The Landscape of Long Non-Coding RNA Dysregulation and Clinical Relevance in Muscle Invasive Bladder Urothelial Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
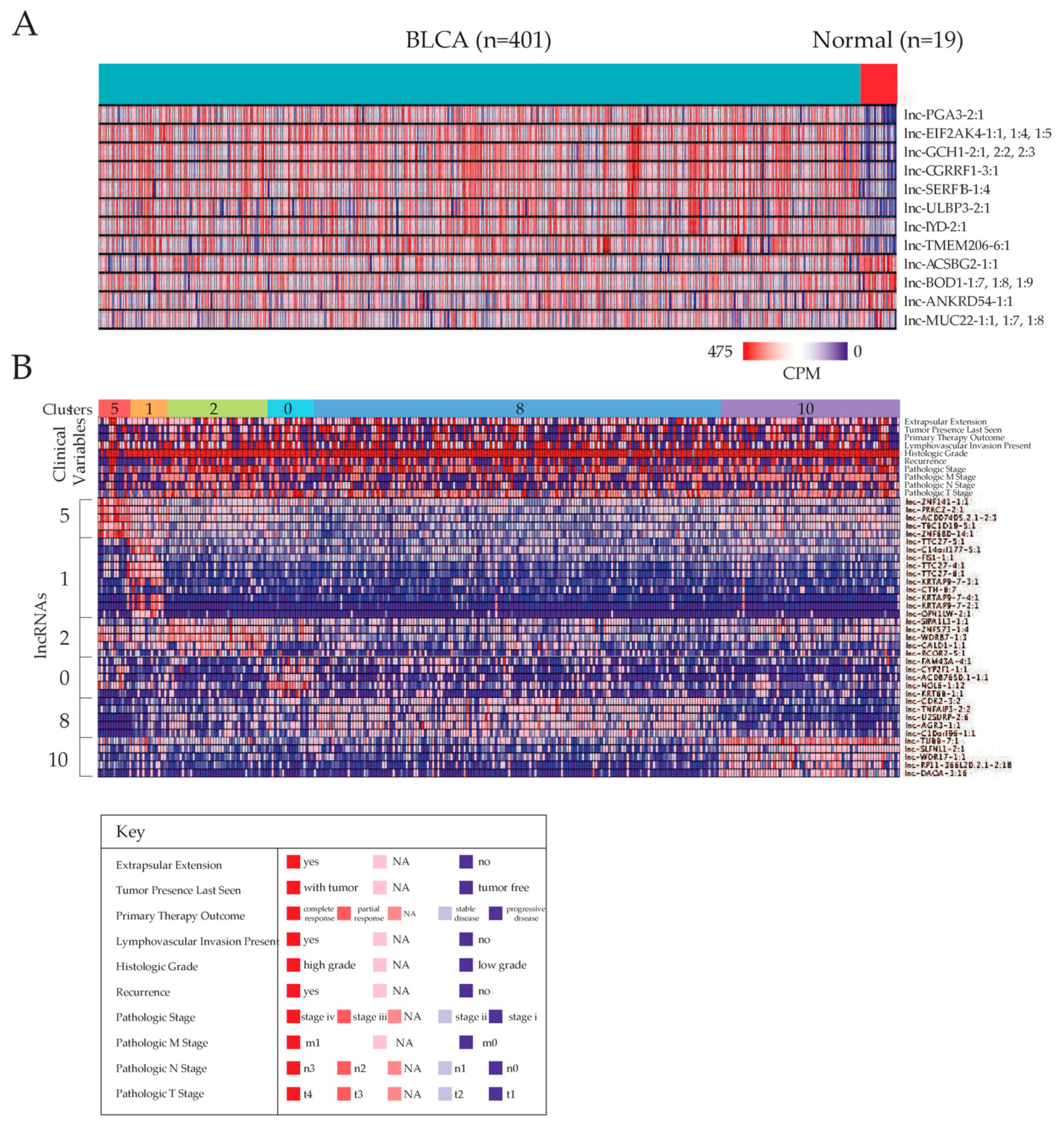
2.1. Identifying Dysregulated lncRNAs in Bladder Carcinoma
