Molecular Targeting Therapy against EGFR Family in Breast Cancer: Progress and Future Potentials

 , ,
, , 

Abstract
:1. Introduction
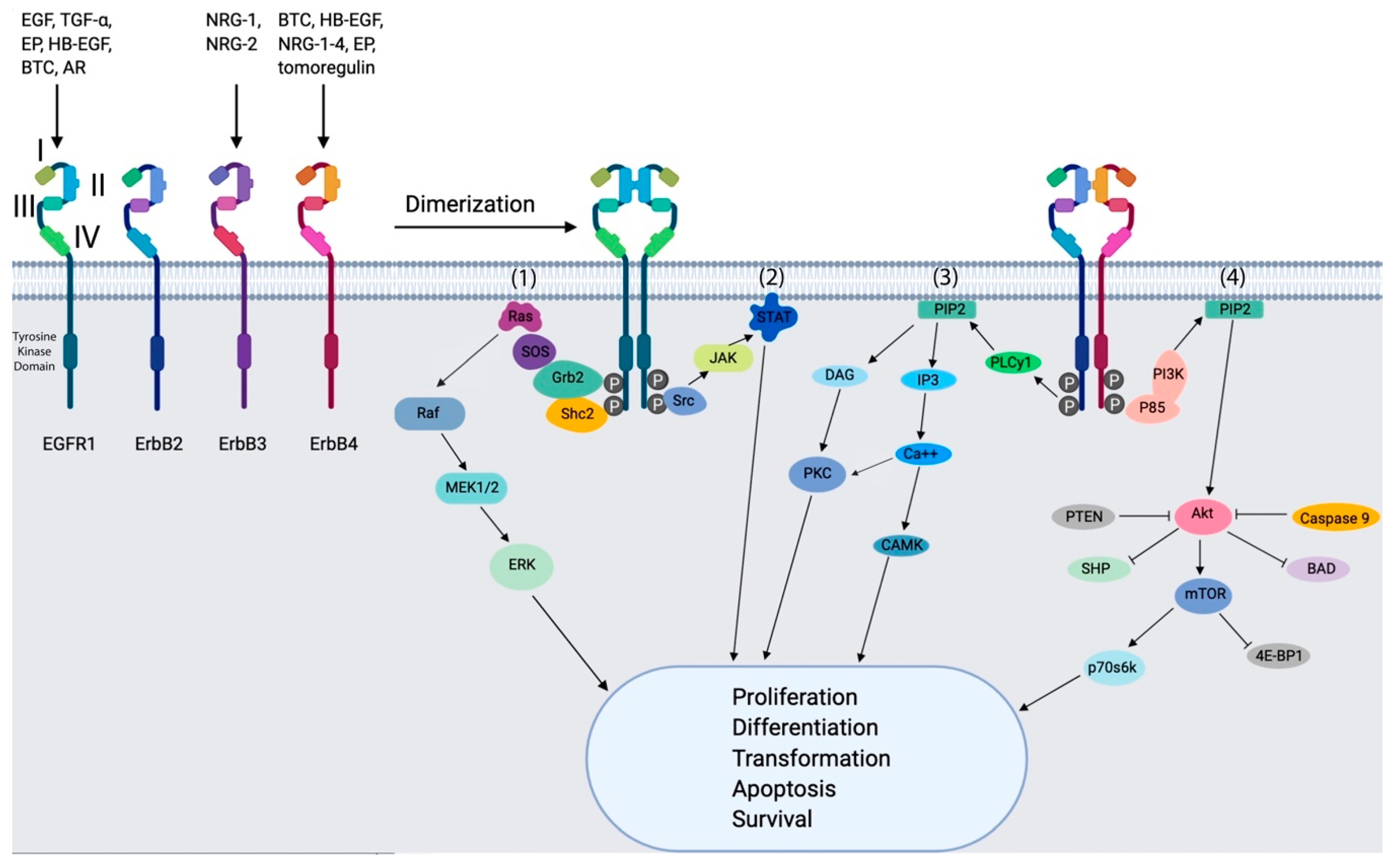
2. EGFRs in Breast Cancer
2.1. EGFR1
2.2. ErbB2/Her2
2.3. ErbB3/Her3
2.4. ErbB4/Her4
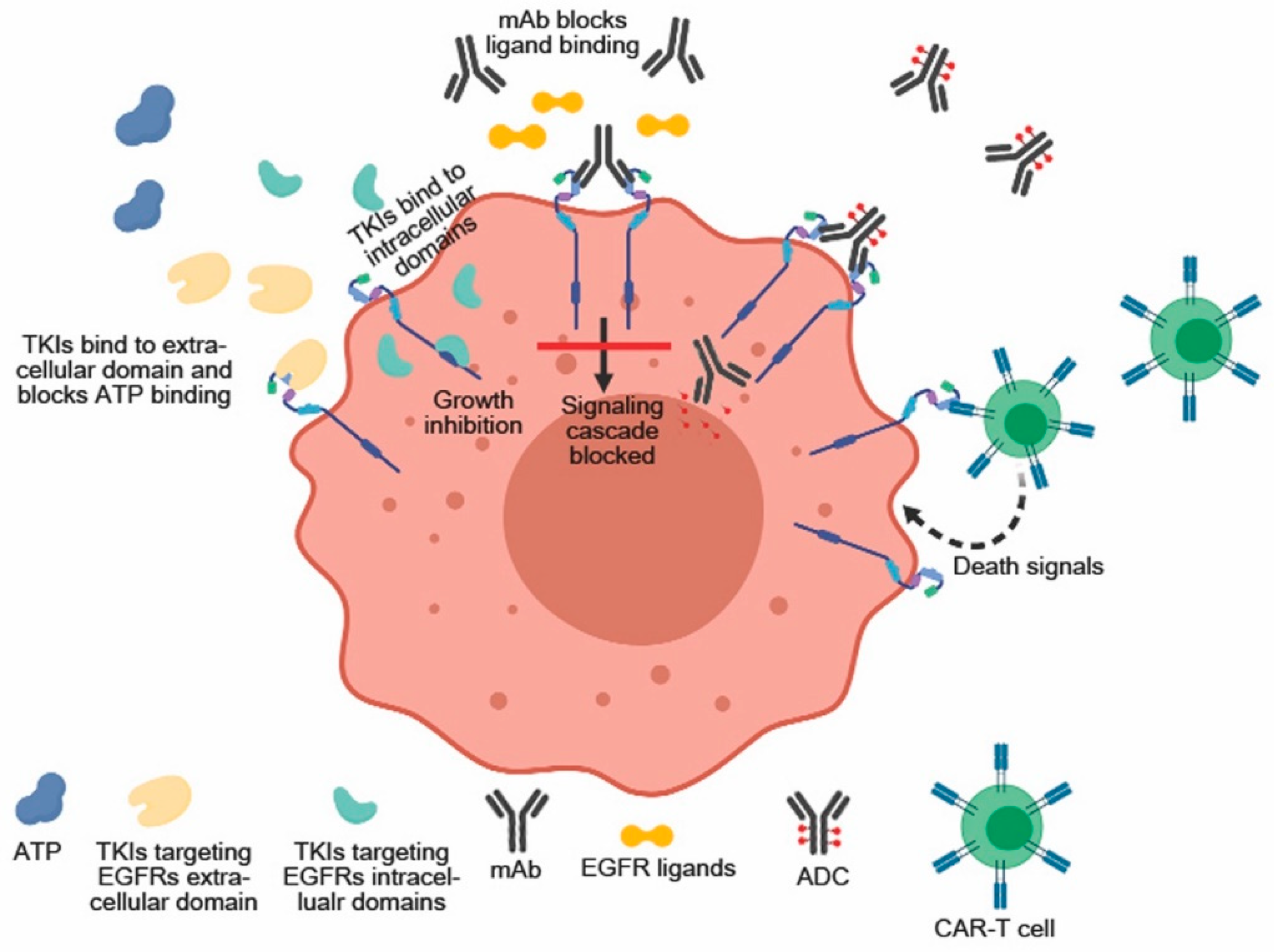
3. Anti-EGFRs Monoclonal Antibodies (mAbs)
3.1. mAbs against EGFR1
3.1.1. Cetuximab (Erbitux™)
3.1.2. Panitumumab (Vectibix™)
3.1.3. Zalutumumab
3.2. mAbs against Her2
3.2.1. Trastuzumab (Herceptin™)
3.2.2. Pertuzumab (Omnitarg™)
3.2.3. Trastuzumab-emtansine (T-DM1, Kadcyla™)
3.2.4. Trastuzumab Deruxtecan (DS-8201a)
3.2.5. MEDI4276
3.3. mAbs against Her3
3.3.1. Seribantumab (MM-121)
3.3.2. Lumretuzumab (RG7116)
3.3.3. MM-111
3.3.4. MCL-128
3.4. mAs against Her4
4. EGFRs Small Molecule Inhibitors (RTK Inhibitors)
4.1. TKIs against EGFR1
4.1.1. Gefitinib
4.1.2. Erlotinib (Tarceva™)
4.2. TKIs against Her2
4.2.1. Lapatinib (Tykerb or Tyverb™)
4.2.2. Neratinib (Nerlynx™)
4.3. TKIs against Her3
4.4. TKIs against Her4
4.4.1. Canertinib (CI-1033)
4.4.2. Afatinib (Gilotrif™)
5. Chimeric Antigen Receptor (CAR) Cell-Based EGFRs Targeting Therapy
6. Anti-EGFR Agent-Associated Toxicity (Side Effects of EGFR Inhibitors)
7. Novel Strategies
7.1. Cetuximab-IR700
7.2. Cytotoxicity and Immunotherapy
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Xiang, L.; Li, T.; Bai, Z. Cancer Hallmarks, Biomarkers and Breast Cancer Molecular Subtypes. J. Cancer 2016, 7, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Hung, M.-C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar]
- Nielsen, T.O.; Hsu, F.D.; Jensen, K.; Cheang, M.; Karaca, G.; Hu, Z.; Hernandez-Boussard, T.; Livasy, C.; Cowan, D.; Dressler, L.; et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin. Cancer Res. 2004, 10, 5367–5374. [Google Scholar] [CrossRef]
- Choccalingam, C.; Rao, L.; Rao, S. Clinico-Pathological Characteristics of Triple Negative and Non Triple Negative High Grade Breast Carcinomas with and without Basal Marker (CK5/6 and EGFR) Expression at a Rural Tertiary Hospital in India. Breast Cancer 2012, 6, 21–29. [Google Scholar] [CrossRef]
- Gelmon, K.; Dent, R.; Mackey, J.R.; Laing, K.; McLeod, D.; Verma, S. Targeting triple-negative breast cancer: Optimising therapeutic outcomes. Ann. Oncol. 2012, 23, 2223–2234. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Pinheiro, C.; Lambros, M.B.; Milanezi, F.; Carvalho, S.; Savage, K.; Simpson, P.T.; Jones, C.; Swift, S.; Mackay, A.; et al. EGFR amplification and lack of activating mutations in metaplastic breast carcinomas. J. Pathol. 2006, 209, 445–453. [Google Scholar] [CrossRef]
- Yatabe, Y.; Kosaka, T.; Takahashi, T.; Mitsudomi, T. EGFR mutation is specific for terminal respiratory unit type adenocarcinoma. Am. J. Surg. Pathol. 2005, 29, 633–639. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Yarden, Y. EGF-ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Madshus, I.H.; Stang, E. Internalization and intracellular sorting of the EGF receptor: A model for understanding the mechanisms of receptor trafficking. J. Cell Sci. 2009, 122, 3433–3439. [Google Scholar] [CrossRef]
