PAK4-NAMPT Dual Inhibition as a Novel Strategy for Therapy Resistant Pancreatic Neuroendocrine Tumors

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results
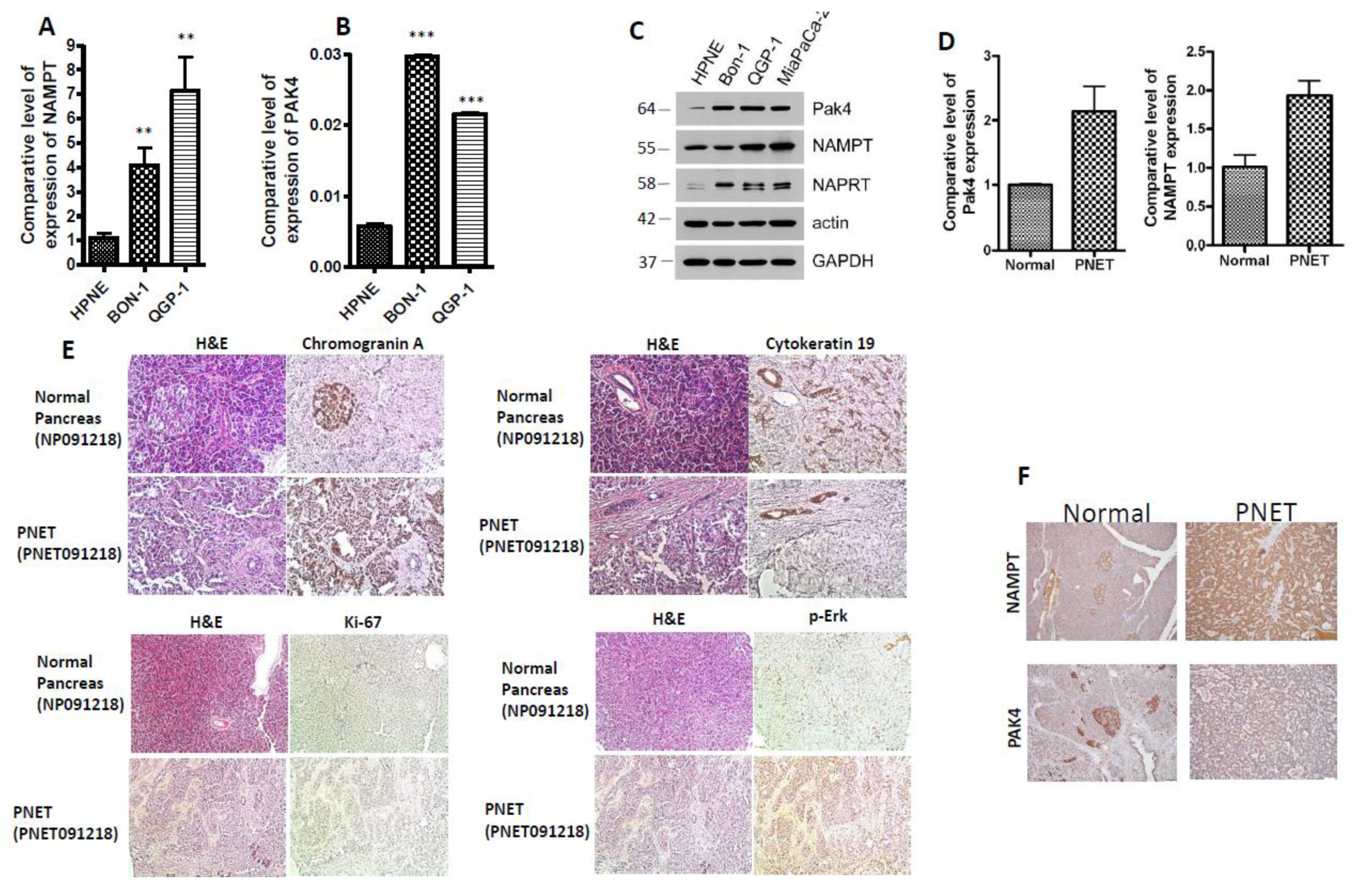
