesenchymal transition |
| FAK | focal adhesion kinase |
| FGF | fibroblast growth factor |
| HCC | hepatocellular carcinoma |
| HGF | hepatocyte growth factor |
| HSC | hepatic stellate cells |
| IFN-γ | interferon-gamma |
| IFN | interferons |
| IL | interleukins |
| miRNAs | microRNAs |
| NK | natural killer cells |
| NKT | natural killer T cells |
| NOG mice | NOD/Shi-Scid/IL-2Rγ null |
| PDGF | platelet-derived growth factor |
| PD-L1/ 2 | programmed death ligand-1 and 2 |
| SDF | stromal-derived factor |
| TAM | tumor-associated macrophages |
| TGF | transforming growth factor |
| TGFβR1 | Type I dimeric receptor |
| TGFβR2 | Type II serine/threonine kinase dimeric receptor |
| TNF-α | tumor necrosis factor-α |
| Treg | regulatory T |
| VEGF | vascular endothelial growth factor |
| α-SMA | α smooth muscle actin |
| DMSO | dimethylsulfoxide |
| PEG | polyethylene glycol |
| EtOH | ethanol |
References
- Derynck, R.; Zhang, Y. Intracellular Signalling: The Mad Way to Do It. Curr. Biol. 1996, 6, 1226–1229. [Google Scholar] [CrossRef]
- Clark, D.A.; Coker, R. Transforming Growth Factor-Beta (TGF-beta). Int. J. Biochem. Cell Biol. 1998, 30, 293–298. [Google Scholar] [CrossRef]
- Ozaki, I.; Hamajima, H.; Matsuhashi, S.; Mizuta, T. Regulation of TGF-?1-Induced Pro-Apoptotic Signaling by Growth Factor Receptors and Extracellular Matrix Receptor Integrins in the Liver. Front. Physiol. 2011, 2, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, N. TGF-Beta: From Latent to Active. Microbes Infect. 1999, 1, 1255–1263. [Google Scholar] [CrossRef]
- Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Transforming Growth Factor-β Gene Expression Signature in Mouse Hepatocytes Predicts Clinical Outcome in Human Cancer. Hepatology 2008, 47, 2059–2067. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of Epithelial to Mesenchymal Transition in Hepatocellular Carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Hu, S.-Q.; Xiao, L. The Cancer-Associated Fibroblasts and Drug Resistance. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2112–2119. [Google Scholar]
- Lau, E.Y.T.; Lo, J.; Cheng, B.Y.L.; Ma, M.K.F.; Lee, J.M.F.; Ng, J.K.Y.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016, 15, 1175–1189. [Google Scholar] [CrossRef] [Green Version]
- Fernando, J.; Malfettone, A.; Cepeda, E.B.; Vilarrasa-Blasi, R.; Bertran, E.; Raimondi, G.; Fabra, À.; Alvarez-Barrientos, A.; Fernández-Salguero, P.; Fernández-Rodríguez, C.M.; et al. A Mesenchymal-Like Phenotype and Expression of CD44 Predict Lack of Apoptotic Response to Sorafenib in Liver Tumor Cells. Int. J. Cancer 2015, 136, E161–E172. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A. Hallmarks of Cancer: Interactions with the Tumor Stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal Fibroblasts in Cancer Initiation and Progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Dou, C.; Jia, Y.; Tu, K.; Zheng, X. TIMP-1 Activated Carcinoma-Associated Fibroblasts Inhibit Tumor Apoptosis by Activating SDF1/CXCR4 Signaling in Hepatocellular Carcinoma. Oncotarget 2015, 6, 12061–12079. [Google Scholar] [CrossRef] [PubMed]