- Hanada, N.; Lo, H.W.; Day, C.P.; Pan, Y.; Nakajima, Y.; Hung, M.C. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol. Carcinog. 2006, 45, 10–17. [Google Scholar] [CrossRef]
- Lin, S.Y.; Makino, K.; Xia, W.; Matin, A.; Wen, Y.; Kwong, K.Y.; Bourguignon, L.; Hung, M.C. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat. Cell Biol. 2001, 3, 802–808. [Google Scholar] [CrossRef]
- Lo, H.W.; Hsu, S.C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.Y.; Hung, M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef]
- Offterdinger, M.; Schofer, C.; Weipoltshammer, K.; Grunt, T.W. c-erbB-3: A nuclear protein in mammary epithelial cells. J. Cell Biol. 2002, 157, 929–939. [Google Scholar] [CrossRef]
- Wang, S.C.; Lien, H.C.; Xia, W.; Chen, I.F.; Lo, H.W.; Wang, Z.; Ali-Seyed, M.; Lee, D.F.; Bartholomeusz, G.; Ou-Yang, F.; et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell 2004, 6, 251–261. [Google Scholar] [CrossRef]
- Baselga, J.; Albanell, J. Epithelial growth factor receptor interacting agents. Hematol. Oncol. Clin. N. Am. 2002, 16, 1041–1063. [Google Scholar] [CrossRef]
- Jin, Q.; Esteva, F.J. Cross-talk between the ErbB/HER family and the type I insulin-like growth factor receptor signaling pathway in breast cancer. J. Mammary Gland Biol. Neoplasia 2008, 13, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Appert-Collin, A.; Hubert, P.; Cremel, G.; Bennasroune, A. Role of ErbB Receptors in Cancer Cell Migration and Invasion. Front. Pharmacol. 2015, 6, 283. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [PubMed]
- Jorgensen, J.T. Role of human epidermal growth factor receptor 2 in gastric cancer: Biological and pharmacological aspects. World J. Gastroenterol. 2014, 20, 4526–4535. [Google Scholar] [CrossRef] [PubMed]
- Landi, L.; Cappuzzo, F. HER2 and lung cancer. Expert Rev. Anticancer Ther. 2013, 13, 1219–1228. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Kim, H.; Muller, W.J. The role of the epidermal growth factor receptor family in mammary tumorigenesis and metastasis. Exp. Cell Res. 1999, 253, 78–87. [Google Scholar] [CrossRef]
- Al-Kuraya, K.; Schraml, P.; Torhorst, J.; Tapia, C.; Zaharieva, B.; Novotny, H.; Spichtin, H.; Maurer, R.; Mirlacher, M.; Kochli, O.; et al. Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res. 2004, 64, 8534–8540. [Google Scholar] [CrossRef]
- Weber, F.; Fukino, K.; Sawada, T.; Williams, N.; Sweet, K.; Brena, R.M.; Plass, C.; Caldes, T.; Mutter, G.L.; Villalona-Calero, M.A.; et al. Variability in organ-specific EGFR mutational spectra in tumour epithelium and stroma may be the biological basis for differential responses to tyrosine kinase inhibitors. Br. J. Cancer 2005, 92, 1922–1926. [Google Scholar] [CrossRef]