2.1. PAK4 and NAMPT Are Overexpressed in PNET
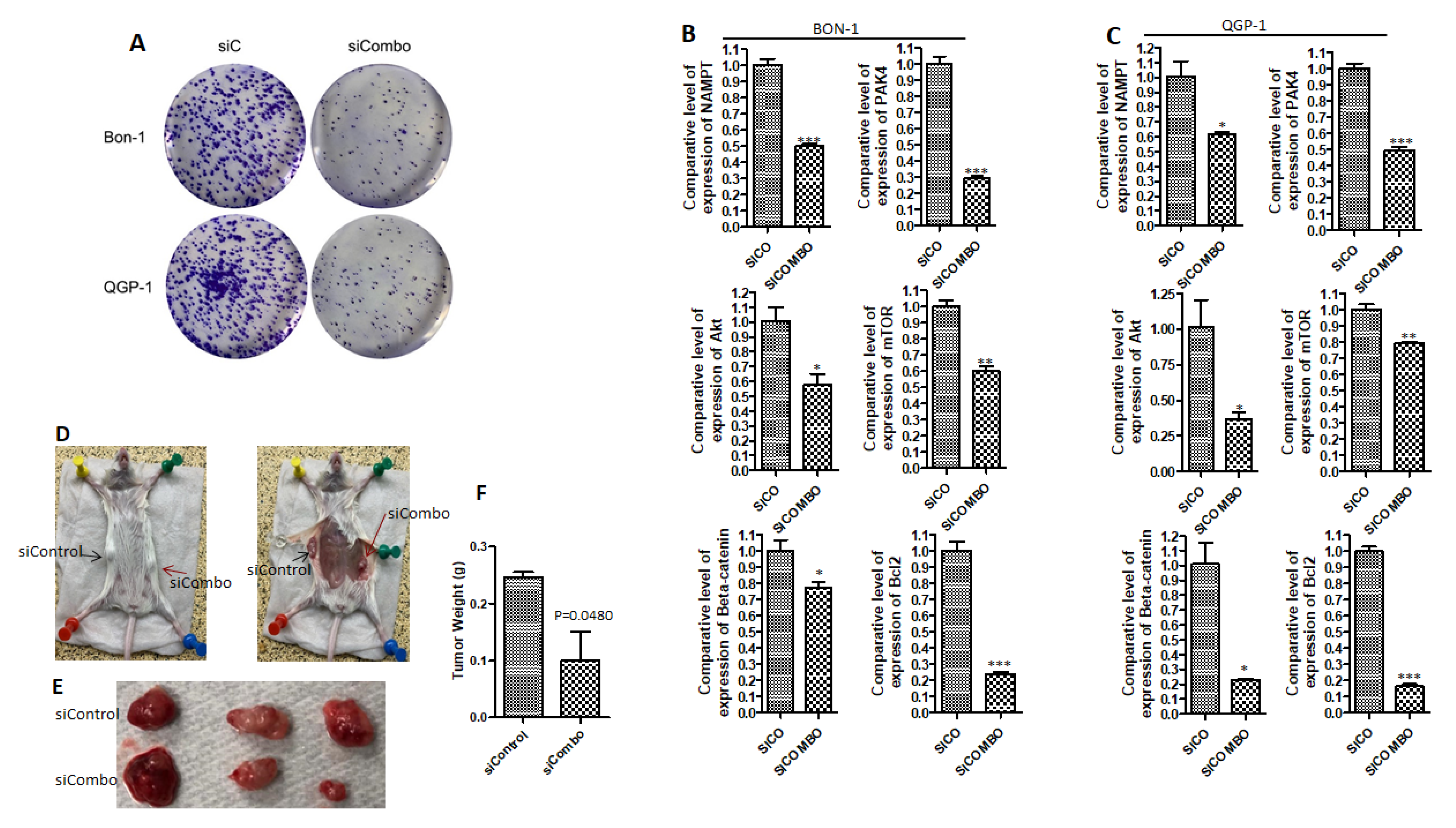
2.2. PAK4 and NAMPT Promote PNET Survival
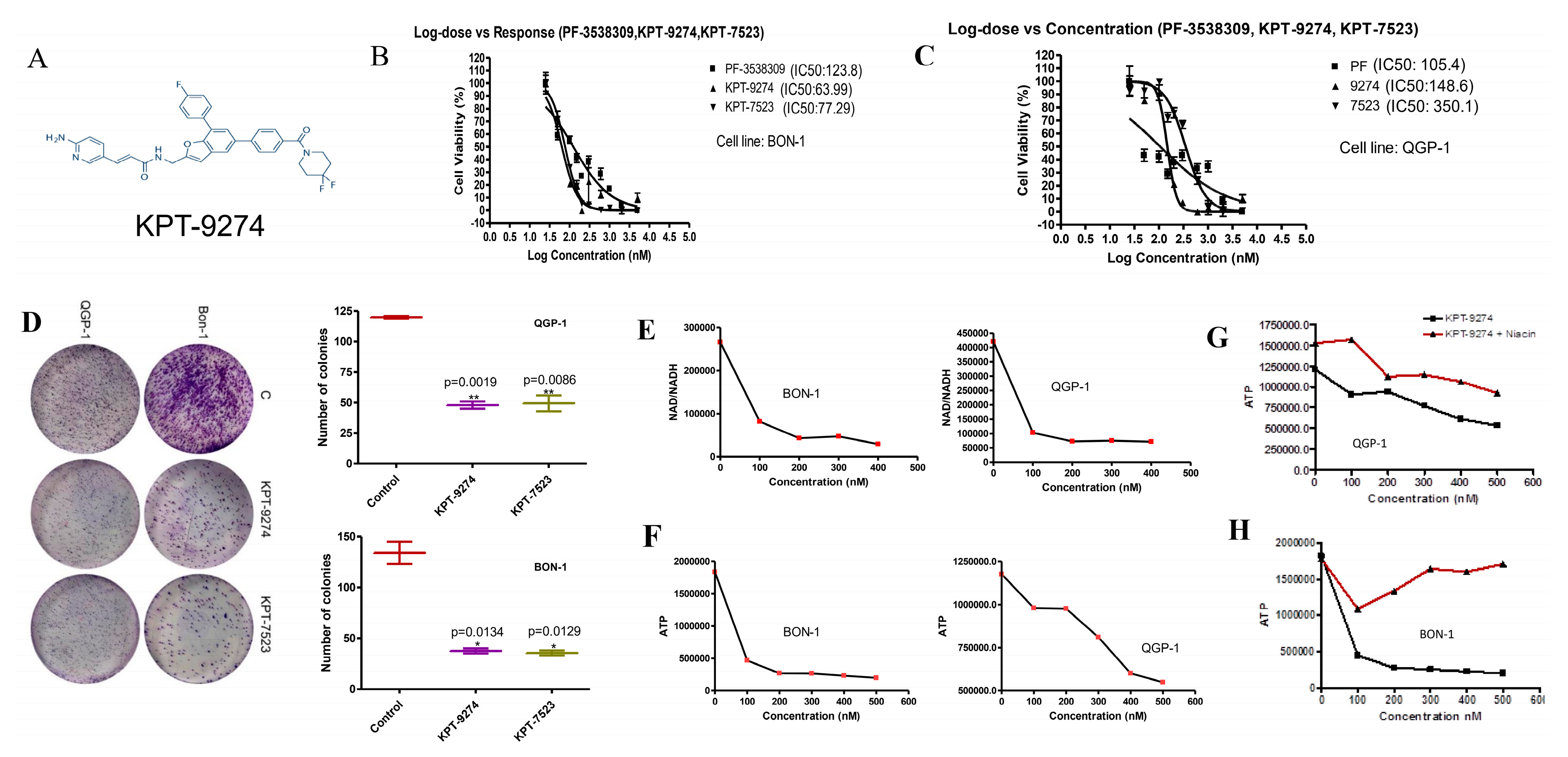
