Inhibition of TFF3 Enhances Sensitivity—and Overcomes Acquired Resistance—to Doxorubicin in Estrogen Receptor-Positive Mammary Carcinoma


 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
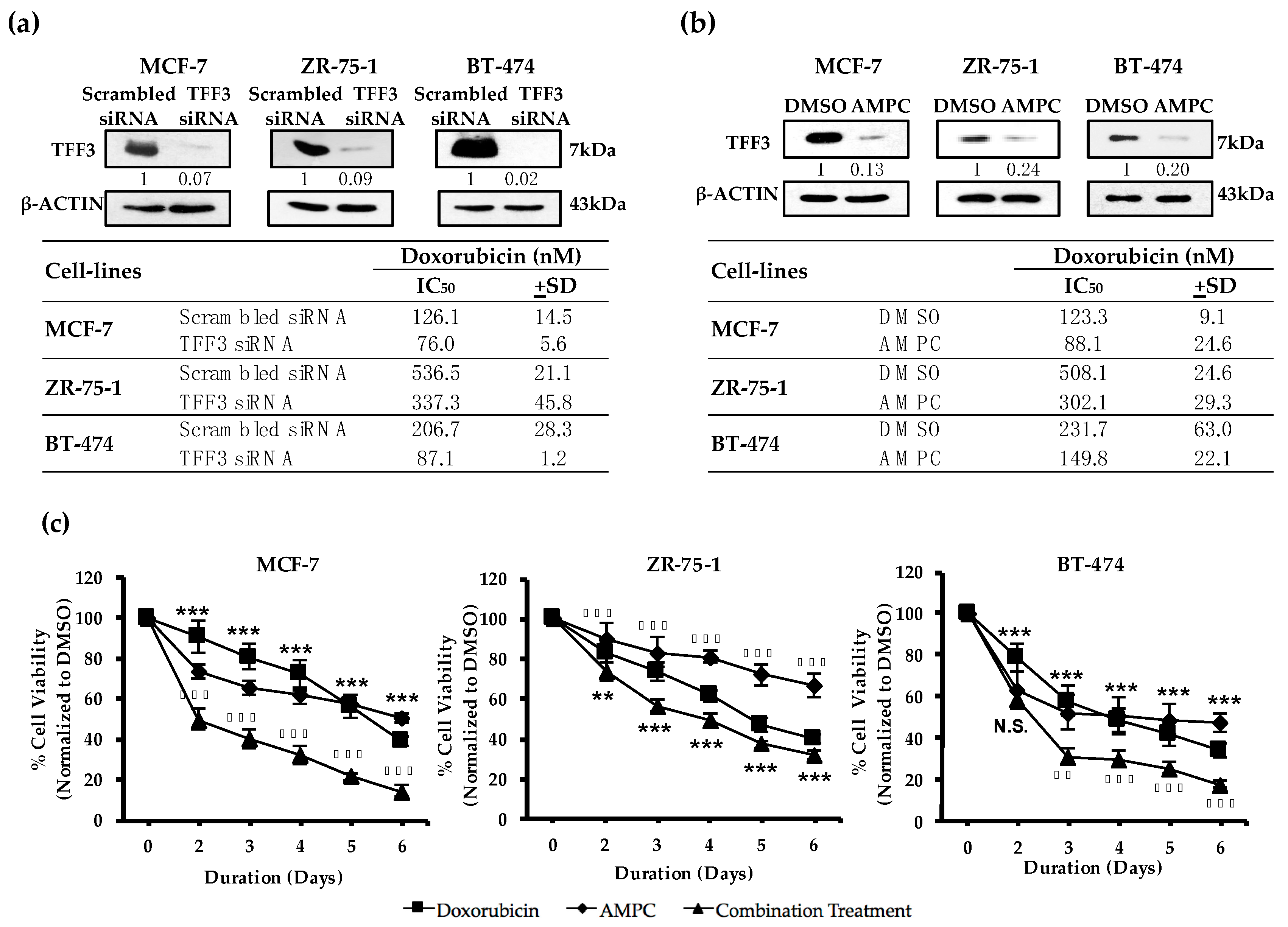
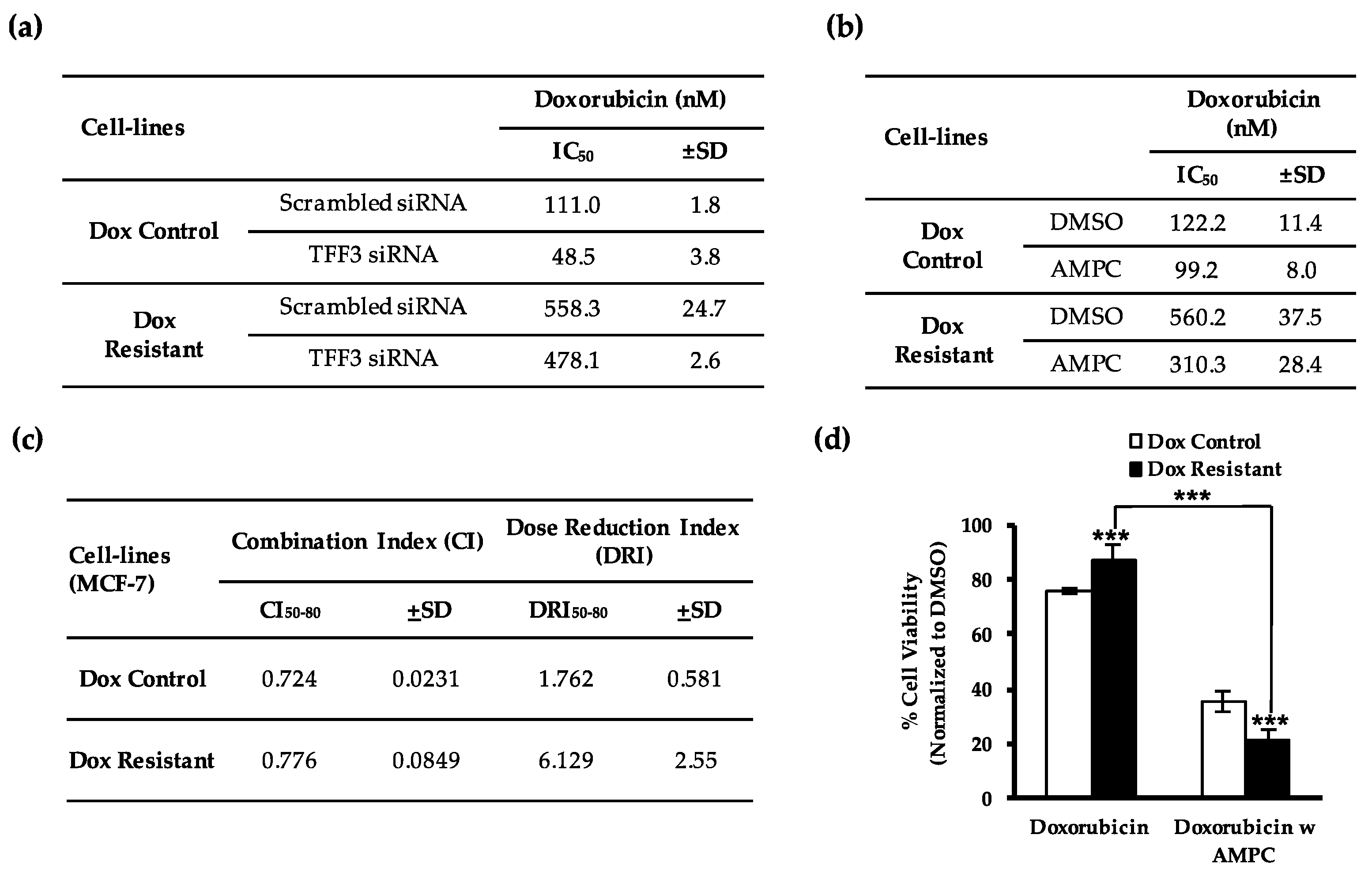
2.1. TFF3 Regulates ER+MC Cell Sensitivity to Doxorubicin
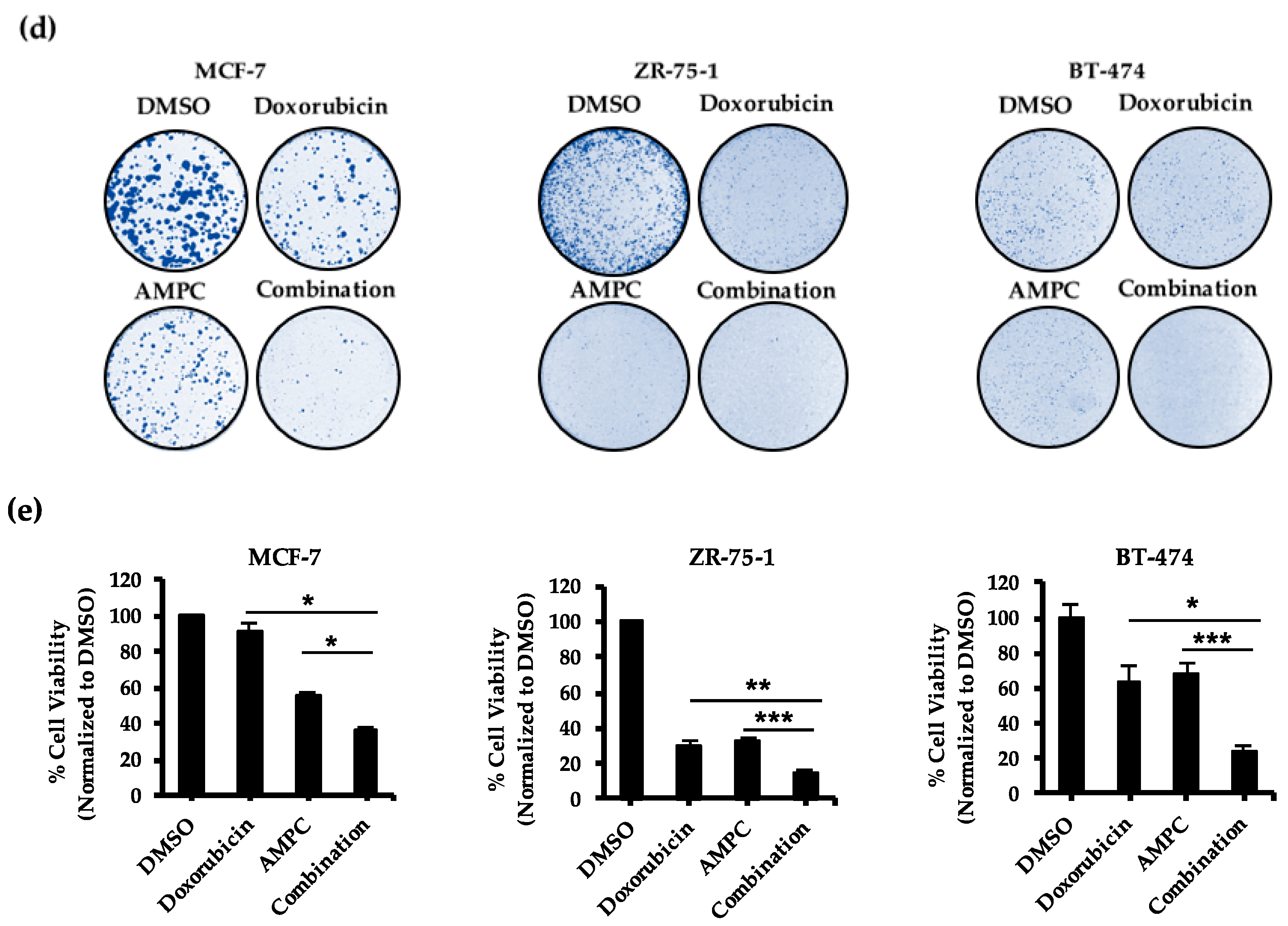
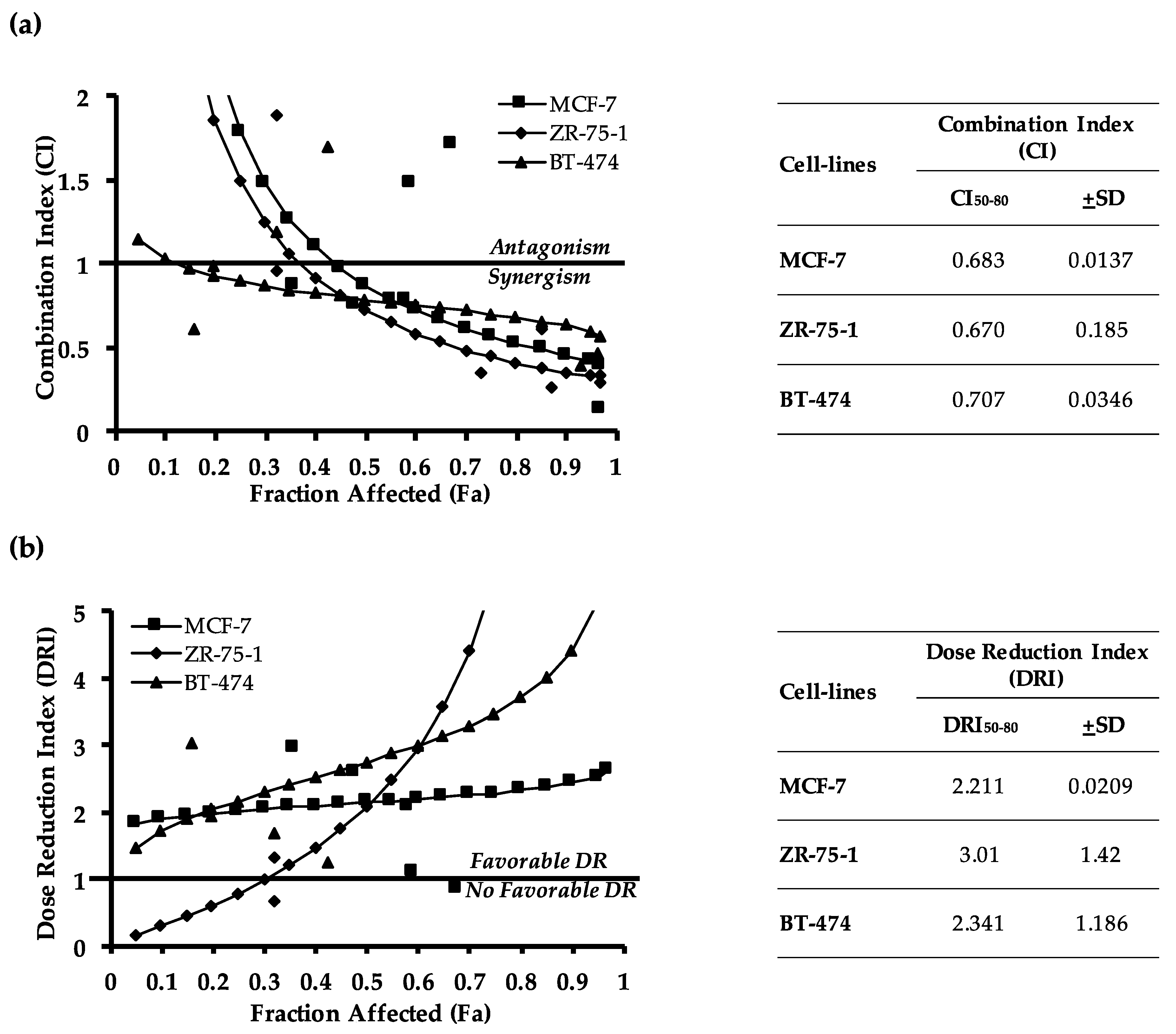
2.2. Pharmacological Inhibition of TFF3 Enhances Cellular Sensitivity to Doxorubicin
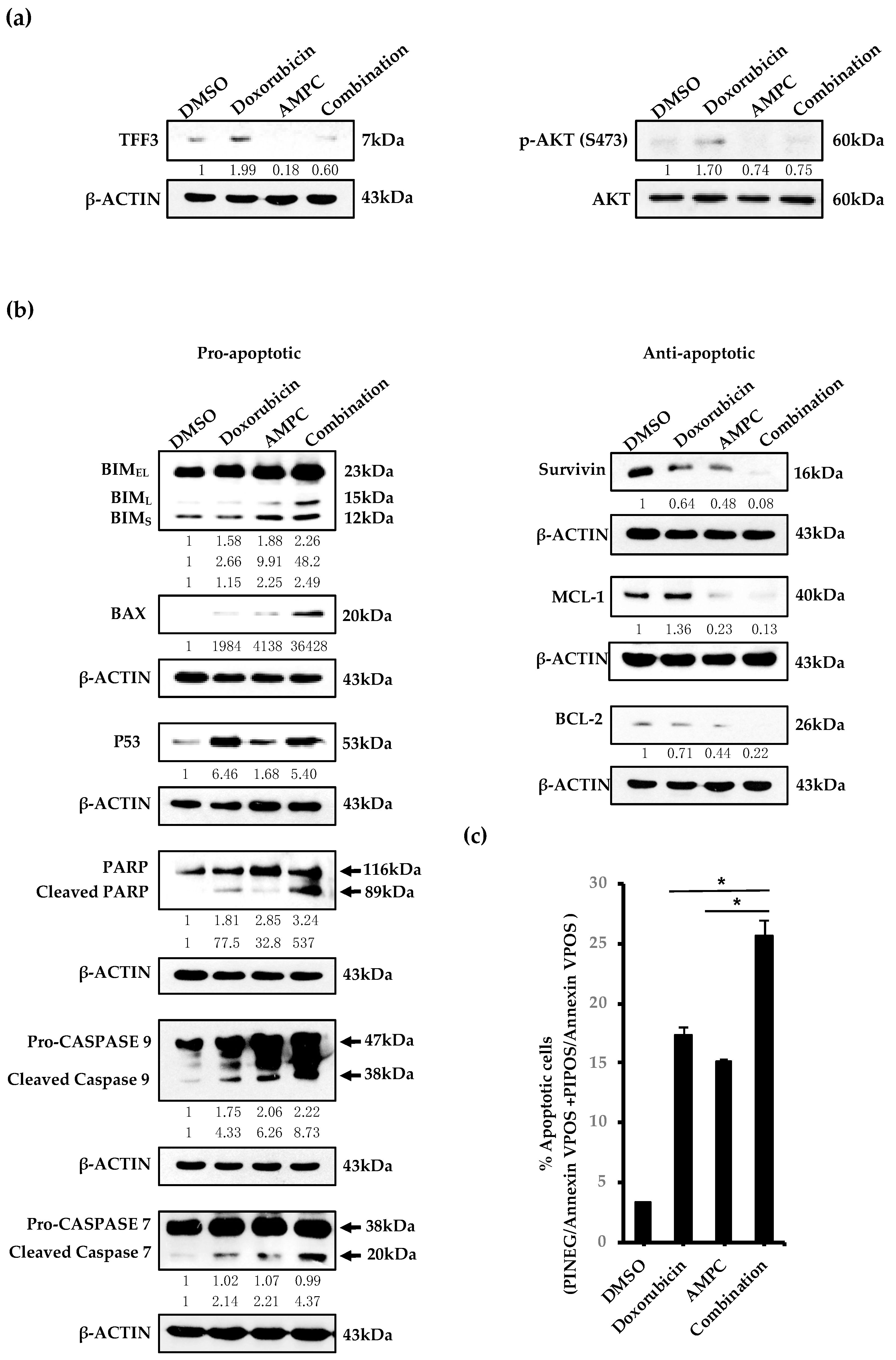
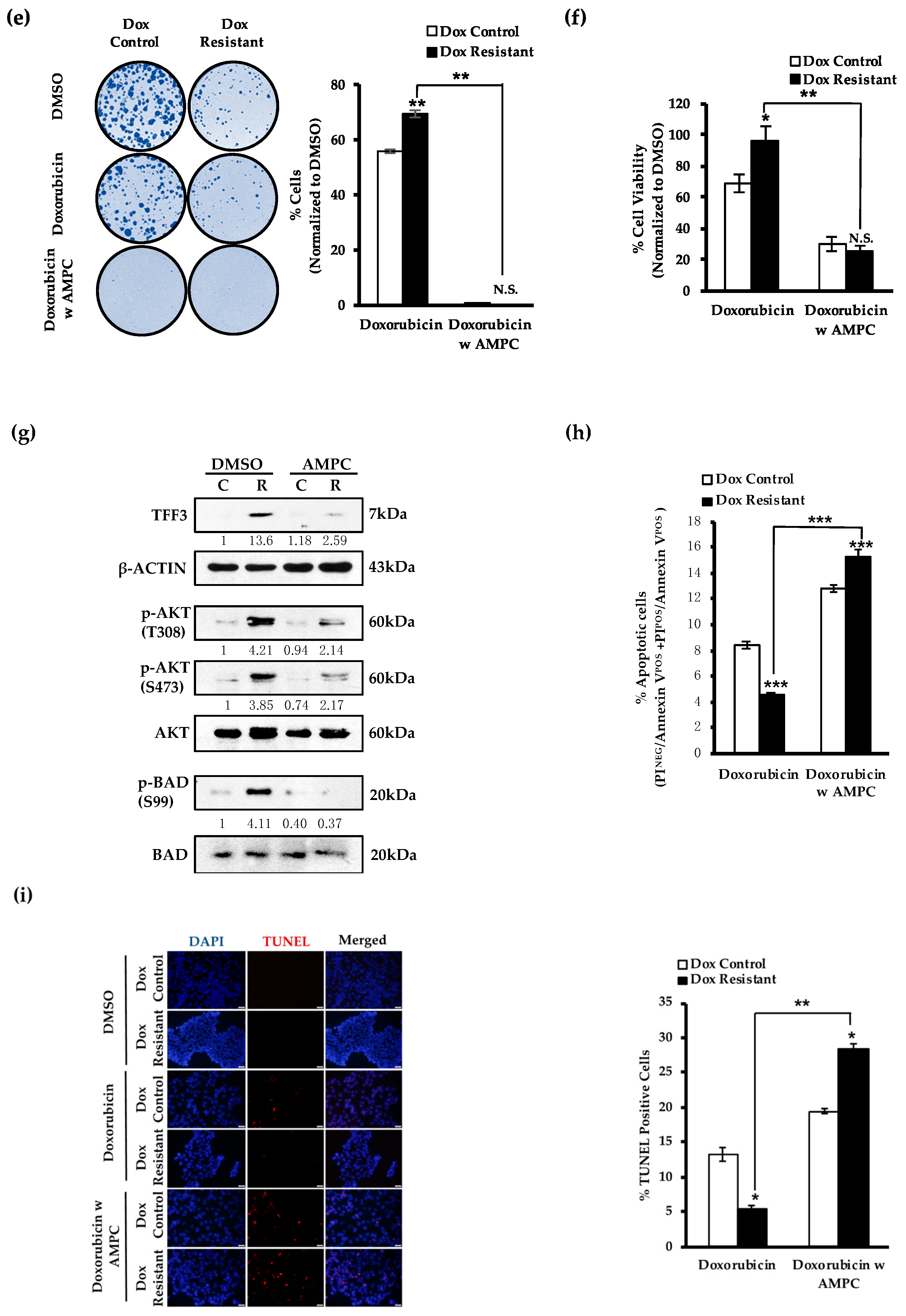
2.3. AMPC Inhibits Doxorubicin-Induced AKT Activation and Enhances Doxorubicin-Induced Apoptosis
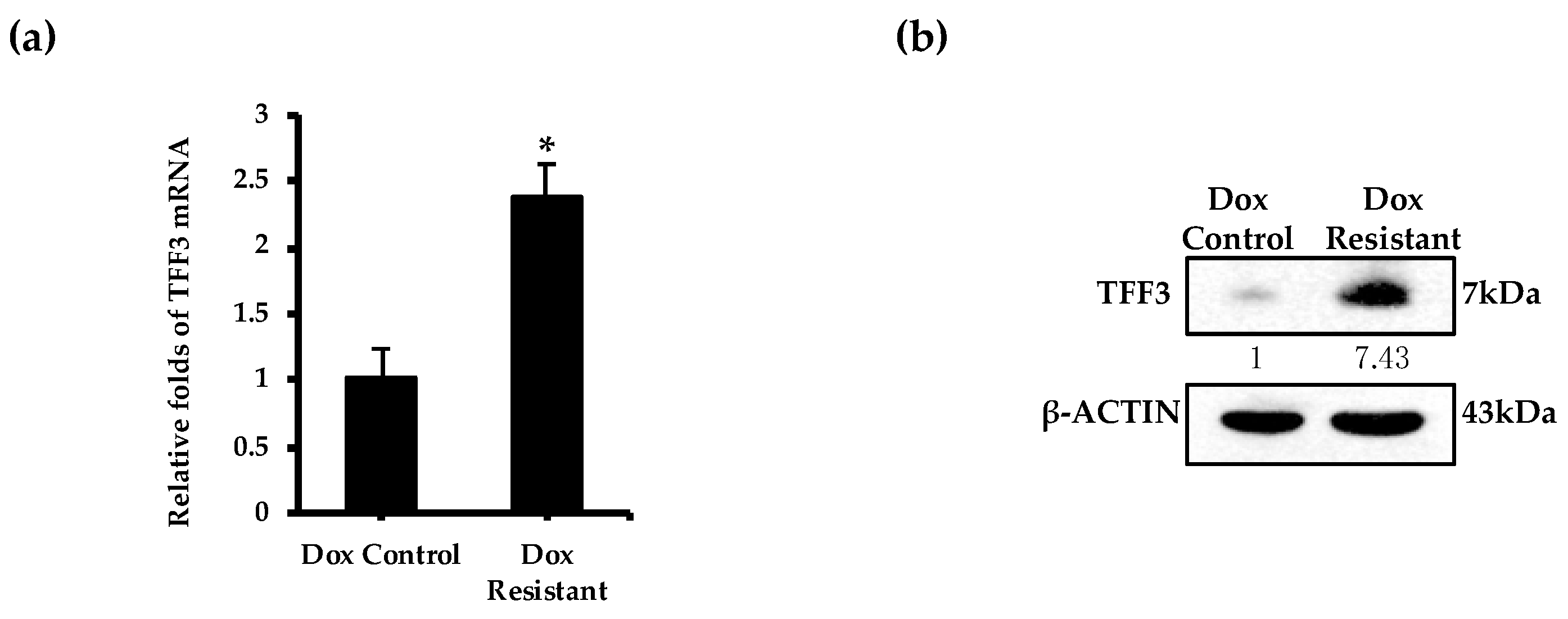
2.4. TFF3 Expression Is Elevated in Doxorubicin Resistant ER+MC Cells
2.5. AMPC Partially Re-Sensitizes Doxorubicin Resistant ER+MC Cells to Doxorubicin
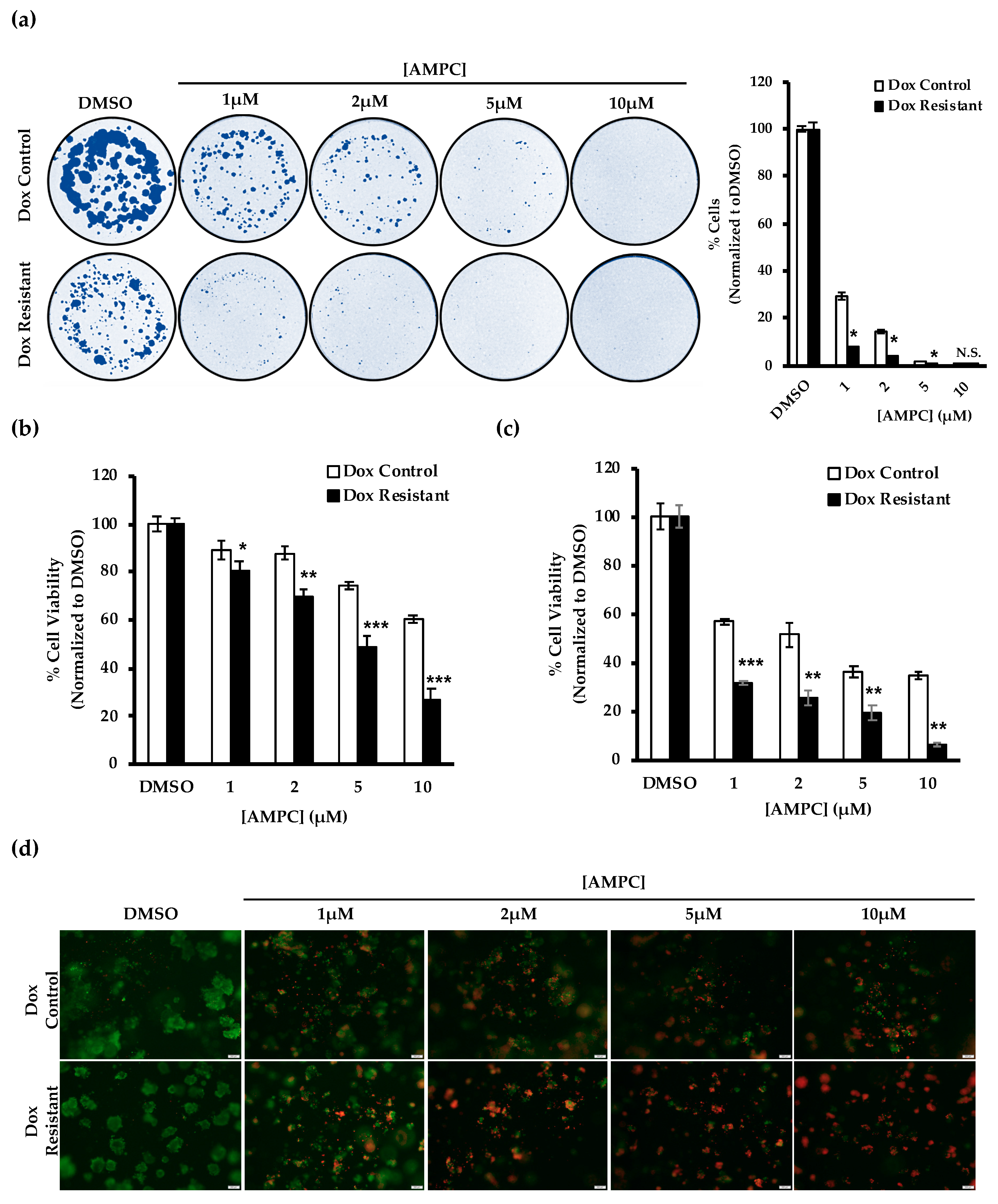
2.6. Potential Use of AMPC as a Monotherapy in Doxorubicin Resistant ER+MC
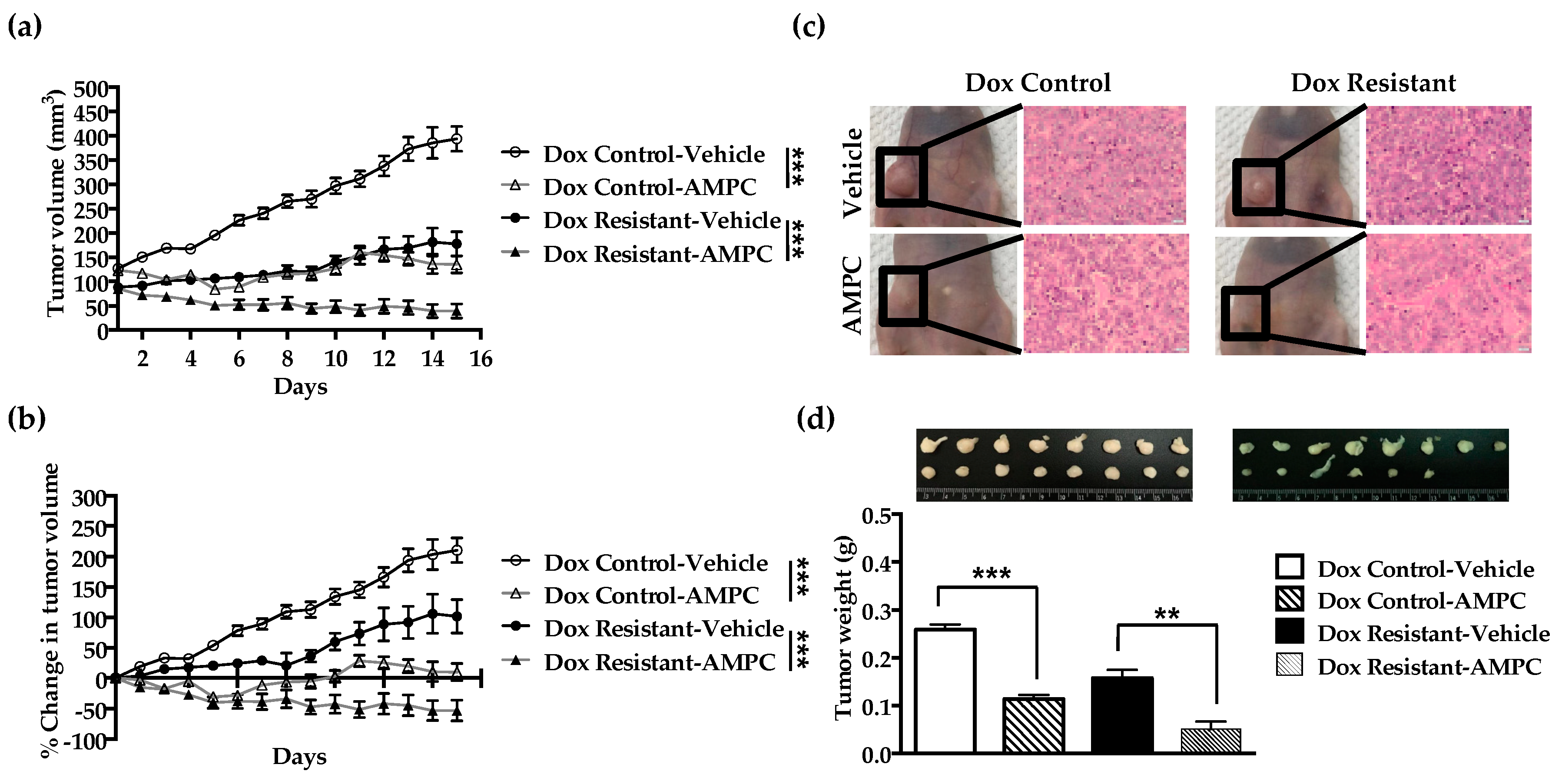
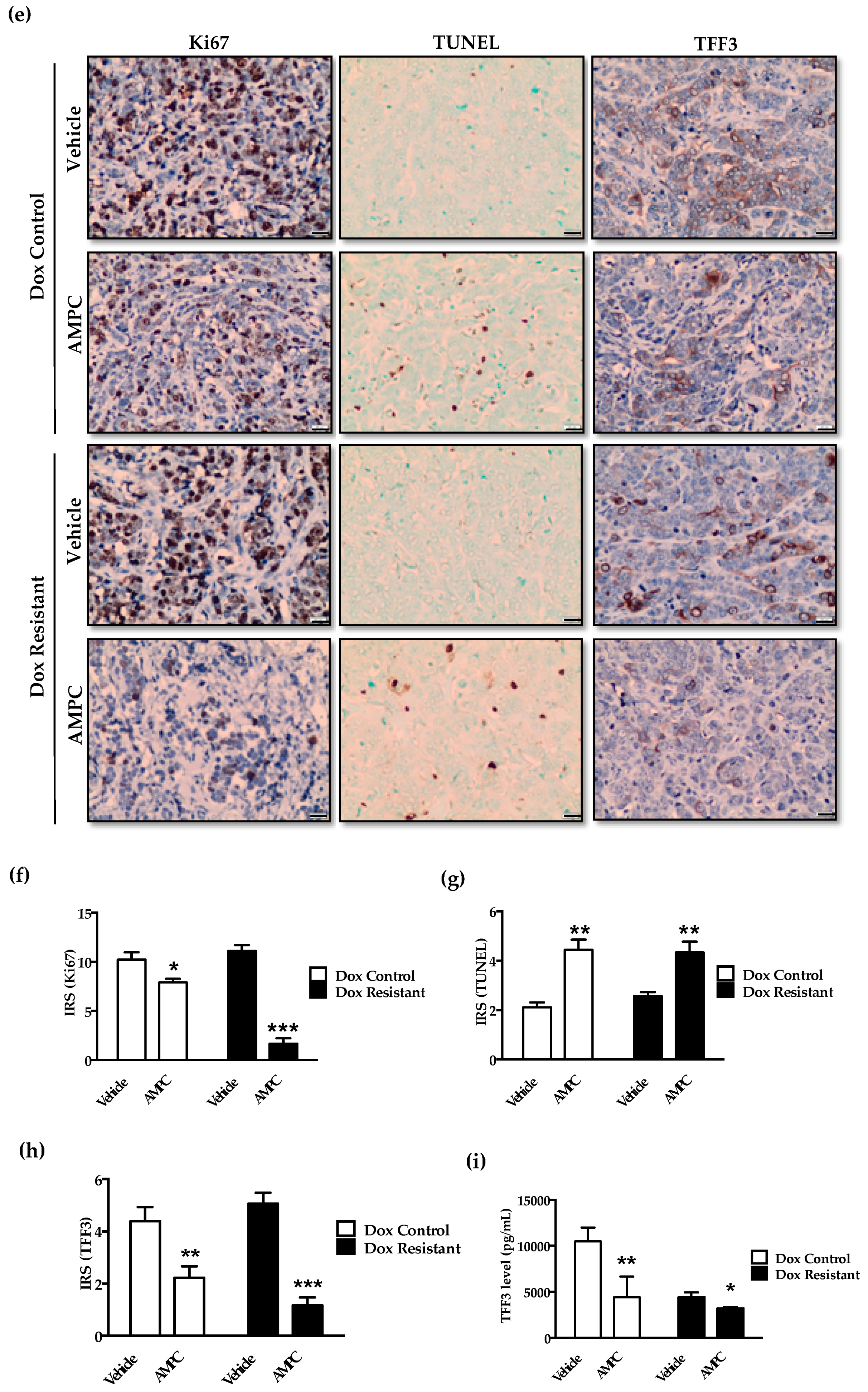
2.7. Inhibition of TFF3 Produces Regression of Doxorubicin Resistant ER+MC xenografts
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Generation of Doxorubicin Resistant Cell Lines
4.3. Cell Viability Assay
4.4. Foci-Formation Assay
4.5. mRNA Expression Analysis
4.6. Protein Expression Analysis
4.7. Annexin-V/ Propidium Iodide (PI) Staining
4.8. TUNEL Assay
4.9. Analysis of Combination Index (CI)
4.10. In Vivo Tumour Xenograft
4.11. Quantification of Serum TFF3 Levels
4.12. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Santa-Maria, C.A.; Gradishar, W.J. Adjuvant and Neoadjuvant Systemic Therapies for Early-Stage Breast Cancer; Elsevier: Amsterdam, The Netherlands, 2018; pp. 752–762. [Google Scholar]
- Pathak, M.; Dwivedi, S.N.; Deo, S.V.S.; Thakur, B.; Sreenivas, V.; Rath, G.K. Neoadjuvant chemotherapy regimens in treatment of breast cancer: A systematic review and network meta-analysis protocol. Syst. Rev. 2018, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- De Andrade, D.A.P.; Zucca-Matthes, G.; Vieira, R.A.D.C.; Andrade, C.T.D.A.E.D.; Da Costa, A.M.; Monteiro, A.J.D.C.; Lago, L.D.; Nunes, J.S. Neoadjuvant chemotherapy and pathologic response: A retrospective cohort. Einstein (São Paulo) 2013, 11, 446–450. [Google Scholar]