- Carambia, A.; Freund, B.; Schwinge, D.; Heine, M.; Laschtowitz, A.; Huber, S.; Wraith, D.C.; Korn, T.; Schramm, C.; Lohse, A.W.; et al. TGF-β-Dependent Induction of CD4+CD25+Foxp3+ Tregs by Liver Sinusoidal Endothelial Cells. J. Hepatol. 2014, 61, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, D.M.W.; van Loosdregt, J.; Gorlani, A.; Bekker, C.P.J.; Gröne, A.; Sibilia, M.; van Bergen en Henegouwen, P.M.P.; Roovers, R.C.; Coffer, P.J.; Sijts, A.J.A.M. Amphiregulin Enhances Regulatory T Cell-Suppressive Function via the Epidermal Growth Factor Receptor. Immunity 2013, 38, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baglieri, J.; Brenner, D.; Kisseleva, T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1723. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, L.; Iacobazzi, R.; Di Fonte, R.; Serratì, S.; Intini, A.; Solimando, A.; Brunetti, O.; Calabrese, A.; Leonetti, F.; Azzariti, A.; et al. CAFs and TGF-β Signaling Activation by Mast Cells Contribute to Resistance to Gemcitabine/Nabpaclitaxel in Pancreatic Cancer. Cancers 2019, 11, 330. [Google Scholar] [CrossRef]
- Giannelli, G.; Villa, E.; Lahn, M. Transforming Growth Factor-β as a Therapeutic Target in Hepatocellular Carcinoma. Cancer Res. 2014, 74, 1890–1894. [Google Scholar] [CrossRef]
- Yu, F.; Chen, B.; Fan, X.; Li, G.; Dong, P.; Zheng, J. Epigenetically-Regulated MicroRNA-9-5p Suppresses the Activation of Hepatic Stellate Cells via TGFBR1 and TGFBR2. Cell. Physiol. Biochem. 2017, 43, 2242–2252. [Google Scholar] [CrossRef] [Green Version]
- Feili, X.; Wu, S.; Ye, W.; Tu, J.; Lou, L. MicroRNA-34a-5p Inhibits Liver Fibrosis by Regulating TGF-β1/Smad3 Pathway in Hepatic Stellate Cells. Cell Biol. Int. 2018, 42, 1370–1376. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, B.; Jiao, A.; Li, F.; Sun, N.; Zhang, G.; Zhang, J. miR-663a Inhibits Tumor Growth and Invasion by Regulating TGF-β1 in Hepatocellular Carcinoma. BMC Cancer 2018, 18, 1179. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Mazzocca, A.; Peidrò, F.J.; Papappicco, P.; Fabregat, I.; De Santis, F.; Paradiso, A.; Sabbà, C.; Giannelli, G. Differential Inhibition of the TGF-β Signaling Pathway in HCC Cells Using the Small Molecule Inhibitor LY2157299 and the D10 Monoclonal Antibody against TGF-β Receptor Type II. PLoS ONE 2013, 8, e67109. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.; Kim, H.; Park, S.; Lee, M.; Yoon, J.; Kim, Y. Transforming Growth Factor-β Decreases Side Population Cells in Hepatocellular Carcinoma in�vitro. Oncol. Lett. 2018, 15, 8723–8728. [Google Scholar] [CrossRef] [PubMed]