2.3. KPT-9274 and Analog KPT-7523 Decrease PNET Cell Survival and Growth
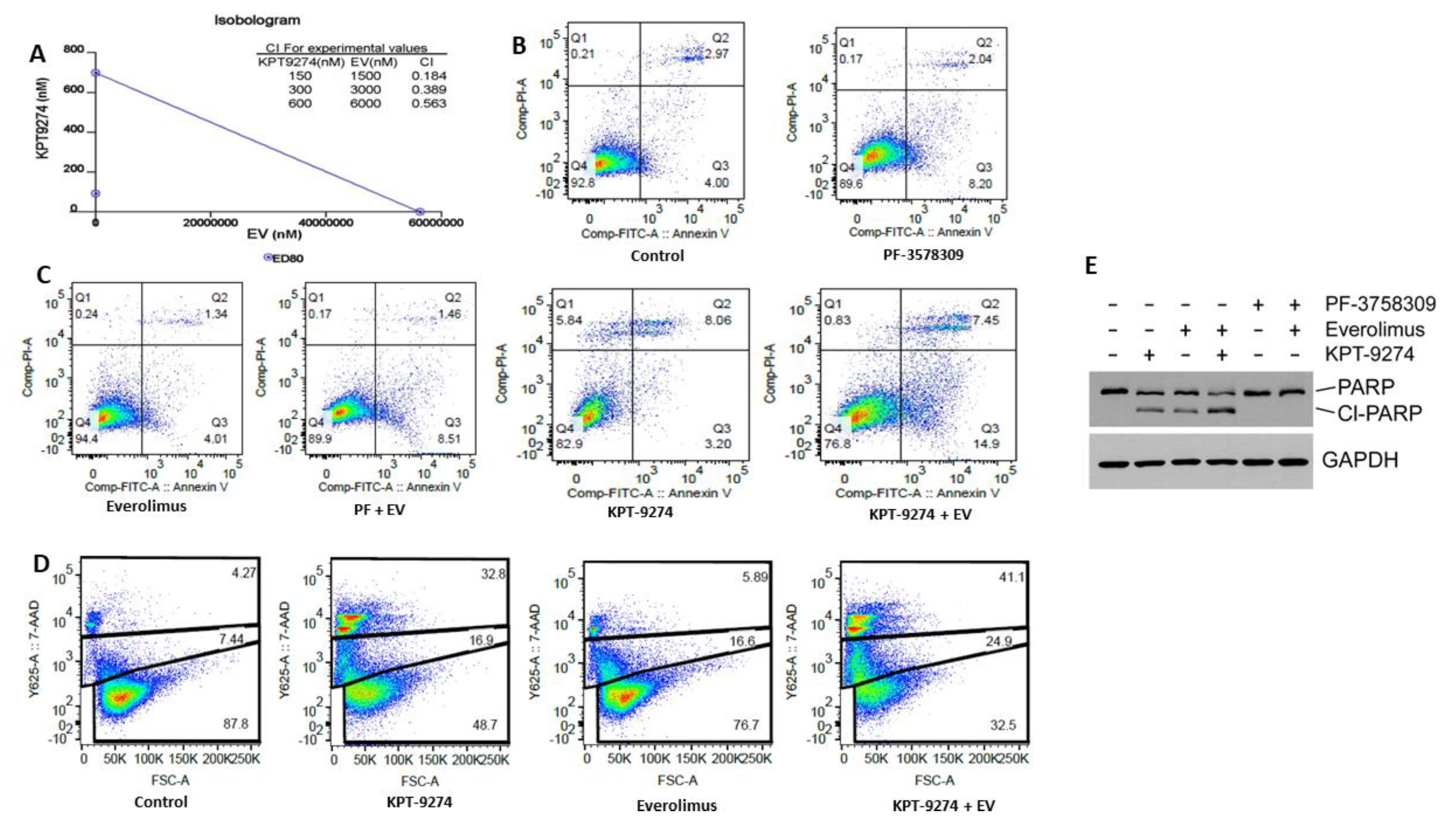
2.4. KPT-9274 Synergizes with Everolimus in PNET Cellular Models
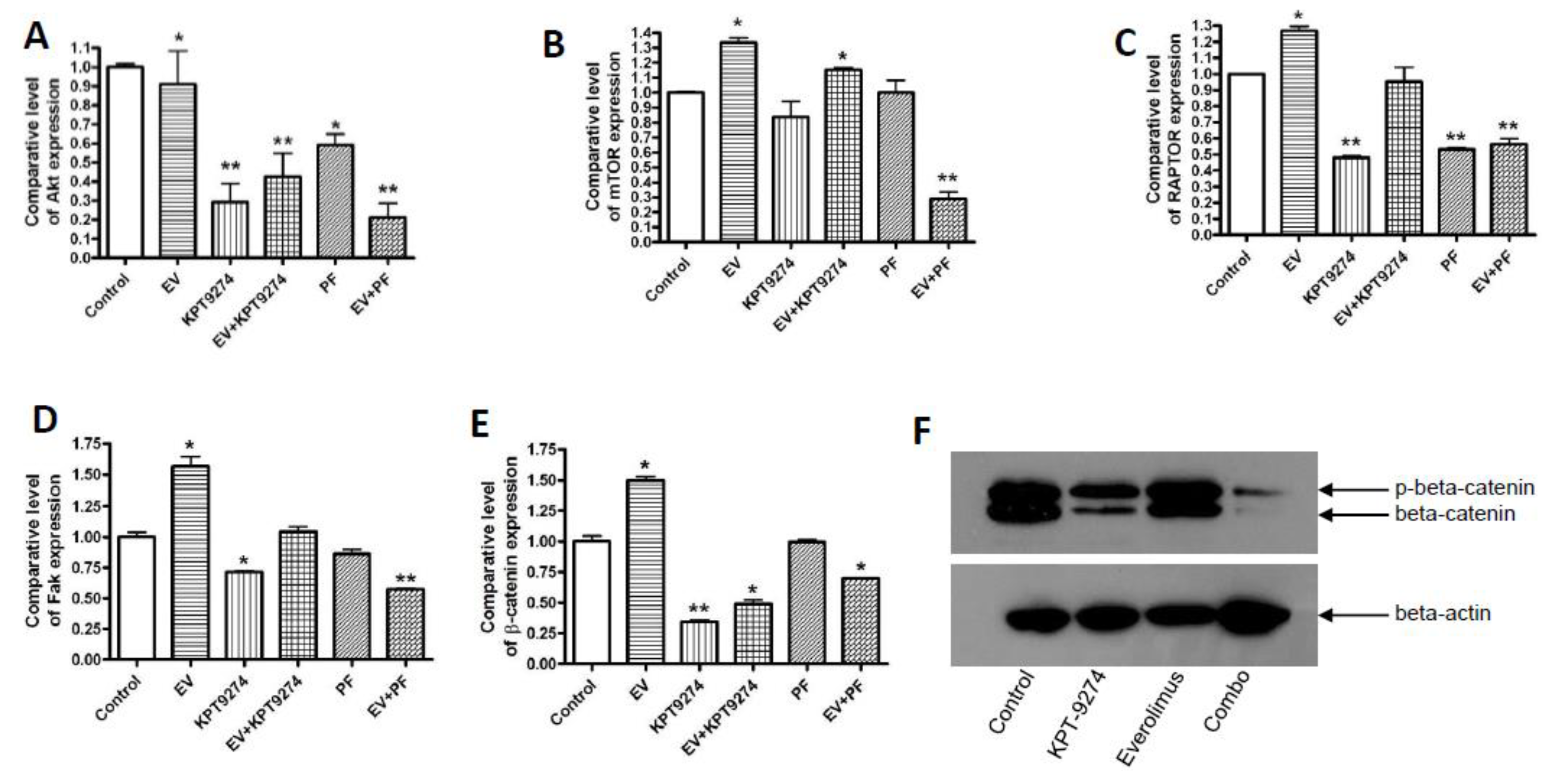