- Group, E.B. Comparisons between different polychemotherapy regimens for early breast cancer: Meta-analyses of long-term outcome among 100 000 women in 123 randomised trials. Lancet 2012, 379, 432–444. [Google Scholar]
- Kaklamani, V.G.; Gradishar, W.J. Epirubicin versus doxorubicin: Which is the anthracycline of choice for the treatment of breast cancer? Clin. Breast Cancer 2003, 4, S26–S33. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.; Santos, M.S.; Moreira, P. Doxorubicin: The Good, the Bad and the Ugly Effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L.; Rozencweig, M.; Muggia, F.M. Risk Factors for Doxorubicin-lnduced Congestive Heart Failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Volkova, M.; Russell, R., 3rd. Anthracycline cardiotoxicity: Prevalence, pathogenesis and treatment. Curr. Cardiol. Rev. 2011, 7, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Shan, K.; Lincoff, A.M.; Young, J.B. Anthracycline-Induced Cardiotoxicity. Ann. Intern. Med. 1996, 125, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Weinman, S. Mechanisms of doxorubicin resistance in hepatocellular carcinoma. Hepatic Oncol. 2016, 3, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.K.; Kannan, N.; Grandison, P.M.; Mitchell, M.D.; Lobie, P.E. Are trefoil factors oncogenic? Trends Endocrinol. Metab. 2008, 19, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Aamann, L.; Vestergaard, E.M.; Grønbæk, H. Trefoil factors in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, A.; Smitha, C.; Suresh, D. Trefoils: An unexplored natural protective shield of oral cavity. J. Oral Boil. Craniofacial Res. 2015, 5, 226–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, W.; Jagla, W.; Wiede, A. Molecular medicine of TFF-peptides: From gut to brain. Histol. Histopathol. 2001, 16, 319–334. [Google Scholar] [PubMed]
- Pandey, V.; Wu, Z.S.; Zhang, M.; Li, R.; Zhang, J.; Zhu, T.; Lobie, P.E. Trefoil factor 3 promotes metastatic seeding and predicts poor survival outcome of patients with mammary carcinoma. Breast Cancer Res. 2014, 16, 429. [Google Scholar] [CrossRef] [PubMed]