2.2. Stratification of Patients Based on lncRNA Expression Landscapes
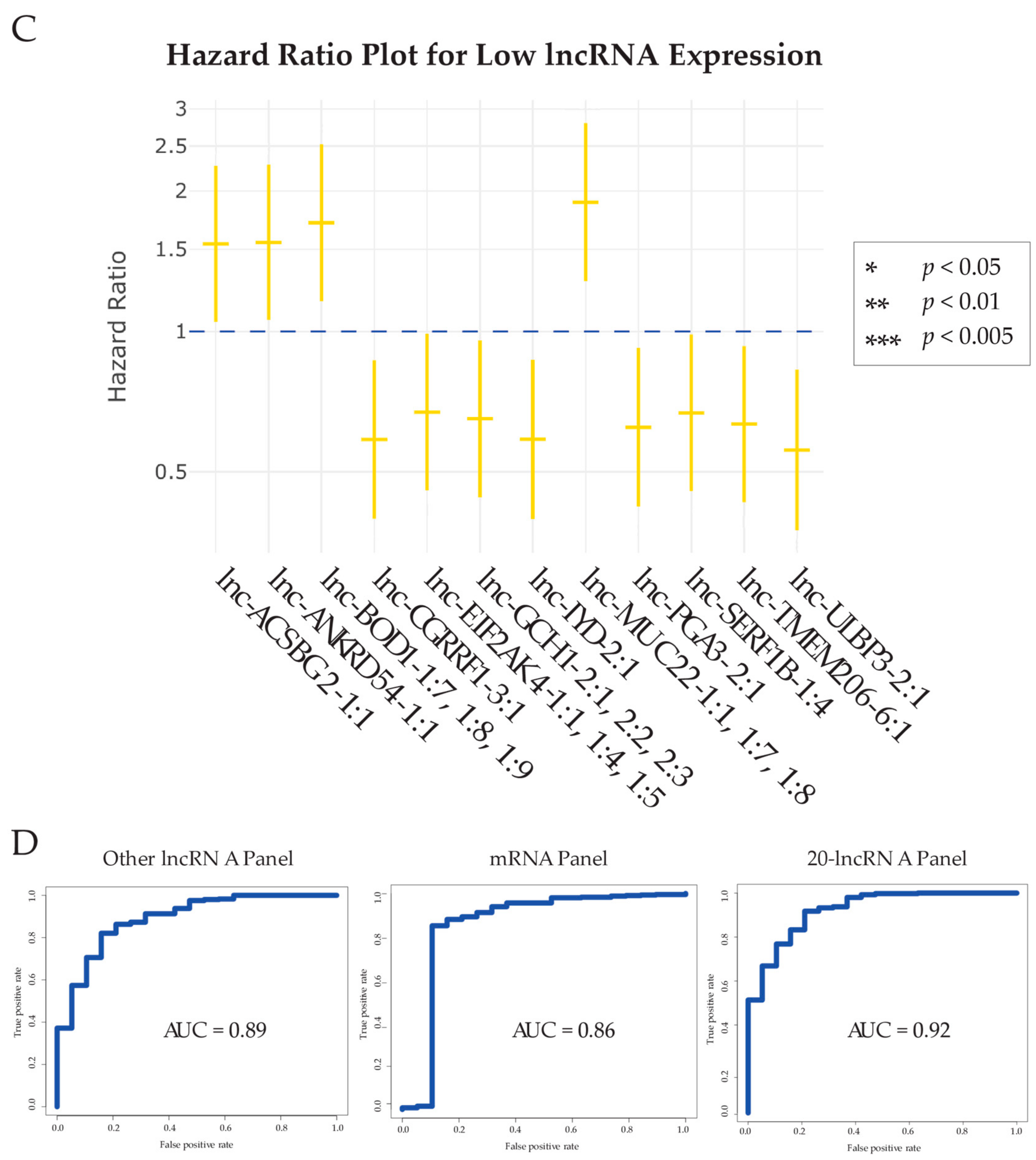
2.3. Analyzing Association between lncRNA Dysregulation and Bladder Carcinoma Patient Survival