2.5. Molecular Analysis of KPT-9274-Everolimus Combination
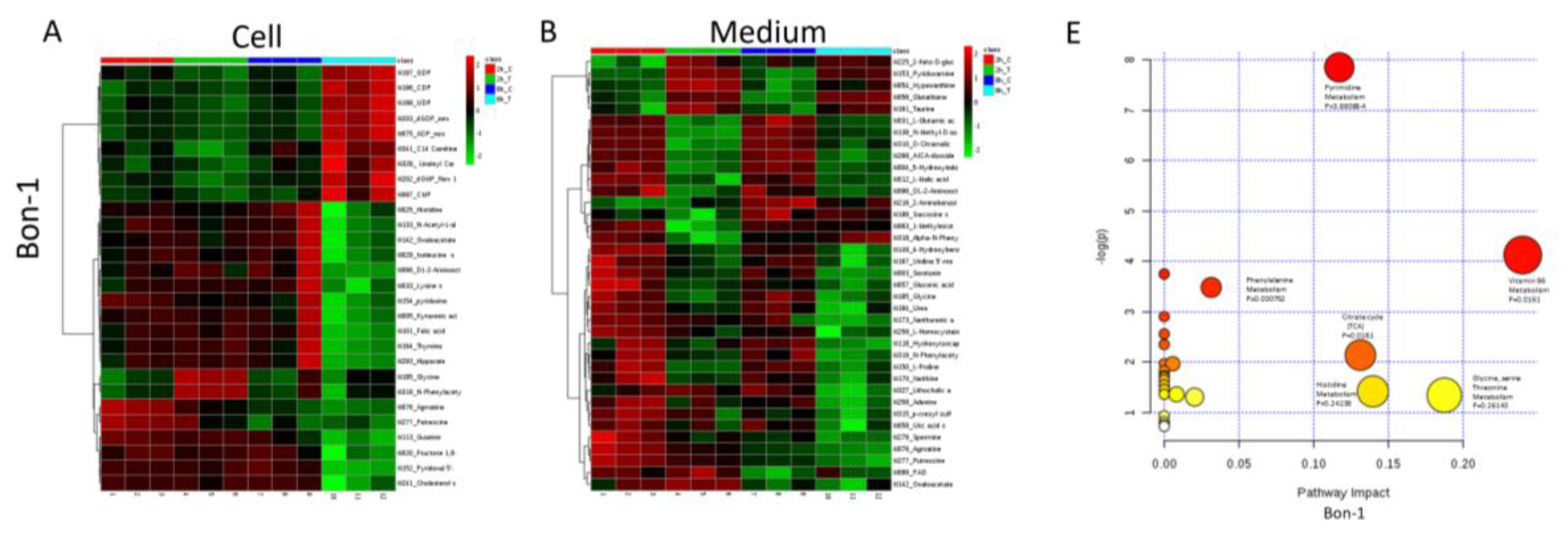
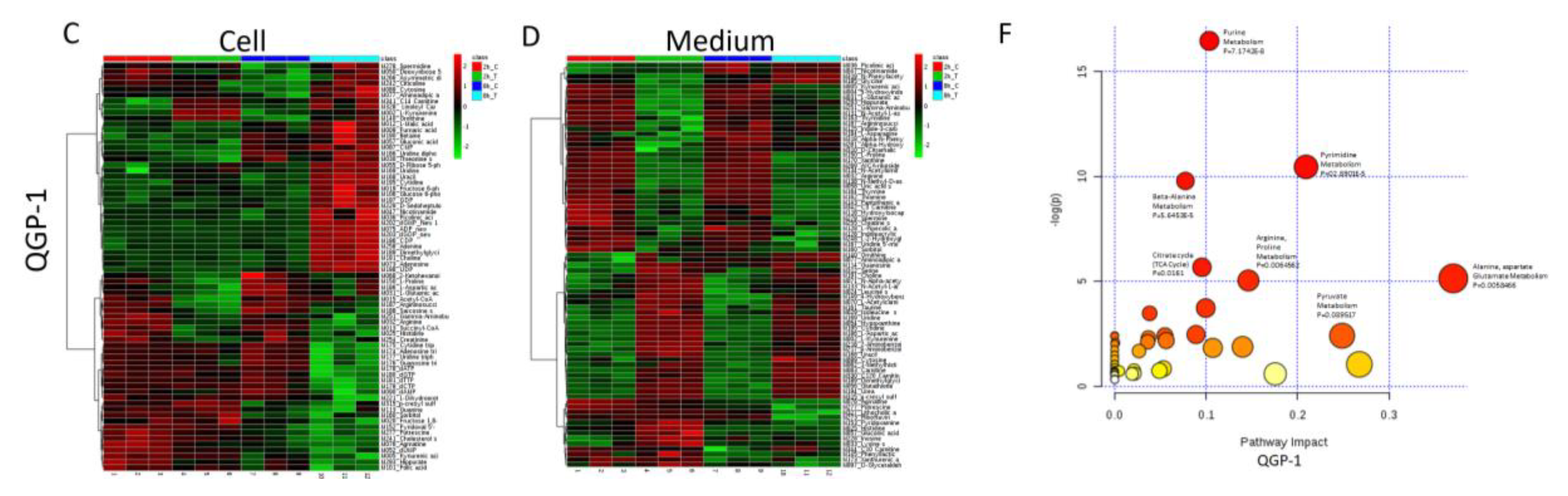
2.6. Metabolomic Analysis of PNET Cells Treated with KPT-9274