- Kannan, N.; Kang, J.; Kong, X.; Tang, J.; Perry, J.K.; Mohankumar, K.M.; Miller, L.D.; Liu, E.T.; Mertani, H.C.; Zhu, T.; et al. Trefoil Factor 3 Is Oncogenic and Mediates Anti-Estrogen Resistance in Human Mammary Carcinoma. Neoplasia 2010, 12, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Ohtsu, H.; Ikemura, M.; Kikuchi, Y.; Niwa, T.; Nishioka, K.; Uchida, Y.; Miura, H.; Aikou, S.; Gunji, T.; et al. Serum TFF1 and TFF3 but not TFF2 are higher in women with breast cancer than in women without breast cancer. Sci. Rep. 2017, 7, 4846. [Google Scholar] [CrossRef]
- Mhawech-Fauceglia, P.; Wang, D.; Samrao, D.; Liu, S.; Dupont, N.C.; Pejovic, T. Trefoil factor family 3 (TFF3) expression and its interaction with estrogen receptor (ER) in endometrial adenocarcinoma. Gynecol. Oncol. 2013, 130, 174–180. [Google Scholar] [CrossRef]
- Terry, S.; Nicolaiew, N.; Basset, V.; Semprez, F.; Soyeux, P.; Maille, P.; Vacherot, F.; Ploussard, G.; Londono-Vallejo, A.; De La Taille, A.; et al. Clinical value of ERG, TFF3, and SPINK1 for molecular subtyping of prostate cancer. Cancer 2015, 121, 1422–1430. [Google Scholar] [CrossRef]
- Perera, O.; Evans, A.; Pertziger, M.; Macdonald, C.; Chen, H.; Liu, D.X.; Lobie, P.E.; Perry, J.K. Trefoil factor 3 (TFF3) enhances the oncogenic characteristics of prostate carcinoma cells and reduces sensitivity to ionising radiation. Cancer Lett. 2015, 361, 104–111. [Google Scholar] [CrossRef]
- Yuan, Z.; Chen, D.; Chen, X.; Yang, H.; Wei, Y. Overexpression of trefoil factor 3 (TFF3) contributes to the malignant progression in cervical cancer cells. Cancer Cell Int. 2017, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- You, M.L.; Chen, Y.J.; Chong, Q.Y.; Wu, M.M.; Pandey, V.; Chen, R.M.; Liu, L.; Ma, L.; Wu, Z.S.; Zhu, T.; et al. Trefoil factor 3 mediation of oncogenicity and chemoresistance in hepatocellular carcinoma is AKT-BCL-2 dependent. Oncotarget 2017, 8, 39323–39344. [Google Scholar] [CrossRef] [PubMed]