- Parsa, Y.; Mirmalek, S.A.; Kani, F.E.; Aidun, A.; Salimi-Tabatabaee, S.A.; Yadollah-Damavandi, S.; Jangholi, E.; Parsa, T.; Shahverdi, E. A Review of the Clinical Implications of Breast Cancer Biology. Electron. Physician 2016, 8, 2416–2424. [Google Scholar] [CrossRef] [PubMed]
- Ponde, N.; Brandao, M.; El-Hachem, G.; Werbrouck, E.; Piccart, M. Treatment of advanced HER2-positive breast cancer: 2018 and beyond. Cancer Treat. Rev. 2018, 67, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, A.; Niemiec, J.; Janecka, A.; Harazin-Lechowska, A.; Ambicka, A.; Grela-Wojewoda, A.; Domagala-Haduch, M.; Cedrych, I.; Majchrzyk, K.; Kruczak, A.; et al. Prognostic value of PIK3CA mutation status, PTEN and androgen receptor expression for metastasis-free survival in HER2-positive breast cancer patients treated with trastuzumab in adjuvant setting. Pol. J. Pathol. 2015, 66, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loibl, S.; Gianni, L. HER2-positive breast cancer. Lancet 2017, 389, 2415–2429. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef] [Green Version]
- Karachaliou, N.; Lazzari, C.; Verlicchi, A.; Sosa, A.E.; Rosell, R. HER3 as a Therapeutic Target in Cancer. BioDrugs 2017, 31, 63–73. [Google Scholar] [CrossRef]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; Network, C.G.A.; Network, T.C.G.A.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Hutcheson, I.R.; Goddard, L.; Barrow, D.; McClelland, R.A.; Francies, H.E.; Knowlden, J.M.; Nicholson, R.I.; Gee, J.M. Fulvestrant-induced expression of ErbB3 and ErbB4 receptors sensitizes oestrogen receptor-positive breast cancer cells to heregulin beta1. Breast Cancer Res. 2011, 13, R29. [Google Scholar] [CrossRef] [Green Version]
- Travis, A.; Pinder, S.E.; Robertson, J.F.R.; Bell, J.A.; Wencyk, P.; Gullick, W.J.; Nicholson, R.I.; Poller, D.N.; Blamey, R.W.; Elston, C.W.; et al. C-erbB-3 in human breast carcinoma: Expression and relation to prognosis and established prognostic indicators. Br. J. Cancer 1996, 74, 229. [Google Scholar] [CrossRef] [Green Version]
- Balko, J.M.; Miller, T.W.; Morrison, M.M.; Hutchinson, K.; Young, C.; Rinehart, C.; Sanchez, V.; Jee, D.; Polyak, K.; Prat, A.; et al. The receptor tyrosine kinase ErbB3 maintains the balance between luminal and basal breast epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, 221–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Hoeflich, S.T.; Crocker, L.; Yao, E.; Pham, T.; Munroe, X.; Hoeflich, K.P.; Sliwkowski, M.X.; Stern, H.M. A central role for HER3 in HER2-amplified breast cancer: Implications for targeted therapy. Cancer Res. 2008, 68, 5878–5887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett, J.T.; Olivares, M.G.; Rinehart, C.; Granja-Ingram, N.D.; Sanchez, V.; Chakrabarty, A.; Dave, B.; Cook, R.S.; Pao, W.; McKinely, E.; et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc. Natl. Acad. Sci. USA 2011, 108, 5021–5026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, B.S.; Kljavin, N.M.; Stawiski, E.W.; Chan, E.; Parikh, C.; Durinck, S.; Chaudhuri, S.; Pujara, K.; Guillory, J.; Edgar, K.A.; et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell 2013, 23, 603–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witton, C.J.; Reeves, J.R.; Going, J.J.; Cooke, T.G.; Bartlett, J.M. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J. Pathol. 2003, 200, 290–297. [Google Scholar] [CrossRef]
- Tovey, S.M.; Witton, C.J.; Bartlett, J.M.; Stanton, P.D.; Reeves, J.R.; Cooke, T.G. Outcome and human epidermal growth factor receptor (HER) 1-4 status in invasive breast carcinomas with proliferation indices evaluated by bromodeoxyuridine labelling. Breast Cancer Res. 2004, 6, R246–R251. [Google Scholar] [CrossRef] [Green Version]
- Junttila, T.T.; Sundvall, M.; Lundin, M.; Lundin, J.; Tanner, M.; Härkönen, P.; Joensuu, H.; Isola, J.; Elenius, K. Cleavable ErbB4 Isoform in Estrogen Receptor–Regulated Growth of Breast Cancer Cells. Cancer Res. 2005, 65, 1384–1393. [Google Scholar] [CrossRef] [Green Version]
- Sartor, C.I.; Zhou, H.; Kozlowska, E.; Guttridge, K.; Kawata, E.; Caskey, L.; Harrelson, J.; Hynes, N.; Ethier, S.; Calvo, B.; et al. Her4 mediates ligand-dependent antiproliferative and differentiation responses in human breast cancer cells. Mol. Cell Biol. 2001, 21, 4265–4275. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.E.; Trout, L.J.; Gallo, R.M.; Pitfield, S.E.; Bryant, I.; Penington, D.J.; Riese, D.J. A constitutively active ErbB4 mutant inhibits drug-resistant colony formation by the DU-145 and PC-3 human prostate tumor cell lines. Cancer Lett. 2003, 192, 67–74. [Google Scholar] [CrossRef]
- Yarden, Y. The EGFR family and its ligands in human cancer: Signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37, 3–8. [Google Scholar] [CrossRef]
- Montor, W.R.; Salas, A.; Melo, F.H.M. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: The current arsenal of inhibitors. Mol. Cancer 2018, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Pernas, S.; Tolaney, S.M. HER2-positive breast cancer: New therapeutic frontiers and overcoming resistance. Ther. Adv. Med. Oncol. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—An emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef] [PubMed]
- (FDA), U.S.F.D.A. Information on Cetuximab (Marketed as Erbitux). Available online: https://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm113714.htm (accessed on 9 December 2018).
- Modi, S.; D’Andrea, G.; Norton, L.; Jyun Yao, T.; Caravelli, J.; Rosen, P.P.; Hudis, C.; Seidman, A.D. A Phase I Study of Cetuximab/Paclitaxel in Patients with Advanced-Stage Breast Cancer. Clin. Breast Cancer 2006, 7, 270–277. [Google Scholar] [CrossRef]
- (FDA), U.S.F.D.A. FDA Approved Drug Products. Approval Letter for Vectibix. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2006/125147LTR.pdf (accessed on 9 December 2018).