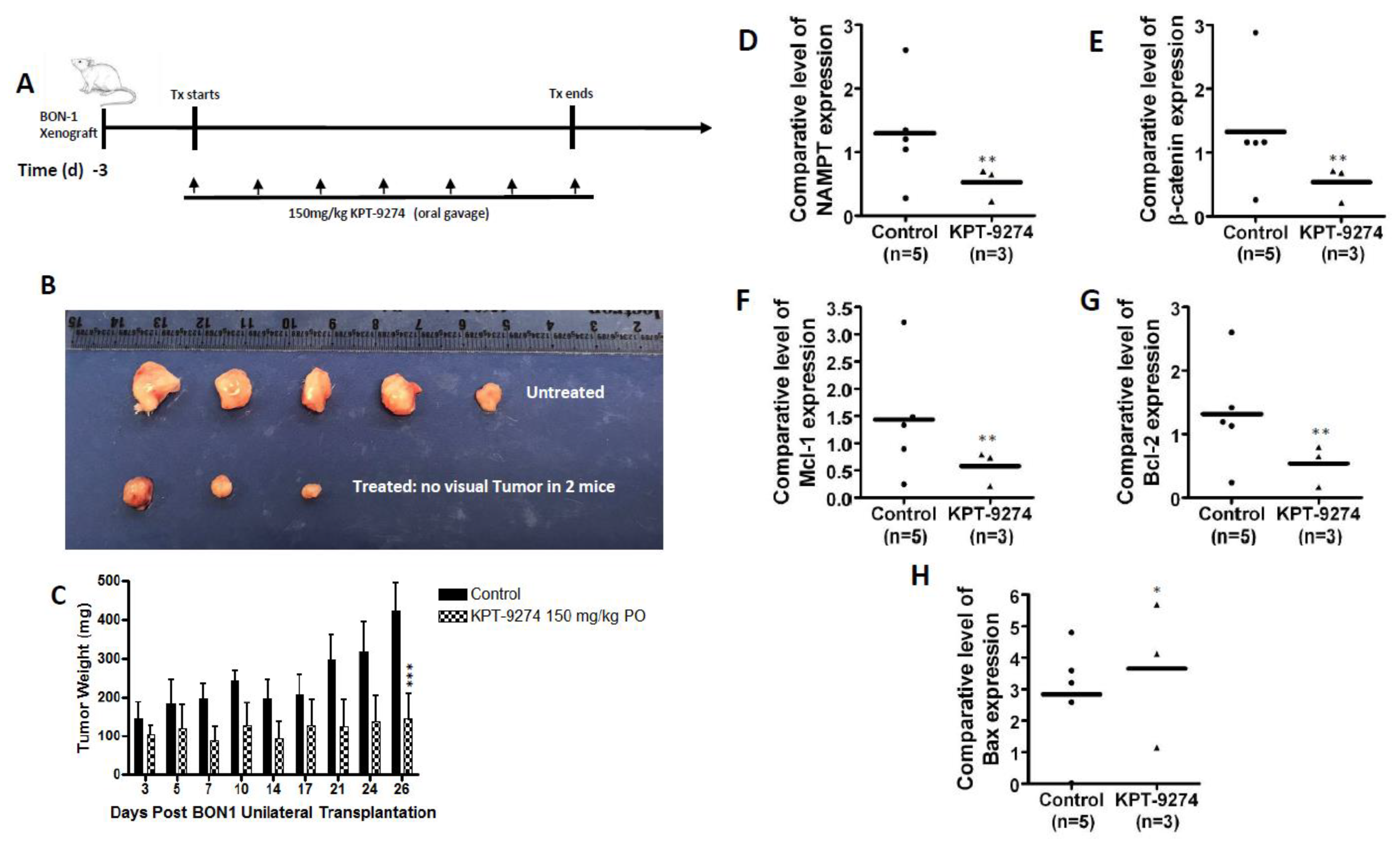
2.7. KPT-9274 Shows Single Agent Anti-Tumor Activity in PNET
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Small Interference RNA and Transfection