- Vocka, M.; Langer, D.; Petrtýl, J.; Vočková, P.; Hanus, T.; Kalousova, M.; Zima, T.; Petruzelka, L. Trefoil factor family (TFF) proteins as potential serum biomarkers in patients with metastatic colorectal cancer. Neoplasma 2015, 62, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Gao, J.; Li, H.; Guo, W.; Mao, Q.; Gao, E.; Zhu, Y.-Q. Trefoil factor 3 peptide regulates migration via a Twist-dependent pathway in gastric cell. Biochem. Biophys. Res. Commun. 2013, 438, 6–12. [Google Scholar] [CrossRef]
- Qu, Y.; Yang, Y.; Ma, D.; Xiao, W. Increased trefoil factor 3 levels in the serum of patients with three major histological subtypes of lung cancer. Oncol. Rep. 2012, 27, 1277–1283. [Google Scholar] [CrossRef]
- Bezler, M.; Hengstler, J.G.; Ullrich, A. Inhibition of doxorubicin-induced HER3-PI3K-AKT signalling enhances apoptosis of ovarian cancer cells. Mol. Oncol. 2012, 6, 516–529. [Google Scholar] [CrossRef]
- Chan, V.Y.; Chan, M.W.; Leung, W.K.; Leung, P.S.; Sung, J.J.; Chan, F.K. Intestinal trefoil factor promotes invasion in non-tumorigenic Rat-2 fibroblast cell. Regul. Pept. 2005, 127, 87–94. [Google Scholar] [CrossRef]
- Lau, W.H.; Pandey, V.; Kong, X.; Wang, X.N.; Wu, Z.; Zhu, T.; Lobie, P.E. Trefoil Factor-3 (TFF3) Stimulates De Novo Angiogenesis in Mammary Carcinoma Both Directly and Indirectly via IL-8/CXCR2. PLoS ONE 2015, 10, e0141947. [Google Scholar] [CrossRef]
- Rodrigues, S.; Aken, E.V.; Bocxlaer, S.V.; Attoub, S.; Nguyen, Q.D.; Bruyneel, E.; Westley, B.R.; May, F.E.; Thim, L.; Mareel, M.; et al. Trefoil peptides as proangiogenic factors in vivo and in vitro- implication of cyclooxygenase-2 and EGF receptor signaling. FASEB J. 2017, 17, 7–16. [Google Scholar] [CrossRef]
- Xiao, L.; Liu, Y.P.; Xiao, C.X.; Ren, J.L.; Guleng, B. Serum TFF3 may be a pharamcodynamic marker of responses to chemotherapy in gastrointestinal cancers. BMC Clin. Pathol. 2014, 14, 26. [Google Scholar] [CrossRef]
- Chong, Q.Y.; You, M.L.; Pandey, V.; Banerjee, A.; Chen, Y.J.; Poh, H.M.; Zhang, M.; Ma, L.; Zhu, T.; Basappa, S.; et al. Release of HER2 repression of trefoil factor 3 (TFF3) expression mediates trastuzumab resistance in HER2+/ER+ mammary carcinoma. Oncotarget 2017, 8, 74188–74208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, M.W.; Chan, V.Y.; Leung, W.K.; Chan, K.K.; To, K.F.; Sung, J.J.; Chan, F.K. Anti-sense trefoil factor family-3 (intestinal trefoil factor) inhibits cell growth and induces chemosensitivity to adriamycin in human gastric cancer cells. Life Sci. 2005, 76, 2581–2592. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Wei, Y.; Park, J.; Hung, L.Y.; Young, T.; Herbine, K.; Oniskey, T.; Pastore, C.; Nieves, W.; Somsouk, M.; et al. TFF3 is a ligand for LINGO2 that de-represses EGFR to control disease outcome during colitis and gastrointestinal nematode infection. BioRxiv 2018. [Google Scholar] [CrossRef]
- Dieckow, J.; Brandt, W.; Hattermann, K.; Schob, S.; Schulze, U.; Mentlein, R.; Ackermann, P.; Sel, S.; Paulsen, F.P. CXCR4 and CXCR7 Mediate TFF3-Induced Cell Migration Independently from the ERK1/2 Signaling Pathway. Investig. Opthalmology Vis. Sci. 2016, 57, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Taupin, D.; Wu, D.-C.; Jeon, W.-K.; Devaney, K.; Wang, T.C.; Podolsky, D.K. The trefoil gene family are coordinately expressed immediate-early genes: EGF receptor– and MAP kinase–dependent interregulation. J. Clin. Investig. 1999, 103, R31–R38. [Google Scholar] [CrossRef] [PubMed]
- Le, J.; Zhang, D.Y.; Zhao, Y.; Qiu, W.; Wang, P.; Sun, Y. ITF promotes migration of intestinal epithelial cells through crosstalk between the ERK and JAK/STAT3 pathways. Sci. Rep. 2016, 6, 33014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivat, C.; Christine, R.; Rodrigues, S.; Sylvie, R.; Bruyneel, E.; Erik, B.; Piétu, G.; Geneviève, P.; Robert, A.; Amélie, R.; et al. Implication of STAT3 signaling in human colonic cancer cells during intestinal trefoil factor 3 (TFF3)—And vascular endothelial growth factor-mediated cellular invasion and tumor growth. Cancer Res. 2005, 65, 195–202. [Google Scholar]
- Lin, N.; Xu, L.F.; Sun, M. The protective effect of trefoil factor 3 on the intestinal tight junction barrier is mediated by toll-like receptor 2 via a PI3K/Akt dependent mechanism. Biochem. Biophys. Res. Commun. 2013, 440, 143–149. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, H.; Yang, Z.; Shao, D.; Zhang, W.; Ren, Y.; Sun, B.; Lin, J.; Xu, M.; Nie, S. Intestinal trefoil factor activates the PI3K/Akt signaling pathway to protect gastric mucosal epithelium from damage. Int. J. Oncol. 2014, 45, 1123–1132. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef]