- Rani, B.; Malfettone, A.; Dituri, F.; Soukupova, J.; Lupo, L.; Mancarella, S.; Fabregat, I.; Giannelli, G. Galunisertib Suppresses the Staminal Phenotype in Hepatocellular Carcinoma by Modulating CD44 Expression. Cell Death Dis. 2018, 9, 373. [Google Scholar] [CrossRef] [PubMed]
- Serova, M.; Tijeras-Raballand, A.; Dos Santos, C.; Albuquerque, M.; Paradis, V.; Neuzillet, C.; Benhadji, K.A.; Raymond, E.; Faivre, S.; de Gramont, A. Effects of TGF-Beta Signalling Inhibition with Galunisertib (LY2157299) in Hepatocellular Carcinoma Models and in ex vivo Whole Tumor Tissue Samples from Patients. Oncotarget 2015, 6, 21614–21627. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Agarwal, R.; Dituri, F.; Lupo, L.; Trerotoli, P.; Mancarella, S.; Winter, P.; Giannelli, G. NGS-based Transcriptome Profiling Reveals Biomarkers for Companion Diagnostics of the TGF-β Receptor Blocker Galunisertib in HCC. Cell Death Dis. 2017, 8, e2634. [Google Scholar] [CrossRef]
- Smith, C.L.; Blake, J.A.; Kadin, J.A.; Richardson, J.E.; Bult, C.J. Mouse Genome Database (MGD)-2018: Knowledgebase for the Laboratory Mouse. Nucleic Acids Res. 2018, 46, D836–D842. [Google Scholar] [CrossRef] [PubMed]
- Santos, N.P.; Colaço, A.A.; Oliveira, P.A. Animal Models as a Tool in Hepatocellular Carcinoma Research: A Review. Tumor Biol. 2017, 39, 101042831769592. [Google Scholar] [CrossRef]
- Mazzocca, A.; Dituri, F.; De Santis, F.; Filannino, A.; Lopane, C.; Betz, R.C.; Li, Y.-Y.; Mukaida, N.; Winter, P.; Tortorella, C.; et al. Lysophosphatidic Acid Receptor LPAR6 Supports the Tumorigenicity of Hepatocellular Carcinoma. Cancer Res. 2015, 75, 532–543. [Google Scholar] [CrossRef]
- Heindryckx, F.; Colle, I.; Van Vlierberghe, H. Experimental Mouse Models for Hepatocellular Carcinoma Research. Int. J. Exp. Pathol. 2009, 90, 367–386. [Google Scholar] [CrossRef]
- Tang, Z.Y.; Ye, S.L.; Liu, Y.K.; Qin, L.X.; Sun, H.C.; Ye, Q.H.; Wang, L.; Zhou, J.; Qiu, S.J.; Li, Y.; et al. A Decade’s Studies on Metastasis of Hepatocellular Carcinoma. J. Cancer Res. Clin. Oncol. 2004, 130, 187–196. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Tian, D.-A.; Li, P.-Y.; He, X.-X. Mouse models of liver cancer: Progress and recommendations. Oncotarget 2015, 6, 23307. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Herlyn, M. Defining Microenvironments within Mouse Models that Enhance Tumor Aggressiveness. Cancer Biol. Ther. 2009, 8, 380–381. [Google Scholar] [CrossRef] [PubMed]