4.3. MTT Assay
4.4. Colonogenic Assay
4.5. Annexin V and 7AAD Apoptosis Analysis
4.6. Immunohistochemistry Analysis
4.7. Western Blot Analysis
4.8. Quantitative Real-Time PCR
4.9. Metabolomic Analysis.
4.10. Animal Studies
4.11. Statistical Consideration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Modlin, I.M.; Oberg, K.; Chung, D.C.; Jensen, R.T.; de Herder, W.W.; Thakker, R.V.; Caplin, M.; Delle, F.G.; Kaltsas, G.A.; Krenning, E.P.; et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008, 9, 61–72. [Google Scholar] [CrossRef]
- Modlin, I.M.; Lye, K.D.; Kidd, M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer 2003, 97, 934–959. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; de Braud, F.; Festinese, F.; Bregant, C.; Lorenzoni, A.; Maccauro, M.; Milione, M.; Concas, L.; Formisano, B.; Leuzzi, L.; et al. Evolution in the treatment of gastroenteropancreatic-neuroendocrine neoplasms, focus on systemic therapeutic options: A systematic review. Future Oncol. 2015, 11, 1947–1959. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, M.; Caterina, I.; de Divitiis, C.; von Arx, C.; Maiolino, P.; Tatangelo, F.; Cavalcanti, E.; di Girolamo, E.; Iaffaioli, R.V.; Scala, S.; et al. Everolimus and pancreatic neuroendocrine tumors (PNETs): Activity, resistance and how to overcome it. Int. J. Surg. 2015, 21 (Suppl. 1), S89–S94. [Google Scholar] [CrossRef] [PubMed]
- Djukom, C.; Porro, L.J.; Mrazek, A.; Townsend, C.M., Jr.; Hellmich, M.R.; Chao, C. Dual inhibition of PI3K and mTOR signaling pathways decreases human pancreatic neuroendocrine tumor metastatic progression. Pancreas 2014, 43, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Francois, R.A.; Maeng, K.; Nawab, A.; Kaye, F.J.; Hochwald, S.N.; Zajac-Kaye, M. Targeting Focal Adhesion Kinase and Resistance to mTOR Inhibition in Pancreatic Neuroendocrine Tumors. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Aristizabal, P.E.T.; Auernhammer, C.J. Targeted therapy of gastroenteropancreatic neuroendocrine tumours: Preclinical strategies and future targets. Endocr. Connect. 2018, 7, R1–R25. [Google Scholar] [CrossRef]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef]
- Dirckx, N.; Tower, R.J.; Mercken, E.M.; Vangoitsenhoven, R.; Moreau-Triby, C.; Breugelmans, T.; Nefyodova, E.; Cardoen, R.; Mathieu, C.; van der Schueren, B.; et al. Vhl deletion in osteoblasts boosts cellular glycolysis and improves global glucose metabolism. J. Clin. Investig. 2018, 128, 1087–1105. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Dart, A.E.; Box, G.M.; Court, W.; Gale, M.E.; Brown, J.P.; Pinder, S.E.; Eccles, S.A.; Wells, C.M. PAK4 promotes kinase-independent stabilization of RhoU to modulate cell adhesion. J. Cell Biol. 2015, 211, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Hakoshima, T.; Shimizu, T.; Maesaki, R. Structural basis of the Rho GTPase signaling. J. Biochem. 2003, 134, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.P.; Crossley, L.; Zhan, Q.; Huang, R.; Robinson, D.; Badwey, J.A. The P21-activated protein kinases (Paks) receive and integrate messages from a variety of signaling pathways. Adv. Exp. Med. Biol. 2002, 507, 497–502. [Google Scholar] [PubMed]
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 2014, 14, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C.; Hezel, A.F.; Aguirre, A.J.; Zheng, H.; Paik, J.H.; Ying, H.; Chu, G.C.; Zhang, J.X.; Sahin, E.; Yeo, G.; et al. Genomic alterations link Rho family of GTPases to the highly invasive phenotype of pancreas cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 19372–19377. [Google Scholar] [CrossRef] [PubMed]
- Mahlamaki, E.H.; Kauraniemi, P.; Monni, O.; Wolf, M.; Hautaniemi, S.; Kallioniemi, A. High-resolution genomic and expression profiling reveals 105 putative amplification target genes in pancreatic cancer. Neoplasia 2004, 6, 432–439. [Google Scholar] [CrossRef]
- Beauchamp, R.L.; James, M.F.; DeSouza, P.A.; Wagh, V.; Zhao, W.N.; Jordan, J.T.; Stemmer-Rachamimov, A.; Plotkin, S.R.; Gusella, J.F.; Haggarty, S.J.; et al. A high-throughput kinome screen reveals serum/glucocorticoid-regulated kinase 1 as a therapeutic target for NF2-deficient meningiomas. Oncotarget 2015, 6, 16981–16997. [Google Scholar] [CrossRef]
- Yun, C.Y.; You, S.T.; Kim, J.H.; Chung, J.H.; Han, S.B.; Shin, E.Y.; Kim, E.G. p21-activated kinase 4 critically regulates melanogenesis via activation of the CREB/MITF and beta-catenin/MITF pathways. J. Investig. Dermatol. 2015, 135, 1385–1394. [Google Scholar] [CrossRef]
- Noda, K.; Nakajima, S.; Godo, S.; Saito, H.; Ikeda, S.; Shimizu, T.; Enkhjargal, B.; Fukumoto, Y.; Tsukita, S.; Yamada, T.; et al. Rho-kinase inhibition ameliorates metabolic disorders through activation of AMPK pathway in mice. PLoS ONE 2014, 9, e110446. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Li, Y.; Muqbil, I.; Aboukameel, A.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Philip, P.A.; Azmi, A.S. Targeting Rho GTPase effector p21 activated kinase 4 (PAK4) suppresses p-Bad-microRNA drug resistance axis leading to inhibition of pancreatic ductal adenocarcinoma proliferation. Small GTPase 2019, 5, 367–377. [Google Scholar] [CrossRef]