- Kim, D.; Dan, H.C.; Park, S.; Yang, L.; Liu, Q.; Kaneko, S.; Ning, J.; He, L.; Yang, H.; Sun, M.; et al. AKT/PKB signaling mechanisms in cancer and chemoresistance. Front Biosci 2005, 10, 975–987. [Google Scholar] [CrossRef]
- Li, X.; Lu, Y.; Liang, K.; Liu, B.; Fan, Z. Differential responses to doxorubicin-induced phosphorylation and activation of Akt in human breast cancer cells. Breast Cancer Res. 2005, 7, R589–R597. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.S.; West, K.; Streicher, S.; Dennis, P.A. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol. Cancer Ther. 2002, 1, 707–717. [Google Scholar] [PubMed]
- Steelman, L.S.; Navolanic, P.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Martelli, A.M.; Cocco, L.; Stivala, F.; Libra, M.; Nicoletti, F.; et al. Involvement of Akt and mTOR in chemotherapeutic and hormonal-based drug resistance and response to radiation in breast cancer cells. Cell Cycle 2011, 10, 3003–3015. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, K.; Taupin, D.R.; Itoh, H.; Podolsky, D.K. Distinct Pathways of Cell Migration and Antiapoptotic Response to Epithelial Injury: Structure-Function Analysis of Human Intestinal Trefoil Factor. Mol. Cell. Boil. 2000, 20, 4680–4690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raja, S.B.; Murali, M.R.; Devaraj, H.; Devaraj, S.N. Differential expression of gastric MUC5AC in colonic epithelial cells: TFF3-wired IL1 /Akt crosstalk-induced mucosal immune response against Shigella dysenteriae infection. J. Cell Sci. 2012, 125, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Lobie, P.E.; Pandey, V.; Kanchugarakoppal, S.R.; Basappa; Chakrabhavi, D.M.; Shobith, R. Compounds Useful in Inhibiting Human Trefoil Factor 3. Patent WO2018226155, 13 December 2018. [Google Scholar]
- Hoffman, R.M. In vitro sensitivity assays in cancer: A review, analysis, and prognosis. J. Clin. Lab. Anal. 1991, 5, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-Dimensional Cell Culture Systems and Their Applications in Drug Discovery and Cell-Based Biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Eom, Y.W.; Kim, M.A.; Park, S.S.; Goo, M.J.; Kwon, H.J.; Sohn, S.; Kim, W.H.; Yoon, G.; Choi, K.S. Two distinct modes of cell death induced by doxorubicin: Apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 2005, 24, 4765–4777. [Google Scholar] [CrossRef]