- Speroni, L.; de los Angeles Bustuoabad, V.; Gasparri, J.; Chiaramoni, N.S.; Taira, M.C.; Ruggiero, R.A.; del Valle Alonso, S. Alternative Site of Implantation Affects Tumor Malignancy and Metastatic Potential in Mice: Its Comparison to the Flank Model. Cancer Biol. Ther. 2009, 8, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Spear, S.; Candido, J.B.; McDermott, J.R.; Ghirelli, C.; Maniati, E.; Beers, S.A.; Balkwill, F.R.; Kocher, H.M.; Capasso, M. Discrepancies in the Tumor Microenvironment of Spontaneous and Orthotopic Murine Models of Pancreatic Cancer Uncover a New Immunostimulatory Phenotype for B Cells. Front. Immunol. 2019, 10, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, Q.; You, A.; Guo, Z.; Zuo, B.; Gao, X.; Zhang, T.; Du, Z.; Wu, C.; Yin, H.F. Correction: Intrahepatic Tissue Implantation Represents a Favorable Approach for Establishing Orthotopic Transplantation Hepatocellular Carcinoma Mouse Models. PLoS ONE 2018, 11, e0148263. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, G.J.; Dong, X.; Whitehorne, M.; Grehan, A.; Seddon, M.; Shah, A.M.; Zhang, X.; Fabre, J.W. Cardiovascular Function Following Acute Volume Overload for Hydrodynamic Gene Delivery to the Liver. Gene Ther. 2007, 14, 1208–1217. [Google Scholar] [CrossRef]
- Suda, T.; Liu, D. Hydrodynamic Gene Delivery: Its Principles and Applications. Mol. Ther. 2007, 15, 2063–2069. [Google Scholar] [CrossRef]
- Chen, X.; Calvisi, D.F. Hydrodynamic Transfection for Generation of Novel Mouse Models for Liver Cancer Research. Am. J. Pathol. 2014, 184, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.; Ju, H.-L.; Chung, S.I.; Cho, K.J.; Eun, J.W.; Nam, S.W.; Han, K.-H.; Calvisi, D.F.; Ro, S.W. Transforming Growth Factor-β Promotes Liver Tumorigenesis in Mice via Up-regulation of Snail. Gastroenterology 2017, 153, 1378–1391. [Google Scholar] [CrossRef]
- Chabicovsky, M.; Wastl, U.; Taper, H.; Grasl-Kraupp, B.; Schulte-Hermann, R.; Bursch, W. Induction of Apoptosis in Mouse Liver Adenoma and Carcinoma in vivo by Transforming Growth Factor-β1. J. Cancer Res. Clin. Oncol. 2003, 129, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Baek, H.J.; Lim, S.C.; Kitisin, K.; Jogunoori, W.; Tang, Y.; Marshall, M.B.; Mishra, B.; Kim, T.H.; Cho, K.H.; Kim, S.S.; et al. Hepatocellular Cancer Arises from Loss of Transforming Growth Factor Beta Signaling Adaptor Protein Embryonic Liver Fodrin through Abnormal Angiogenesis. Hepatology 2008, 48, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Inokuchi, S.; Roh, Y.S.; Song, J.; Loomba, R.; Park, E.J.; Seki, E. Transforming Growth Factor–β Signaling in Hepatocytes Promotes Hepatic Fibrosis and Carcinogenesis in Mice With Hepatocyte-Specific Deletion of TAK1. Gastroenterology 2013, 144, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Han, J.; Hou, B.; Deng, C.; Wu, H.; Shen, L. Sulforaphane inhibits TGF-β-Induced Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma Cells via the Reactive Oxygen Species-Dependent Pathway. Oncol. Rep. 2016, 35, 2977–2983. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Kim, M.J.; Park, S.Y.; Kim, J.S.; Lim, W.; Nam, J.S.; Sheen, Y.Y. TIMP-1 Mediates TGF-β-Dependent Crosstalk between Hepatic Stellate and Cancer Cells via FAK Signaling. Sci. Rep. 2015, 5, 16492. [Google Scholar] [CrossRef] [PubMed]