- Chini, C.C.; Guerrica, A.M.; Nin, V.; Camacho-Pereira, J.; Escande, C.; Barbosa, M.T.; Chini, E.N. Targeting of NAD metabolism in pancreatic cancer celss: Potential novel therapy for pancreatic tumors. Clin. Cancer Res. 2014, 20, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Moratta, V.; Zatelli, M.C.; Sciammarella, C.; Ambrosio, M.R.; Bondanelli, M.; Colao, A.; Faggiano, A. Chromogranin A as circulating marker for diagnosis and management of neuroendocrine neoplasms: More flaws than fame. Emdocr. Relat. Cancer 2018, 1, R11–R29. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Yang, Z.; Liu, Z.; Miao, X.; Yang, L.; Li, D.; Zou, Q.; Yuan, Y. Glypican-3 and KRT19 are markers associating with metastasis and poor prognosis of pancreatic ductal adenocarcinoma. Cancer Biomark. 2016, 4, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Abu, A.O.; Chen, C.H.; Senapedis, W.; Baloglu, E.; Argueta, C.; Weiss, R.H. Dual and Specific Inhibition of NAMPT and PAK4 By KPT-9274 Decreases Kidney Cancer Growth. Mol. Cancer Ther. 2016, 9, 2119–2129. [Google Scholar]
- Mitchell, S.R.; Larkin, K.; Grieselhuber, N.R.; Lai, T.H.; Cannon, M.; Orwick, S.; Sharma, P.; Asemelash, Y.; Zhang, P.; Goettl, V.M. Selective targeting of NAMPT by KPT-9274 in acute myeloid leukemia. Blood Adv. 2019, 3, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lopez, M.A.; Linares, M.; Kumar, S.; Oliva, S.; Martinez-Lopez, J.; Xu, L.; Xu, Y.; Perini, T.; Senapedis, W.; et al. Dual PAK4-NAMPT Inhibition Impacts Growth and Survival, and Increases Sensitivity to DNA-Damaging Agents in Waldenstrom Macroglobulinemia. Clin. Cancer Res. 2019, 25, 369–377. [Google Scholar] [CrossRef]
- Takao, S.; Chien, W.; Madan, V.; Lin, D.C.; Ding, L.W.; Sun, Q.Y.; Mayakonda, A.; Sudo, M.; Xu, L.; Chen, Y.; et al. Targeting the vulnerability to NAD(+) depletion in B-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 616–625. [Google Scholar] [CrossRef]
- Rane, C.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Crochiere, M.; Das-Gupta, S.; Minden, A. A novel orally bioavailable compound KPT-9274 inhibits PAK4, and blocks triple-negative breast cancer tumor growth. Sci. Rep. 2017, 7, 42555. [Google Scholar] [CrossRef]
- Aboukameel, A.; Muqbil, I.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Shacham, S.; Kauffman, M.; Philip, P.A.; Mohammad, R.M.; Azmi, A.S. Novel p21-Activated Kinase 4 (PAK4) Allosteric Modulators Overcome Drug Resistance and Stemness in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 76–87. [Google Scholar] [CrossRef]
- Yao, J.C.; Phan, A.T.; Jehl, V.; Shah, G.; Meric-Bernstam, F. Everolimus in advanced pancreatic neuroendocrine tumors: The clinical experience. Cancer Res. 2013, 73, 1449–1453. [Google Scholar] [CrossRef]
- Won, S.Y.; Park, J.J.; Shin, E.Y.; Kim, E.G. PAK4 signaling in health and disease: Defining the PAK4-CREB axis. Exp. Mol. Med. 2019, 51, 11. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Lim, S.M.; Yoo, Y.I.; Jung, J.; Park, J.W.; Kim, G.J. Microenvironmental Interaction Between Hypoxia and Endothelial Cells Controls the Migration Ability of Placenta-Derived Mesenchymal Stem Cells via alpha4 Integrin and Rho Signaling. J. Cell Biochem. 2016, 117, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Cea, M.; Cagnetta, A.; Fulciniti, M.; Tai, Y.T.; Hideshima, T.; Chauhan, D.; Roccaro, A.; Sacco, A.; Calimeri, T.; Cottini, F.; et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood 2012, 120, 3519–3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, S.; Penke, M.; Gorski, T.; Gebhardt, R.; Weiss, T.S.; Kiess, W.; Garten, A. FK866-induced NAMPT inhibition activates AMPK and downregulates mTOR signaling in hepatocarcinoma cells. Biochem. Biophys. Res. Commun. 2015, 458, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.J.; Subramaniam, P.S.; Tang, L.H.; Grunn, A.; Aburi, M.; Rieckhof, G.; Komissarova, E.V.; Hagan, E.A.; Bodei, L.; Clemons, P.A. A precision oncology approach to pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 2018, 50, 979–989. [Google Scholar] [CrossRef] [PubMed]
- de Figueiredo, L.F.; Gossmann, T.I.; Ziegler, M.; Schuster, S. Pathway analysis of NAD+ metabolism. Biochem. J. 2011, 439, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Khan, J.A.; Tao, X.; Tong, L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat. Struct. Mol. Biol. 2006, 13, 582–588. [Google Scholar] [CrossRef]
- Garten, A.; Petzold, S.; Korner, A.; Imai, S.; Kiess, W. Nampt: Linking NAD biology, metabolism and cancer. Trends Endocrinol. Metab. 2009, 20, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Shames, D.S.; Elkins, K.; Walter, K.; Holcomb, T.; Du, P.; Mohl, D.; Xiao, Y.; Pham, T.; Haverty, P.M.; Liederer, B. Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin. Cancer Res. 2013, 19, 6912–6923. [Google Scholar] [CrossRef] [Green Version]