- Konorev, E.A.; Kotamraju, S.; Kalivendi, S.; Kalyanaraman, B.; Joseph, J.; Wang, S. Doxorubicin Induces Apoptosis in Normal and Tumor Cells via Distinctly Different Mechanisms. J. Boil. Chem. 2004, 279, 25535–25543. [Google Scholar]
- Al-Salam, S.; Sudhadevi, M.; Awwad, A.; Al Bashir, M. Trefoil factors peptide-3 is associated with residual invasive breast carcinoma following neoadjuvant chemotherapy. BMC Cancer 2019, 19, 135. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Xie, L.; Xing, H. PI3K Inhibitor Combined with Chemotherapy Can Enhance the Apoptosis of Neuroblastoma Cells In Vitro and In Vivo. Technol. Cancer Res. Treat. 2016, 15, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Marklein, D.; Graab, U.; Naumann, I.; Yan, T.; Ridzewski, R.; Nitzki, F.; Rosenberger, A.; Dittmann, K.; Wienands, J.; Wojnowski, L.; et al. PI3K Inhibition Enhances Doxorubicin-Induced Apoptosis in Sarcoma Cells. PLoS ONE 2012, 7, e52898. [Google Scholar] [CrossRef]
- Fodale, V.; Pierobon, M.; Liotta, L.; Petricoin, E. Mechanism of cell adaptation: When and how do cancer cells develop chemoresistance? Cancer J. 2011, 17, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-M.; Bai, J.-Y.; Yang, K.-X. Effect of resveratrol on doxorubicin resistance in breast neoplasm cells by modulating PI3K/Akt signaling pathway. IUBMB Life 2018, 70, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Storesund, T.; Schenck, K.; Osmundsen, H.; Røed, A.; Helgeland, K.; Kolltveit, K.M. Signal transduction and gene transcription induced by TFF3 in oral keratinocytes. Eur. J. Oral Sci. 2009, 117, 511–517. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Outmezguine, V.R.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef]