2.4. Comparison of the Diagnostic Power of our lncRNA Panel with Other Expression Panels
2.5. Correlating Dysregulated lncRNA Expression Levels with Clinical Variables
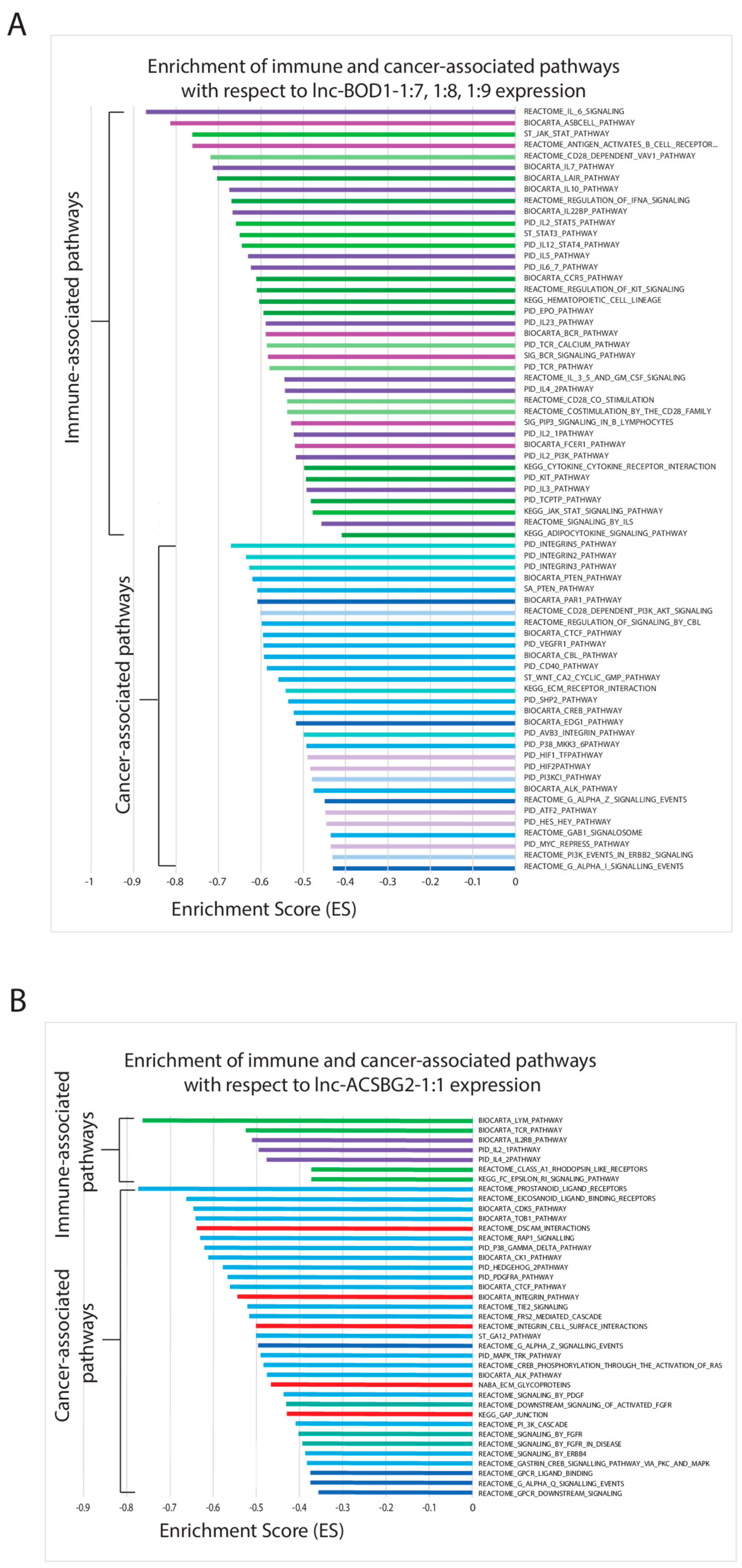
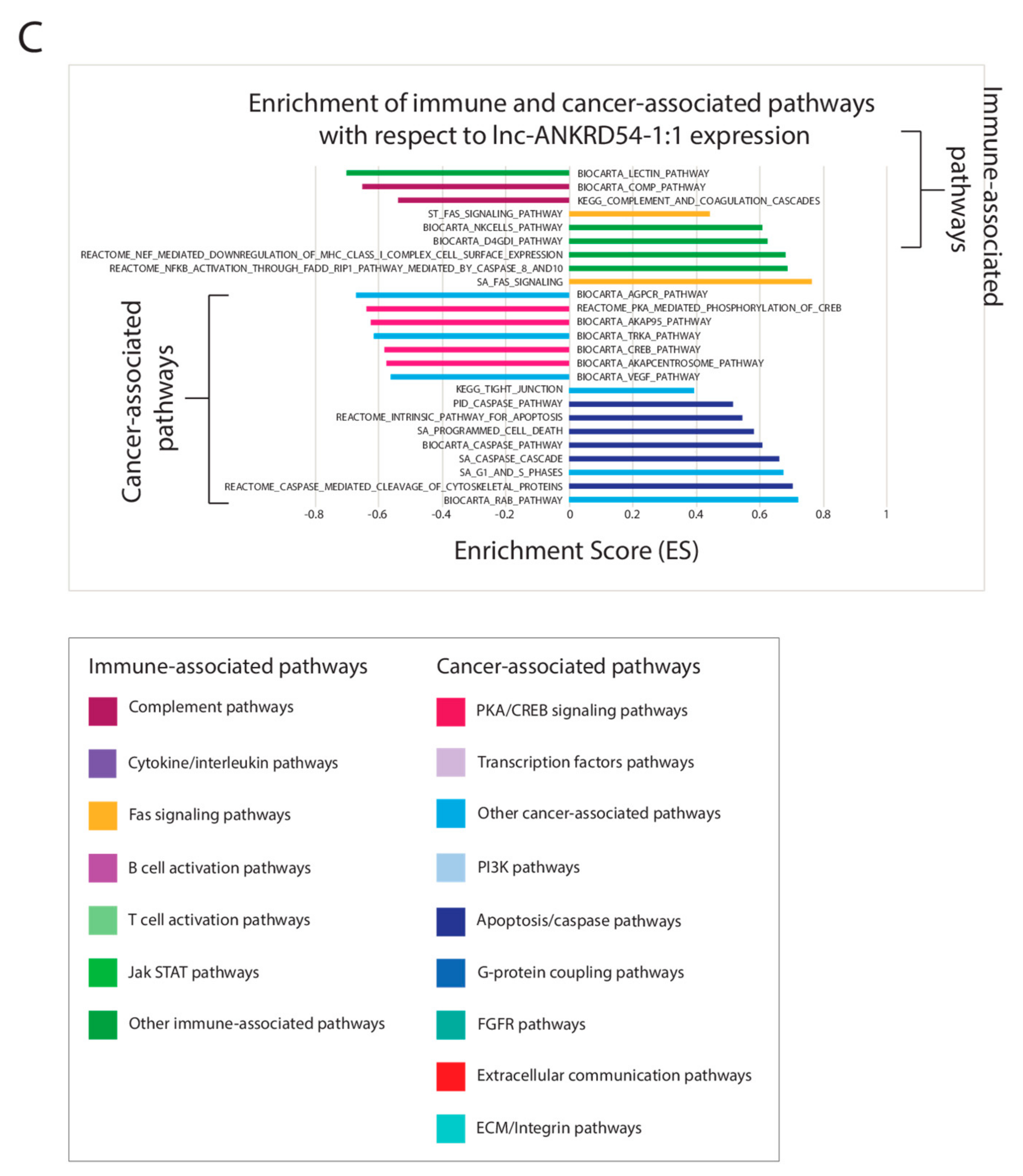