- Nabholtz, J.; Weber, B.; Mouret-Reynier, M.; Gligorov, J.; Coudert, B.P.; Vanlemmens, L.; Petit, T.; Tredan, O.; Praagh-Doreau, I.V.; Dubray-Longeras, P.; et al. Panitumumab in combination with FEC 100 (5-fluorouracil, epidoxorubicin, cyclophosphamide) followed by docetaxel (T) in patients with operable, triple-negative breast cancer (TNBC): Preliminary results of a multicenter neoadjuvant pilot phase II study. J. Clin. Oncol. 2011, 29, e11574. [Google Scholar] [CrossRef]
- Nabholtz, J.M.; Mouret-Reynier, M.A.; Van-Praagh, I.; Servent, V.; Jacquin, J.-P.; Bourcier, A.-V.; Del Piano, F.; Dubray-Longeras, P.; Nayl, B.; Abrial, C.; et al. Cetuximab in combination with docetaxel (T) in patients with operable, triple-negative breast cancer (TNBC): Preliminary results of a multicentre neoadjuvant pilot phase II study. ASCO Meet. Abstr. 2013, 31, 1057. [Google Scholar]
- Nabholtz, J.M.; Abrial, C.; Mouret-Reynier, M.A.; Dauplat, M.M.; Weber, B.; Gligorov, J.; Forest, A.M.; Tredan, O.; Vanlemmens, L.; Petit, T.; et al. Multicentric neoadjuvant phase II study of panitumumab combined with an anthracycline/taxane-based chemotherapy in operable triple-negative breast cancer: Identification of biologically defined signatures predicting treatment impact. Ann. Oncol. 2014, 25, 1570–1577. [Google Scholar] [CrossRef]
- Rivera, F.; Salcedo, M.; Vega, N.; Blanco, Y.; Lopez, C. Current situation of zalutumumab. Expert Opin. Biol. Ther. 2009, 9, 667–674. [Google Scholar] [CrossRef]
- Schick, U.; Gujral, D.M.; Richards, T.M.; Harrington, K.J.; Nutting, C.M. Zalutumumab in head and neck cancer. Expert Opin. Biol. Ther. 2012, 12, 119–125. [Google Scholar] [CrossRef]
- Baselga, J.; Gómez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pego, A.; Chan, A.; et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A.; et al. TBCRC 001: Randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 2615–2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornier, M.; Esteva, F.J.; Seidman, A.D. Trastuzumab in combination with chemotherapy for the treatment of metastatic breast cancer. Semin. Oncol. 2001, 27, 38–45. [Google Scholar]
- Madrid-Paredes, A.; Cañadas-Garre, M.; Sánchez-Pozo, A.; Calleja-Hernández, M.Á. Non-HER2 signaling pathways activated in resistance to anti-HER2 therapy in breast cancer. Breast Cancer Res. Treat. 2015, 153, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Ban, M.; Viculin, J.; Tomic, S.; Capkun, V.; Strikic, A.; Mise, B.P.; Utrobicic, I.; Vrdoljak, E. Retrospective analysis of efficacy of trastuzumab in adjuvant treatment of HER 2 positive early breast cancer—Single institution experience. Neoplasma 2016, 63, 761–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, D.; Piccart-Gebhart, M.J.; Gelber, R.D.; Procter, M.; Goldhirsch, A.; de Azambuja, E.; Castro, G., Jr.; Untch, M.; Smith, I.; Gianni, L.; et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: Final analysis of the HERceptin Adjuvant (HERA) trial. Lancet 2017, 389, 1195–1205. [Google Scholar] [CrossRef] [Green Version]
- Maximiano, S.; Magalhaes, P.; Guerreiro, M.P.; Morgado, M. Trastuzumab in the Treatment of Breast Cancer. BioDrugs 2016, 30, 75–86. [Google Scholar] [CrossRef]
- Figueroa-Magalhaes, M.C.; Jelovac, D.; Connolly, R.; Wolff, A.C. Treatment of HER2-positive breast cancer. Breast 2014, 23, 128–136. [Google Scholar] [CrossRef]
- Laakmann, E.; Muller, V.; Schmidt, M.; Witzel, I. Systemic Treatment Options for HER2-Positive Breast Cancer Patients with Brain Metastases beyond Trastuzumab: A Literature Review. Breast Care 2017, 12, 168–171. [Google Scholar] [CrossRef]
- Tiwari, S.R.; Mishra, P.; Raska, P.; Calhoun, B.; Abraham, J.; Moore, H.; Budd, G.T.; Fanning, A.; Valente, S.; Stewart, R.; et al. Retrospective study of the efficacy and safety of neoadjuvant docetaxel, carboplatin, trastuzumab/pertuzumab (TCH-P) in nonmetastatic HER2-positive breast cancer. Breast Cancer Res. Treat. 2016, 158, 189–193. [Google Scholar] [CrossRef]
- Nahta, R.; Hung, M.C.; Esteva, F.J. The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res. 2004, 64, 2343–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, Q.; Ibrahim, A.; Cohen, M.H.; Johnson, J.; Ko, C.-W.; Sridhara, R.; Justice, R.; Pazdur, R. FDA Drug Approval Summary: Lapatinib in Combination with Capecitabine for Previously Treated Metastatic Breast Cancer That Overexpresses HER-2. Oncologist 2008, 13, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Q.; Gao, M.; Fu, L.; Li, Y.; Quan, H.; Lou, L. STAT3 activation confers trastuzumab-emtansine (T-DM1) resistance in HER2-positive breast cancer. Cancer Sci. 2018, 109, 3305. [Google Scholar] [CrossRef] [PubMed]
- Fathi, N.; Rashidi, G.; Khodadadi, A.; Shahi, S.; Sharifi, S. STAT3 and apoptosis challenges in cancer. Int. J. Biol. Macromol. 2018, 117, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Giehl, N.; Wu, Y.; Vadgama, J.V. STAT3 activation in HER2-overexpressing breast cancer promotes epithelial-mesenchymal transition and cancer stem cell traits. Int. J. Oncol. 2014, 44, 403–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinnerthaler, G.; Gampenrieder, S.P.; Greil, R. HER2 Directed Antibody-Drug-Conjugates beyond T-DM1 in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oganesyan, V.; Peng, L.; Bee, J.S.; Li, J.; Perry, S.R.; Comer, F.; Xu, L.; Cook, K.; Senthil, K.; Clarke, L.; et al. Structural insights into the mechanism of action of a biparatopic anti-HER2 antibody. J. Biol. Chem. 2018, 293, 8439–8448. [Google Scholar] [CrossRef] [Green Version]
- Schoeberl, B.; Faber, A.C.; Li, D.; Liang, M.-C.; Crosby, K.; Onsum, M.; Burenkova, O.; Pace, E.; Walton, Z.; Nie, L.; et al. An ErbB3 Antibody, MM-121, Is Active in Cancers with Ligand-Dependent Activation. Cancer Res. 2010, 70, 2485–2494. [Google Scholar] [CrossRef]
- Moyo, V.M.; Higgins, M.J.; Aravelo-Araujo, R.; Iannotti, N.; Charu, V.; Dhindsa, N.; Goss, P.E. A randomized, double-blind phase II trial of exemestane with or without MM-121 in postmenopausal women with locally advanced or metastatic estrogen receptor-positive (ER+) and/or progesterone receptor-positive (PR+), HER2-negative breast cancer. J. Clin. Oncol. 2011, 29, TPS112. [Google Scholar] [CrossRef]
- Pharmaceuticals, M. Phase 2 Trial of Seribantumab plus Fulvestrant in Postmenopausal Women with Metastatic Breast Cancer (SHERBOC). Available online: https://clinicaltrials.gov/ct2/show/NCT03241810 (accessed on 9 December 2018).