- Jansen, V.M.; Mayer, I.A.; Arteaga, C.L. Is There a Future for AKT Inhibitors in the Treatment of Cancer? Clin. Cancer Res. 2016, 22, 2599–2601. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Therapeut. 2017, 172, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Wisinski, K.B.; Tevaarwerk, A.J.; Burkard, M.E.; Rampurwala, M.; Eickhoff, J.; Bell, M.C.; Kolesar, J.M.; Flynn, C.; Liu, G. Phase I Study of an AKT Inhibitor (MK-2206) Combined with Lapatinib in Adult Solid Tumors Followed by Dose Expansion in Advanced HER2+ Breast Cancer. Clin. Cancer Res. 2016, 22, 2659–2667. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.H.; Yan, L.; Mezynski, J.; Patnaik, A.; Morena, V.; Papadopoulos, K.P.; Garrett, C.R.; Ong, M.; Shannon, K.A.; Morosky, A.; et al. A phase I dose escalation study of oral MK-2206 (allosteric Akt inhibitor) with oral selumetinib (AZD6244; ARRY-142866) (MEK 1/2 inhibitor) in patients with advanced or metastatic solid tumors. J. Clin. Oncol. 2017, 30, e13599. [Google Scholar]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poh, H.M.; Chiou, Y.S.; Chong, Q.Y.; Chen, R.-M.; Rangappa, K.S.; Ma, L.; Zhu, T.; Kumar, A.P.; Pandey, V.; Basappa; et al. Inhibition of TFF3 Enhances Sensitivity—and Overcomes Acquired Resistance—to Doxorubicin in Estrogen Receptor-Positive Mammary Carcinoma. Cancers 2019, 11, 1528. https://doi.org/10.3390/cancers11101528
Poh HM, Chiou YS, Chong QY, Chen R-M, Rangappa KS, Ma L, Zhu T, Kumar AP, Pandey V, Basappa, et al. Inhibition of TFF3 Enhances Sensitivity—and Overcomes Acquired Resistance—to Doxorubicin in Estrogen Receptor-Positive Mammary Carcinoma. Cancers. 2019; 11(10):1528. https://doi.org/10.3390/cancers11101528
Chicago/Turabian StylePoh, Han Ming, Yi Shiou Chiou, Qing Yun Chong, Ru-Mei Chen, Kanchugarakoppal S. Rangappa, Lan Ma, Tao Zhu, Alan Prem Kumar, Vijay Pandey, Basappa, and et al. 2019. "Inhibition of TFF3 Enhances Sensitivity—and Overcomes Acquired Resistance—to Doxorubicin in Estrogen Receptor-Positive Mammary Carcinoma" Cancers 11, no. 10: 1528. https://doi.org/10.3390/cancers11101528
APA StylePoh, H. M., Chiou, Y. S., Chong, Q. Y., Chen, R. -M., Rangappa, K. S., Ma, L., Zhu, T., Kumar, A. P., Pandey, V., Basappa, Lee, S. -C., & Lobie, P. E. (2019). Inhibition of TFF3 Enhances Sensitivity—and Overcomes Acquired Resistance—to Doxorubicin in Estrogen Receptor-Positive Mammary Carcinoma. Cancers, 11(10), 1528. https://doi.org/10.3390/cancers11101528