- del Mercato, L.L.; Ferraro, M.M.; Baldassarre, F.; Mancarella, S.; Greco, V.; Rinaldi, R.; Leporatti, S. Biological Applications of LbL Multilayer Capsules: From Drug Delivery to Sensing. Adv. Colloid Interface Sci. 2014, 207, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Kubes, P. Immune Surveillance by the Liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Shang, N.; Figini, M.; Shangguan, J.; Wang, B.; Sun, C.; Pan, L.; Ma, Q.; Zhang, Z. Dendritic Cells Based Immunotherapy. Am. J. Cancer Res. 2017, 7, 2091–2102. [Google Scholar]
- Cheng, J.; Deng, Y.; Yi, H.; Wang, G.; Fu, B.; Chen, W.; Liu, W.; Tai, Y.; Peng, Y.; Zhang, Q. Hepatic Carcinoma-Associated Fibroblasts Induce IDO-Producing Regulatory Dendritic Cells through IL-6-Mediated STAT3 Activation. Oncogenesis 2016, 5, e198. [Google Scholar] [CrossRef]
- Maecker, B.; Mougiakakos, D.; Zimmermann, M.; Behrens, M.; Hollander, S.; Schrauder, A.; Schrappe, M.; Welte, K.; Klein, C. Dendritic Cell Deficiencies in Pediatric Acute Lymphoblastic Leukemia Patients. Leukemia 2006, 20, 645–649. [Google Scholar] [CrossRef]
- Wojas, K.; Tabarkiewicz, J.; Jankiewicz, M.; Roliński, J. Dendritic Cells in Peripheral Blood of Patients with Breast and Lung Cancer—A Pilot Study. Folia Histochem. Cytobiol. 2004, 42, 45–48. [Google Scholar] [PubMed]
- Bellik, L.; Gerlini, G.; Parenti, A.; Ledda, F.; Pimpinelli, N.; Neri, B.; Pantalone, D. Role of Conventional Treatments on Circulating and Monocyte-Derived Dendritic Cells in Colorectal Cancer. Clin. Immunol. 2006, 121, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Mancarella, S.; Cigliano, A.; Chieti, A.; Giannelli, G. TGF-β as Multifaceted Orchestrator in HCC Progression: Signaling, EMT, Immune Microenvironment, and Novel Therapeutic Perspectives. Semin. Liver Dis. 2019, 39, 53–69. [Google Scholar]
- Chen, K.-J.; Lin, S.-Z.; Zhou, L.; Xie, H.-Y.; Zhou, W.-H.; Taki-Eldin, A.; Zheng, S.-S. Selective Recruitment of Regulatory T Cell through CCR6-CCL20 in Hepatocellular Carcinoma Fosters Tumor Progression and Predicts Poor Prognosis. PLoS ONE 2011, 6, e24671. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Di Lernia, G.; Solimando, A.G.; Cicco, S.; Saltarella, I.; Lamanuzzi, A.; Ria, R.; Frassanito, M.A.; Ponzoni, M.; Ditonno, P.; et al. Bone Marrow Endothelial Cells Sustain a Tumor-Specific CD8+ T Cell Subset with Suppressive Function in Myeloma Patients. Oncoimmunology 2019, 8, e1486949. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gingold, J.A.; Su, X. Immunomodulatory TGF-β Signaling in Hepatocellular Carcinoma. Trends Mol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Argentiero, A.; De Summa, S.; Di Fonte, R.; Iacobazzi, R.M.; Porcelli, L.; Da Vià, M.; Brunetti, O.; Azzariti, A.; Silvestris, N.; Solimando, A.G. Gene Expression Comparison between the Lymph Node-Positive and -Negative Reveals a Peculiar Immune Microenvironment Signature and a Theranostic Role for WNT Targeting in Pancreatic Ductal Adenocarcinoma: A Pilot Study. Cancers 2019, 11, 942. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhou, L.; Xie, H.; Ahmed, T.-E.; Feng, X.; Zheng, S. Intratumoral Regulatory T Cells Alone or in Combination with Cytotoxic T Cells Predict Prognosis of Hepatocellular Carcinoma after Resection. Med. Oncol. 2012, 29, 1817–1826. [Google Scholar] [CrossRef]
- Tanaka, T.; Nakamura, J.; Noshiro, H. Promising Immunotherapies for Esophageal Cancer. Expert Opin. Biol. Ther. 2017, 17, 723–733. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ Pathway with Galunisertib, a TGFβRI Small Molecule Inhibitor, promotes anti-tumor Immunity Leading to Durable, Complete Responses, as Monotherapy and in Combination with Checkpoint Blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Rong, D.; Zhang, B.; Zheng, W.; Wang, X.; Chen, Z.; Tang, W. Current Perspectives on the Immunosuppressive Tumor Microenvironment in Hepatocellular Carcinoma: Challenges and Opportunities. Mol. Cancer 2019, 18, 130. [Google Scholar] [CrossRef] [PubMed]
- Heindryckx, F. Targeting the Tumor Stroma in Hepatocellular Carcinoma. World J. Hepatol. 2014, 7, 165. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.W.Y.; Pang, R.W.C.; Lau, C.; Sun, C.K.; Yu, W.C.; Fan, S.T.; Poon, R.T.P. Significance of Circulating Endothelial Progenitor Cells in Hepatocellular Carcinoma. Hepatology 2006, 44, 836–843. [Google Scholar] [CrossRef]
- Feng, T.; Yu, H.; Xia, Q.; Ma, Y.; Yin, H.; Shen, Y.; Liu, X. Cross-talk Mechanism between Endothelial Cells and Hepatocellular Carcinoma Cells via Growth Factors and Integrin Pathway Promotes Tumor Angiogenesis and Cell Migration. Oncotarget 2017, 8, 69577. [Google Scholar] [CrossRef] [PubMed]
- Neiva, K.G.; Zhang, Z.; Miyazawa, M.; Warner, K.A.; Karl, E.; Nör, J.E. Cross talk Initiated by Endothelial Cells Enhances Migration and Inhibits Anoikis of Squamous Cell Carcinoma Cells through STAT3/Akt/ERK Signaling. Neoplasia 2009, 11, 583. [Google Scholar] [CrossRef] [PubMed]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic Stellate Cells and Extracellular Matrix in Hepatocellular Carcinoma: More Complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.G.; Ren, X.H.; Wang, S.S.; Liang, X.H.; Tang, Y.L. Immunocompromised and Immunocompetent Mouse Models for Head and Neck Squamous Cell Carcinoma. Onco Targets Ther. 2016, 9, 545–555. [Google Scholar] [PubMed]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The Inflammatory Microenvironment in Hepatocellular Carcinoma: A Pivotal Role for Tumor-Associated Macrophages. Biomed Res. Int. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogure, A.; Kosaka, N.; Ochiya, T. Cross-talk between Cancer Cells and Their Neighbors via miRNA in Extracellular Vesicles: An Emerging Player in Cancer Metastasis. J. Biomed. Sci. 2019, 26, 7. [Google Scholar] [CrossRef] [PubMed]