- Mirschberger, C.; Schiller, C.B.; Schräml, M.; Dimoudis, N.; Friess, T.; Gerdes, C.A.; Reiff, U.; Lifke, V.; Hoelzlwimmer, G.; Kolm, I.; et al. RG7116, a Therapeutic Antibody That Binds the Inactive HER3 Receptor and Is Optimized for Immune Effector Activation. Cancer Res. 2013, 73, 5183–5194. [Google Scholar] [CrossRef] [Green Version]
- Roche, H.-L. A Study to Evaluate Lumretuzumab in Combination with Pertuzumab and Paclitaxel in Participants with Metastatic Breast Cancer Expressing Human Epidermal Growth Factor Receptor (HER) 3 and HER2 Protein. Available online: https://clinicaltrials.gov/ct2/show/NCT01918254 (accessed on 13 September 2018).
- McDonagh, C.F.; Huhalov, A.; Harms, B.D.; Adams, S.; Paragas, V.; Oyama, S.; Zhang, B.; Luus, L.; Overland, R.; Nguyen, S.; et al. Antitumor Activity of a Novel Bispecific Antibody That Targets the ErbB2/ErbB3 Oncogenic Unit and Inhibits Heregulin-Induced Activation of ErbB3. Mol. Cancer Ther. 2012, 11, 582–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, R.; Patel, H.; Alanazi, S.; Yuan, L.; Garrett, J.T. HER3 signaling and targeted therapy in cancer. Oncol. Rev. 2018, 12, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merus, N.V. MCLA-128 with Trastuzumab/Chemotherapy in HER2+ and With Endocrine Therapy in ER+ and Low HER2 Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03321981 (accessed on 21 September 2018).
- Hollmen, M.; Maatta, J.A.; Bald, L.; Sliwkowski, M.X.; Elenius, K. Suppression of breast cancer cell growth by a monoclonal antibody targeting cleavable ErbB4 isoforms. Oncogene 2009, 28, 1309–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, T.T.; Sundvall, M.; Maatta, J.A.; Elenius, K. Erbb4 and its isoforms: Selective regulation of growth factor responses by naturally occurring receptor variants. Trends Cardiovasc. Med. 2000, 10, 304–310. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.; Rubin Grandis, J. Pharmacokinetic and pharmacodynamic properties of EGFR inhibitors under clinical investigation. Cancer Treat. Rev. 2004, 30, 255–268. [Google Scholar] [CrossRef]
- Woodburn, J.R. The Epidermal Growth Factor Receptor and Its Inhibition in Cancer Therapy. Pharmacol. Ther. 1999, 82, 241–250. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Ginther, C.; Press, M.; Forbes, J.; Mackey, J.; French, T.; South, M.; Rupin, M.; Slamon, D.J. ER+ PR- breast cancer defines a unique subtype of breast cancer that is driven by growth factor signaling and may be more likely to respond to EGFR targeted therapies. J. Clin. Oncol. 2006, 24, 514. [Google Scholar]
- Moyer, J.D.; Barbacci, E.; Iwata, K.K.; Arnold, L.; Boman, B.; Cunningham, A.; Soderstrom, C.; Doty, J.; Morin, M.J.; Moyer, M.P.; et al. Induction of Apoptosis and Cell Cycle Arrest by CP-358,774, an Inhibitor of Epidermal Growth Factor Receptor Tyrosine Kinase. Cancer Res. 1997, 57, 4838–4848. [Google Scholar]
- Bareschino, M.A.; Schettino, C.; Troiani, T.; Martinelli, E.; Morgillo, F.; Ciardiello, F. Erlotinib in cancer treatment. Ann. Oncol. 2007, 18, vi35–vi41. [Google Scholar] [CrossRef]
- Kaur, H.; Silverman, P.; Singh, D.; Cooper, B.W.; Fu, P.; Krishnamurthi, S.; Remick, S.; Overmoyer, B. Toxicity and outcome data in a phase II study of weekly docetaxel in combination with erlotinib in recurrent and/or metastatic breast cancer (MBC). J. Clin. Oncol. 2006, 24, 10623. [Google Scholar]
- Corkery, B.; Crown, J.; Clynes, M.; O’Donovan, N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann. Oncol. 2009, 20, 862–867. [Google Scholar] [CrossRef] [PubMed]
- El Guerrab, A.; Bamdad, M.; Kwiatkowski, F.; Bignon, Y.J.; Penault-Llorca, F.; Aubel, C. Anti-EGFR monoclonal antibodies and EGFR tyrosine kinase inhibitors as combination therapy for triple-negative breast cancer. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, K.L.; Burstein, H.J.; Storniolo, A.M.; Rugo, H.; Sledge, G.; Koehler, M.; Ellis, C.; Casey, M.; Vukelja, S.; Bischoff, J.; et al. Randomized study of Lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J. Clin. Oncol. 2010, 28, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Medina, P.J.; Goodin, S. Lapatinib: A dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin. Ther. 2008, 30, 1426–1447. [Google Scholar] [CrossRef]
- Solinas, C.; Ceppi, M.; Lambertini, M.; Scartozzi, M.; Buisseret, L.; Garaud, S.; Fumagalli, D.; de Azambuja, E.; Salgado, R.; Sotiriou, C.; et al. Tumor-infiltrating lymphocytes in patients with HER2-positive breast cancer treated with neoadjuvant chemotherapy plus trastuzumab, lapatinib or their combination: A meta-analysis of randomized controlled trials. Cancer Treat. Rev. 2017, 57, 8–15. [Google Scholar] [CrossRef]
- Xu, Z.-Q.; Zhang, Y.; Li, N.; Liu, P.-J.; Gao, L.; Gao, X.; Tie, X.-J. Efficacy and safety of lapatinib and trastuzumab for HER2-positive breast cancer: A systematic review and meta-analysis of randomised controlled trials. BMJ Open 2017, 7, e013053. [Google Scholar] [CrossRef] [Green Version]
- Wissner, A.; Mansour, T.S. The development of HKI-272 and related compounds for the treatment of cancer. Arch. Pharm. 2008, 341, 465–477. [Google Scholar] [CrossRef]
- Bose, P.; Ozer, H. Neratinib: An oral, irreversible dual EGFR/HER2 inhibitor for breast and non-small cell lung cancer. Expert Opin. Investig. Drugs 2009, 18, 1735–1751. [Google Scholar] [CrossRef]
- (FDA), U.S.F.D.A. FDA Approves Neratinib for Extended Adjuvant Treatment of Early Stage HER2-Positive Breast Cancer. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm567259.htm (accessed on 21 September 2017).