- Neggers, J.E.; Kwanten, B.; Dierckx, T.; Noguchi, H.; Voet, A.; Bral, L.; Minner, K.; Massant, B.; Kint, N.; Delforge, M. Target identification of small molecules using large-scale CRISPR-Cas mutagenesis scanning of essential genes. Nat. Commun. 2018, 9, 502. [Google Scholar] [CrossRef]
- Aboukameel, A.; Muqbil, I.; Baloglu, E.; Senapedis, W.; Landesman, Y.; Argueta, C.; Kauffman, M.; Chang, H.; Kashyap, T.; Shacham, S.; et al. Down-regulation of AR splice variants through XPO1 suppression contributes to the inhibition of prostate cancer progression. Oncotarget 2018, 9, 35327–35342. [Google Scholar] [CrossRef] [PubMed]
- Wall, N.R.; Mohammad, R.M.; Nabha, S.M.; Pettit, G.R.; Al-Katib, A.M. Modulation of cIAP-1 by novel antitubulin agents when combined with bryostatin 1 results in increased apoptosis in the human early pre-B acute lymphoblastic leukemia cell line Reh. Biochem. Biophys. Res. Commun. 1999, 266, 76–80. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolites | Treatment Effect | p-Value |
|---|---|---|
| Kynurenic acid | −2.173 | 0.026 |
| Fructose 1,6-bisphosphate | −1.903 | 0.016 |
| Histidine | −2.356 | 0.023 |
| Phenylalanine | −1.076 | 0.044 |
| Arginine | −1.693 | 0.025 |
| Lysine (s) | −2.729 | 0.021 |
| dUMP | −1.043 | 0.025 |
| 1-Methylhistidine | −1.381 | 0.045 |
| 2-Ketohexanoic acid | 1.938 | 0.001 |
| N-Alpha-acetyllysine | −1.866 | 0.047 |
| ADP_new | 1.410 | 0.001 |
| CMP | 1.610 | 0.023 |
| DL-2-Aminooctanoic acid | −2.158 | 0.008 |
| Folic acid | −2.659 | 0.007 |
| Guanine | −1.380 | 0.010 |
| N-Acetylornithine | −2.706 | 0.046 |
| Phenyllactic acid | −1.514 | 0.047 |
| Pyridoxal 5-phosphate | −2.006 | 6.05 × 10−7 |
| pyridoxine | −1.682 | 0.046 |
| Thymine | −1.959 | 0.027 |
| Sarcosine (s) | −1.121 | 0.018 |
| CDP | 2.040 | 0.003 |
| GDP | 1.496 | 0.002 |
| UDP | 1.875 | 0.001 |
| dGMP_New 1 | 1.465 | 0.015 |
| dGDP_new | 1.452 | 0.001 |
| Cholesterol sulfate | −1.412 | 0.010 |
| Hippurate | −1.911 | 0.027 |
| p-cresyl sulfate | −1.536 | 0.049 |
| Linoleyl Carnitine | 1.224 | 0.033 |
| C14 Carnitine (Myristoyl-L-carnitine) | 1.307 | 0.040 |
| Metabolites | Treatment Effect | p-Value |
|---|---|---|
| Succinyl-CoA | −1.299 | 8.87 × 10−4 |
| Fructose 6-phosphate | 2.483 | 0.008 |
| Arginine | −1.179 | 0.008 |
| Picolinic acid | 1.633 | 0.034 |
| Nicotinamide | 1.695 | 0.033 |
| D-Ribose 5-phosphate | 2.741 | 6.27 × 10−6 |
| Adenosine | 3.164 | 2.51 × 10−6 |
| ADP_new | 1.805 | 5.26 × 10−8 |
| Aminoadipic acid | 1.352 | 0.026 |
| Cytosine | 1.806 | 0.007 |
| Glucose 6-phosphate | 2.506 | 0.008 |
| GMP_New | 2.775 | 2.08 × 10−4 |
| Ornithine | 1.342 | 0.025 |
| Uracil | 3.249 | 0.001 |
| Uridine | 3.731 | 0.001 |
| Adenosine triphosphate | −1.486 | 0.004 |
| Cytidine triphosphate | −1.884 | 0.003 |
| Guanosine triphosphate | −1.819 | 0.006 |
| Uridine triphosphate | −1.394 | 0.008 |
| dATP | −1.520 | 0.001 |
| dCTP | −1.011 | 0.008 |
| dGTP | −1.389 | 0.008 |
| dTTP | −1.052 | 0.002 |
| Argininosuccinic acid | −1.162 | 0.001 |
| Sarcosine (s) | −1.266 | 0.010 |
| Dimethylglycine+M191 | 1.571 | 0.001 |
| Choline | 1.509 | 0.001 |
| Cytidine | 2.923 | 0.001 |
| CDP | 2.720 | 2.06 × 10−6 |
| GDP | 1.622 | 0.004 |
| UDP | 2.185 | 0.001 |
| dGMP_New 1 | 3.419 | 3.40 × 10−8 |
| dGDP_new | 1.811 | 2.91 × 10−6 |
| D-Sedoheptulose 7-phosphate | 1.285 | 0.008 |
| N-Acetyl-glucosamine 1-phosphate | 1.210 | 0.008 |
| Citicoline | 3.320 | 0.005 |
| Adenine | 1.726 | 0.001 |
| Spermidine | 2.009 | 0.013 |
| C16:0 Carnitine (Palmitoyl-L carnitine) | 3.023 | 0.013 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mpilla, G.; Aboukameel, A.; Muqbil, I.; Kim, S.; Beydoun, R.; Philip, P.A.; Mohammad, R.M.; Kamgar, M.; Shidham, V.; Senapedis, W.; et al. PAK4-NAMPT Dual Inhibition as a Novel Strategy for Therapy Resistant Pancreatic Neuroendocrine Tumors. Cancers 2019, 11, 1902. https://doi.org/10.3390/cancers11121902
Mpilla G, Aboukameel A, Muqbil I, Kim S, Beydoun R, Philip PA, Mohammad RM, Kamgar M, Shidham V, Senapedis W, et al. PAK4-NAMPT Dual Inhibition as a Novel Strategy for Therapy Resistant Pancreatic Neuroendocrine Tumors. Cancers. 2019; 11(12):1902. https://doi.org/10.3390/cancers11121902
Chicago/Turabian StyleMpilla, Gabriel, Amro Aboukameel, Irfana Muqbil, Steve Kim, Rafic Beydoun, Philip A. Philip, Ramzi M. Mohammad, Mandana Kamgar, Vinod Shidham, William Senapedis, and et al. 2019. "PAK4-NAMPT Dual Inhibition as a Novel Strategy for Therapy Resistant Pancreatic Neuroendocrine Tumors" Cancers 11, no. 12: 1902. https://doi.org/10.3390/cancers11121902
APA StyleMpilla, G., Aboukameel, A., Muqbil, I., Kim, S., Beydoun, R., Philip, P. A., Mohammad, R. M., Kamgar, M., Shidham, V., Senapedis, W., Baloglu, E., Li, J., Dyson, G., Xue, Y., El-Rayes, B., & Azmi, A. S. (2019). PAK4-NAMPT Dual Inhibition as a Novel Strategy for Therapy Resistant Pancreatic Neuroendocrine Tumors. Cancers, 11(12), 1902. https://doi.org/10.3390/cancers11121902