- Fricke, F.; Lee, J.; Michalak, M.; Warnken, U.; Hausser, I.; Suarez-Carmona, M.; Halama, N.; Schnölzer, M.; Kopitz, J.; Gebert, J. TGFBR2-Dependent Alterations of Exosomal Cargo and Functions in DNA Mismatch Repair-Deficient HCT116 Colorectal Cancer Cells. Cell Commun. Signal. 2017, 15, 14. [Google Scholar] [CrossRef] [PubMed]
- Kogure, T.; Lin, W.-L.; Yan, I.K.; Braconi, C.; Patel, T. Intercellular Nanovesicle-Mediated microRNA Transfer: A Mechanism of Environmental Modulation of Hepatocellular Cancer Cell Growth. Hepatology 2011, 54, 1237–1248. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I.; Basel, M.T.; Narayanan, S.; Ganta, C.; et al. Tumor-Derived Exosomal miR-1247-3p induces Cancer-Associated Fibroblast Activation to Foster Lung Metastasis of Liver Cancer. Hepatology 2018, 8, 139–154. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Strain | Immune Competence | Tumor Formation | Model | Aim of the Study | Results | Reference |
|---|---|---|---|---|---|---|
| C57BL/6 | Competent | Induced | Hydrodynamic tail-vein injection with HRASG12V | Overexpression of SMAD7 or knockdown of SMAD2,3,4 and its influence on TGF-β pathways | TGF-β inhibition reduced formation and growth of liver tumors when RAS, TAZ proteins and short hairpin RNA are expressed | [40] |
| B6C3F1 | Competent | Induced | Injection of DEN. Single injection of TGF-β before sacrifice | Evaluation of liver apoptosis extent by exogenous TGF-β | Apoptosis is high in HCC and increases even more by administration of pro-apoptotic cytokine | [41] |
| ELF (embryonic liver fodrin) knockout | Competent | Spontaneous | ELF knockouts develop HCC in 15 months | ELF as a target for enhancing TGF-β pathway to suppress tumor formation | Loss of ELF causes disruption of TGF-β pathways and HCC development | [42] |
| Tak1ΔHep | Competent | Spontaneous | Tak1ΔHep mice develop HCC in 9 months | TGF-β signaling in TAK1 deleted hepatocytes | TGF-β promotes HCC and expression of anti-apoptotic, pro-oncogenic, and angiogenic factors | [43] |
| Female BALB/C nude | Deficient | Induced | Subcutaneous injection with Hg2 cells | Effects of sulforaphane on TGF-β pathways | Sulforaphane inhibits TGF-β linked EMT transition | [44] |
| Female BALB/C nu/nu | Deficient | Induced | Oral administration of EW-7197 (ALK 5 inhibitor) in orthotopic model/implanted SK-HEP1-Luc cells | ALK 5 inhibition effects TGF-β signaling between Stellate cells and HCC cells | ALK 5 inhibitor interferes with tumor growth | [45] |