- Canonici, A.; Gijsen, M.; Mullooly, M.; Bennett, R.; Bouguern, N.; Pedersen, K.; O’Brien, N.A.; Roxanis, I.; Li, J.-L.; Bridge, E.; et al. Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget 2013, 4, 1592–1605. [Google Scholar] [CrossRef]
- Awada, A.; Dirix, L.; Manso Sanchez, L.; Xu, B.; Luu, T.; Dieras, V.; Hershman, D.L.; Agrapart, V.; Ananthakrishnan, R.; Staroslawska, E. Safety and efficacy of neratinib (HKI-272) plus vinorelbine in the treatment of patients with ErbB2-positive metastatic breast cancer pretreated with anti-HER2 therapy. Ann. Oncol. 2013, 24, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Arribas, J.; Baselga, J.; Pedersen, K.; Parra-Palau, J.L. p95HER2 and Breast Cancer. Cancer Res. 2011, 71, 1515–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslin, S.; Lowry, M.; Driscoll, L. Neratinib resistance and cross-resistance to other HER2-targeted drugs due to increased activity of metabolism enzyme cytochrome P4503A4. Br. J. Cancer 2017, 116, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Winer, E.P.; Wheatley, D.; Carey, L.A.; Houston, S.; Mendelson, D.; Munster, P.; Frakes, L.; Kelly, S.; Garcia, A.A.; et al. A phase II study of afatinib (BIBW 2992), an irreversible ErbB family blocker, in patients with HER2-positive metastatic breast cancer progressing after trastuzumab. Breast Cancer Res. Treat. 2012, 133, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- De Pauw, I.; Lardon, F.; Van den Bossche, J.; Baysal, H.; Fransen, E.; Deschoolmeester, V.; Pauwels, P.; Peeters, M.; Vermorken, J.B.; Wouters, A. Simultaneous targeting of EGFR, HER2, and HER4 by afatinib overcomes intrinsic and acquired cetuximab resistance in head and neck squamous cell carcinoma cell lines. Mol. Oncol. 2018, 12, 830–854. [Google Scholar] [CrossRef] [Green Version]
- Smaill, J.B.; Rewcastle, G.W.; Loo, J.A.; Greis, K.D.; Chan, O.H.; Reyner, E.L.; Lipka, E.; Showalter, H.D.H.; Vincent, P.W.; Elliott, W.L.; et al. Tyrosine Kinase Inhibitors. 17. Irreversible Inhibitors of the Epidermal Growth Factor Receptor: 4-(Phenylamino)quinazoline- and 4-(Phenylamino)pyrido[3,2-d]pyrimidine-6-acrylamides Bearing Additional Solubilizing Functions. J. Med. Chem. 2000, 43, 1380–1397. [Google Scholar] [CrossRef]
- De Pauw, I.; Wouters, A.; Van den Bossche, J.; Peeters, M.; Pauwels, P.; Deschoolmeester, V.; Vermorken, J.B.; Lardon, F. Preclinical and clinical studies on afatinib in monotherapy and in combination regimens: Potential impact in colorectal cancer. Pharmacol. Ther. 2016, 166, 71–83. [Google Scholar] [CrossRef]
- Macha, M.; Rachagani, S.; Qazi, A.; Jahan, R.; Gupta, S.; Patel, A.; Seshacharyulu, P.; Lin, C.; Li, S.; Wang, S.; et al. Afatinib radiosensitizes head and neck squamous cell carcinoma cells by targeting cancer stem cells. Oncotarget 2017, 8. [Google Scholar] [CrossRef]
- Huguet, F.; Fernet, M.; Giocanti, N.; Favaudon, V.; Larsen, A.K. Afatinib, an Irreversible EGFR Family Inhibitor, Shows Activity toward Pancreatic Cancer Cells, Alone and in Combination with Radiotherapy, Independent of KRAS Status. Targeted Oncol. 2016, 11, 371–381. [Google Scholar] [CrossRef]
- Roselli, E.; Frieling, J.S.; Thorner, K.; Ramello, M.C.; Lynch, C.C.; Abate-Daga, D. CAR-T Engineering: Optimizing Signal Transduction and Effector Mechanisms. BioDrugs 2019. [Google Scholar] [CrossRef]
- Kloess, S.; Kretschmer, A.; Stahl, L.; Fricke, S.; Koehl, U. CAR-Expressing Natural Killer Cells for Cancer Retargeting. Transfus. Med. Hemother. 2019, 46, 4–13. [Google Scholar] [CrossRef]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef]
- Wilkie, S.; van Schalkwyk, M.C.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.; Pereira, A.C.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e1411. [Google Scholar] [CrossRef] [Green Version]
- Frejd, F.Y. Novel Alternative Scaffolds and Their Potential Use for Tumor Targeted Radionuclide Therapy. In Targeted Radionuclide Tumor Therapy: Biological Aspects; Stigbrand, T., Carlsson, J., Adams, G.P., Eds.; Springer: Dordrecht, The Netherlands, 2008; pp. 89–116. [Google Scholar] [CrossRef]