| Samples | Intercept | Slope | R% |
|---|---|---|---|
| STDs Solution | 8.65 | 224.73 | 99.94 |
| STDs Plasma | 6.60 | 201.91 | 99.95 |
| Recovery% | 89.80 |
| Concentration | 0.05 | 0.1 | 0.25 | 0.5 | 1 | 2.00 | Intercept | Slope | R% |
|---|---|---|---|---|---|---|---|---|---|
| DAY1 | 0.055 | 0.085 | 0.294 | 0.505 | 0.998 | 1.957 | 2.54 | 207.38 | 98.31 |
| DAY2 | 0.044 | 0.076 | 0.269 | 0.509 | 1.011 | 1.991 | 6.60 | 201.91 | 99.95 |
| DAY3 | 0.053 | 0.090 | 0.286 | 0.495 | 0.992 | 1.962 | 3.19 | 205.36 | 99.16 |
| Mean | 0.05 | 0.08 | 0.28 | 0.50 | 1.00 | 1.97 | 4.11 | 204.88 | 99.14 |
| SD | 0.006 | 0.007 | 0.013 | 0.007 | 0.010 | 0.018 | 2.77 | 0.82 | |
| Precision | 11.565 | 8.480 | 4.511 | 1.434 | 0.971 | 0.932 | 0.83 | ||
| Accuracy | 1.333 | −16.333 | 13.200 | 0.600 | 0.033 | −1.500 |
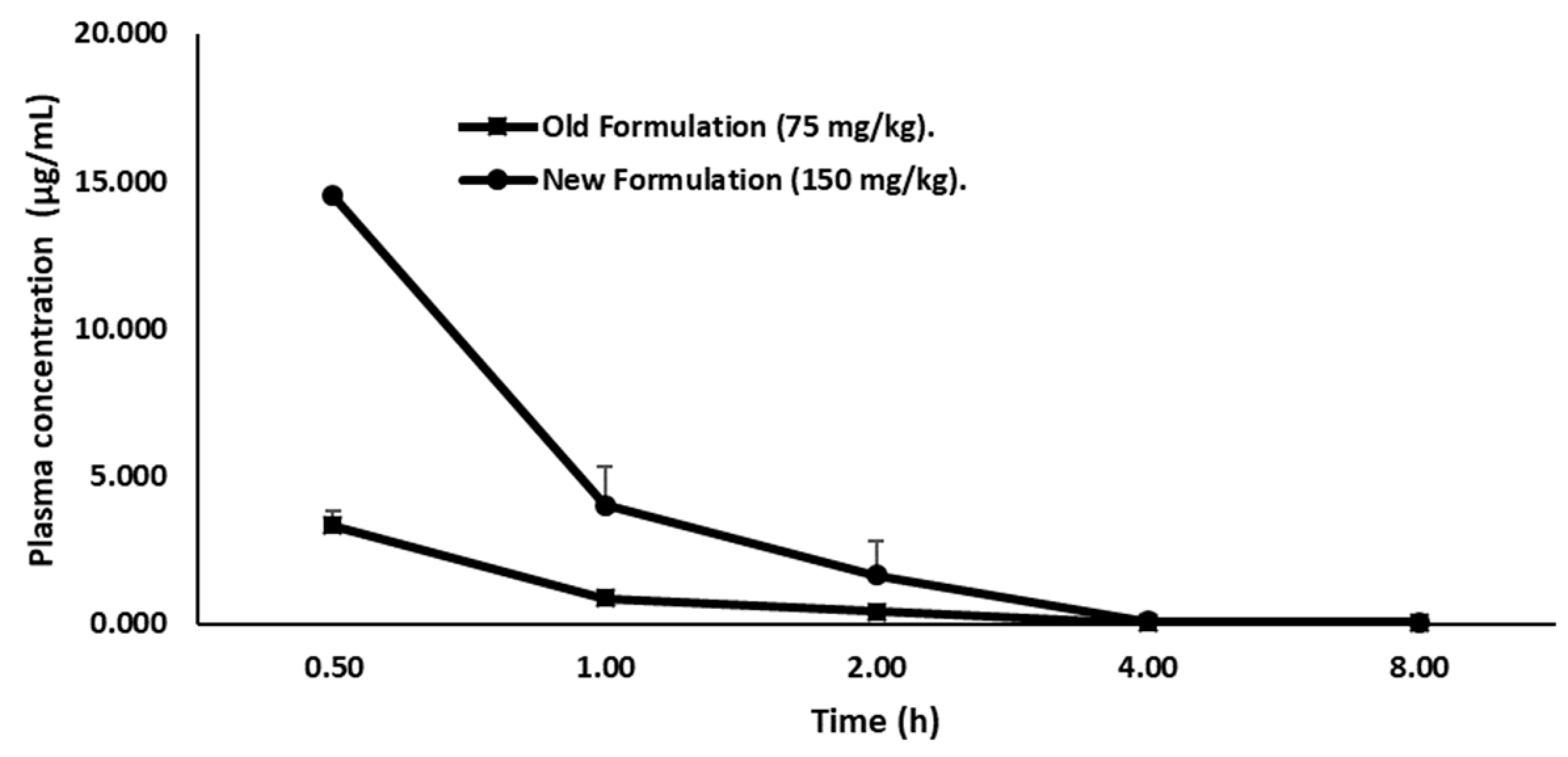
| Parameters | Suspension (75mg/kg) | Solution (150 mg/kg) |
|---|---|---|
| Kelim (h−1) | 0.49 | 0.59 |
| T1/2 (h) | 1.40 | 1.71 |
| Cmax (µg/mL) | 3.33 | 19.01 |
| Tmax (h) | 0.5 | 0.5 |
| AUClast (µg/h/mL) | 3.11 | 15.48 |
| Relative bioavailability (F) | 2.49 |
| Mouse Model | NSG™ | NRG | NSGS | NOD scid | BALB scid | B6 Rag1 | Nude | |
|---|---|---|---|---|---|---|---|---|
| Immune Cells | ||||||||
| Macrophages | defective | defective | defective | defective | present | present | present | |
| Dendritic cells | defective | defective | defective | defective | present | present | present | |
| Mature T-cells | absent | absent | absent | absent | absent | absent | absent | |
| note | Capable of maintaining a human tumor microenvironment after engraftment | |||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancarella, S.; Krol, S.; Crovace, A.; Leporatti, S.; Dituri, F.; Frusciante, M.; Giannelli, G. Validation of Hepatocellular Carcinoma Experimental Models for TGF-β Promoting Tumor Progression. Cancers 2019, 11, 1510. https://doi.org/10.3390/cancers11101510
Mancarella S, Krol S, Crovace A, Leporatti S, Dituri F, Frusciante M, Giannelli G. Validation of Hepatocellular Carcinoma Experimental Models for TGF-β Promoting Tumor Progression. Cancers. 2019; 11(10):1510. https://doi.org/10.3390/cancers11101510
Chicago/Turabian StyleMancarella, Serena, Silke Krol, Alberto Crovace, Stefano Leporatti, Francesco Dituri, Martina Frusciante, and Gianluigi Giannelli. 2019. "Validation of Hepatocellular Carcinoma Experimental Models for TGF-β Promoting Tumor Progression" Cancers 11, no. 10: 1510. https://doi.org/10.3390/cancers11101510
APA StyleMancarella, S., Krol, S., Crovace, A., Leporatti, S., Dituri, F., Frusciante, M., & Giannelli, G. (2019). Validation of Hepatocellular Carcinoma Experimental Models for TGF-β Promoting Tumor Progression. Cancers, 11(10), 1510. https://doi.org/10.3390/cancers11101510