- Beckman, R.A.; Weiner, L.M.; Davis, H.M. Antibody constructs in cancer therapy. Cancer 2007, 109, 170–179. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, L.; Liang, C.; Wang, C.; Peng, R.; Liu, Z. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat. Commun. 2016, 7, 13193. [Google Scholar] [CrossRef]
- Lin, Z.; Liu, Y.; Ma, X.; Hu, S.; Zhang, J.; Wu, Q.; Ye, W.; Zhu, S.; Yang, D.; Qu, D.; et al. Photothermal ablation of bone metastasis of breast cancer using PEGylated multi-walled carbon nanotubes. Sci. Rep. 2015, 5, 11709. [Google Scholar] [CrossRef] [Green Version]
- Photoimmunotherapy (PIT) Study in Recurrent Head/Neck Cancer for Patients Who Have Failed at Least Two Lines of Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT03769506 (accessed on 7 July 2018).
- Nagaya, T.; Sato, K.; Harada, T.; Nakamura, Y.; Choyke, P.L.; Kobayashi, H. Near Infrared Photoimmunotherapy Targeting EGFR Positive Triple Negative Breast Cancer: Optimizing the Conjugate-Light Regimen. PLoS ONE 2015, 10, e0136829. [Google Scholar] [CrossRef]
- Kobayashi, H.; Choyke, P.L. Near-Infrared Photoimmunotherapy of Cancer. Acc. Chem. Res. 2019, 52, 2332–2339. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Drug | Format | Target | Clinical Phase | Literature |
|---|---|---|---|---|
| Cetuximab | Chimeric MAb | EGFR1 | I, II | [44,55] |
| Panitumumab | Humanized MAb | EGFR1 | II | [56,57] |
| Zalutumumab | Human MAb | EGFR1 | I, II | [60,61] |
| Trastuzumab | Humanized MAb | HER2 | approved | [64,65,66] |
| Pertuzumab | Humanized MAb | HER2 | approved | [70,71,72,73] |
| Trastuzumab Emtansin | Antibody-drug conjugate | HER2 | approved | [75,76] |
| Trastuzumab Deruxtecan | Antibody-drug conjugate | HER2/Tubulin | I, II | [78] |
| MEDI4276 | Bispecific antibody | HER2 | I, II | [79] |
| Seribantumab | Humanized MAb | HER3 | I, II | [80,81,82] |
| Lumretuzumab | Humanized MAb | HER3 | I | [83,84] |
| MM-111 | Bispecific antibody | HER3 | I, II | [85,86] |
| MCL-128 | Bispecific antibody | HER2/3 | I, II | [87] |
| Gefitinib | Small-molecule tyrosine kinase inhibitor | EGFR1 | approved | [91,92] |
| Erlotinib | Small-molecule tyrosine kinase inhibitor | EGFR1 | approved | [94,95,98,99] |
| Lapatinib | Small-molecule tyrosine kinase inhibitor | EGFR1/HER2 | approved | [70,100,101] |
| Neratinib | Small-molecule tyrosine kinase inhibitor | HER2 | approved | [105,106,108] |
| Canertinib | Small-molecule tyrosine kinase inhibitor | pan-ERB | I, II | [113] |
| Afatinib | Small-molecule tyrosine kinase inhibitor | EGFR1, HER2, HER4 | approved | [114,115,116] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maennling, A.E.; Tur, M.K.; Niebert, M.; Klockenbring, T.; Zeppernick, F.; Gattenlöhner, S.; Meinhold-Heerlein, I.; Hussain, A.F. Molecular Targeting Therapy against EGFR Family in Breast Cancer: Progress and Future Potentials. Cancers 2019, 11, 1826. https://doi.org/10.3390/cancers11121826
Maennling AE, Tur MK, Niebert M, Klockenbring T, Zeppernick F, Gattenlöhner S, Meinhold-Heerlein I, Hussain AF. Molecular Targeting Therapy against EGFR Family in Breast Cancer: Progress and Future Potentials. Cancers. 2019; 11(12):1826. https://doi.org/10.3390/cancers11121826
Chicago/Turabian StyleMaennling, Amaia Eleonora, Mehmet Kemal Tur, Marcus Niebert, Torsten Klockenbring, Felix Zeppernick, Stefan Gattenlöhner, Ivo Meinhold-Heerlein, and Ahmad Fawzi Hussain. 2019. "Molecular Targeting Therapy against EGFR Family in Breast Cancer: Progress and Future Potentials" Cancers 11, no. 12: 1826. https://doi.org/10.3390/cancers11121826
APA StyleMaennling, A. E., Tur, M. K., Niebert, M., Klockenbring, T., Zeppernick, F., Gattenlöhner, S., Meinhold-Heerlein, I., & Hussain, A. F. (2019). Molecular Targeting Therapy against EGFR Family in Breast Cancer: Progress and Future Potentials. Cancers, 11(12), 1826. https://doi.org/10.3390/cancers11121826