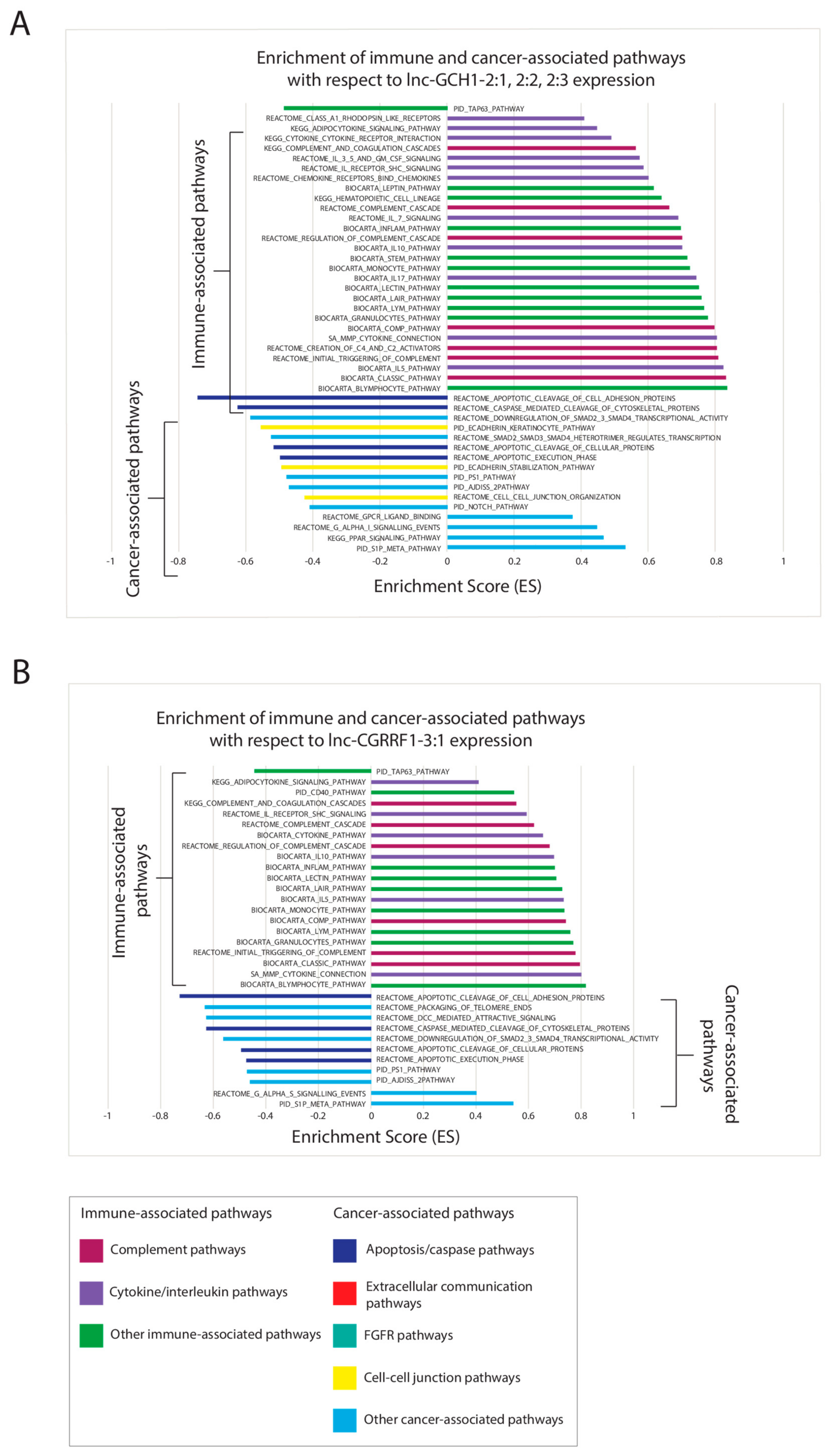
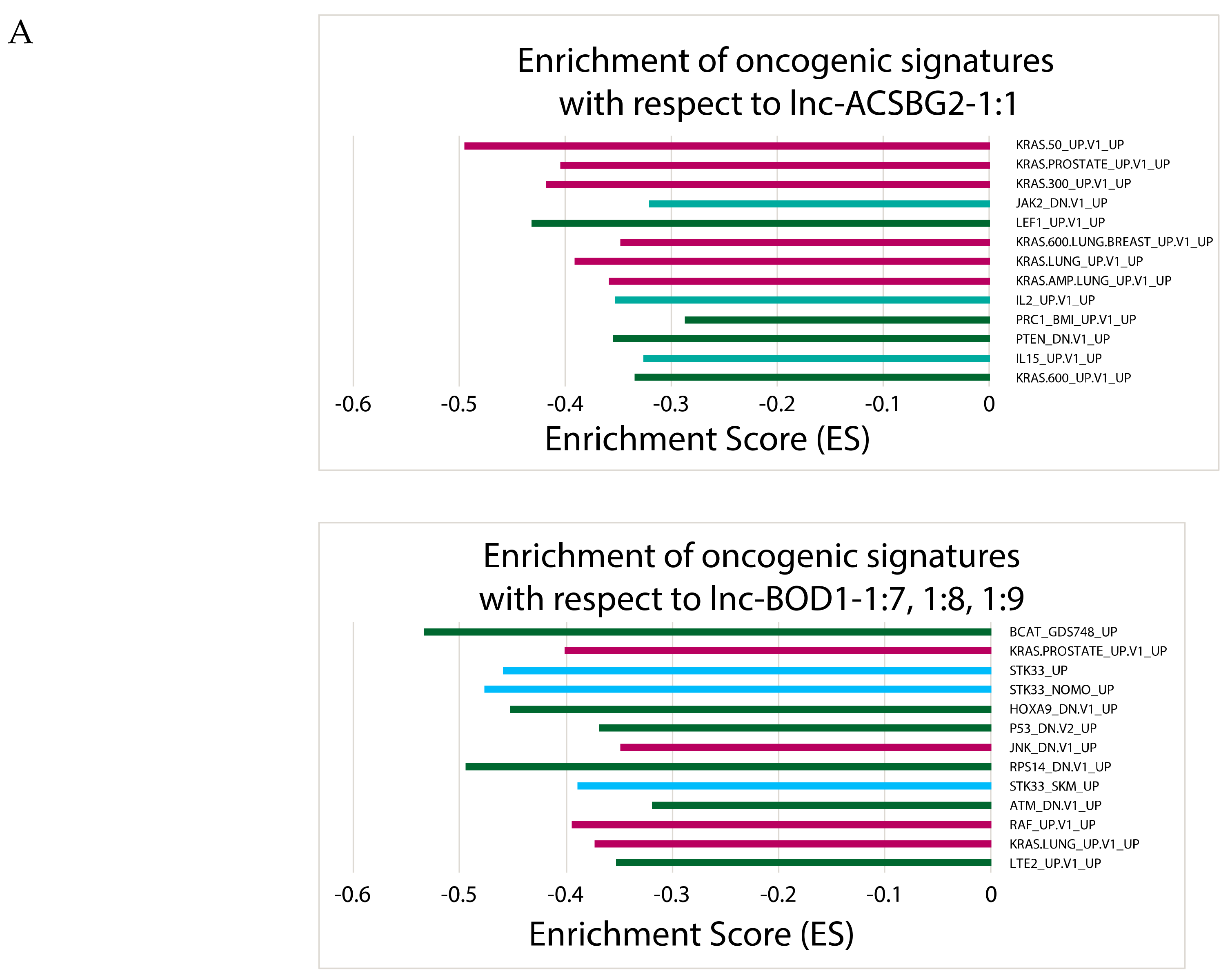
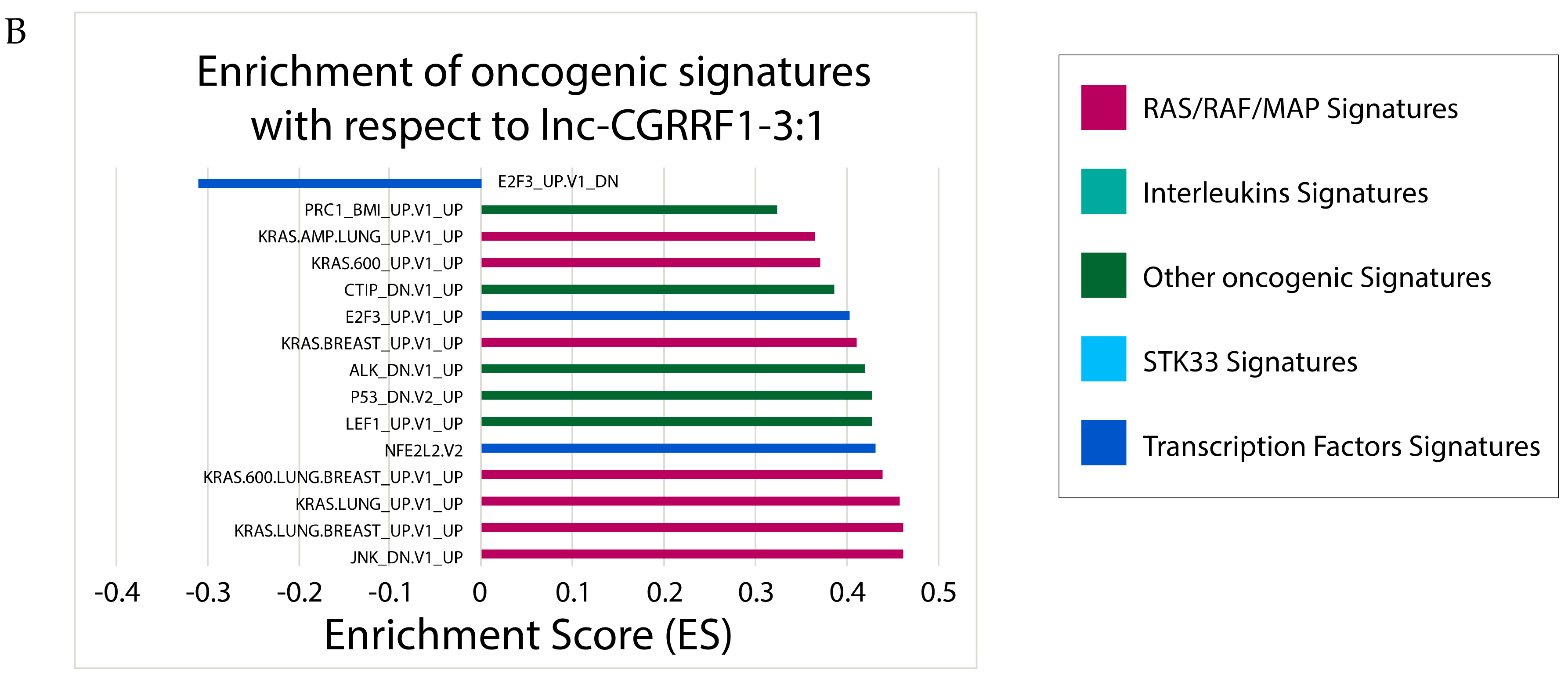
2.6. Correlating Expressions of Key lncRNAs to Cancer-Associated Gene Sets
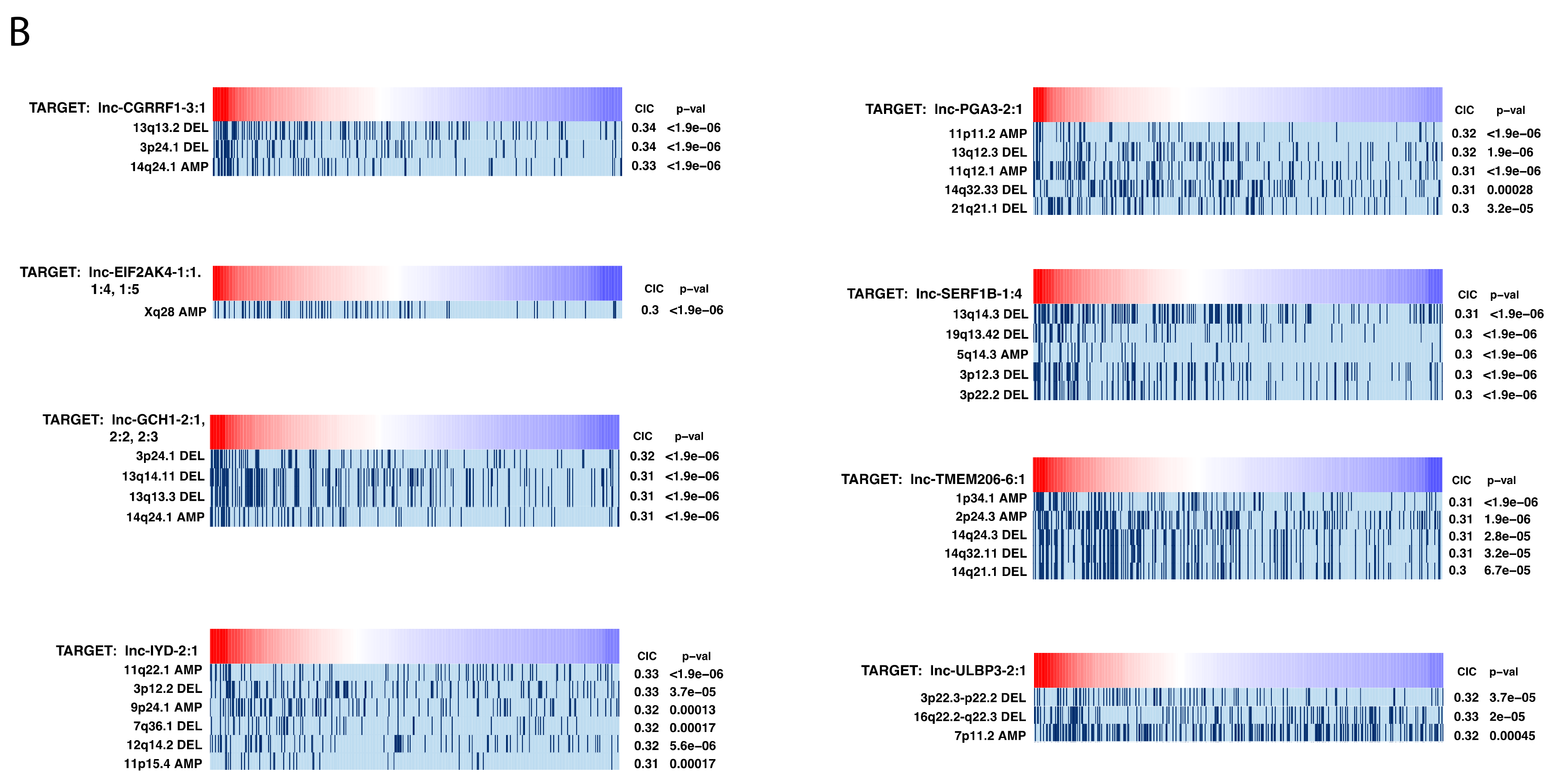
2.7. Investigating Genomic Alterations’ Association with lncRNA Expression
2.8. Investigating the Potential Role of lncRNAs as Inhibitors of MicroRNAs (MiRNAs)
3. Discussion
4. Materials and Methods
4.1. lncRNA-Sequencing Datasets and Clinical Data
4.2. lncRNA Differential Expression Analyses
4.3. Clustering of Patients Using lncRNA Expression Profiles
4.4. Receiver Operating Characteristic Curve Generation for the Diagnostic Abilities of Expression Panels
4.5. Association of Candidate lncRNAs with Patient Survival and Clinical Variables
4.6. Identification of Gene Sets Associated with Clinically Significant lncRNA Expression
4.7. Information Coefficient-Based Correlation of lncRNA Expression with Genomic Alterations
4.8. LncBase Prediction of MiRNA–LncRNA Interaction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA: Cancer J. Clin. 2019, 69, 7–34. [Google Scholar]
- Hussain, M.H.A.; MacVicar, G.R.; Etrylak, D.P.; Vaishampayan, U.; Lara, P.N., Jr.; Chatta, G.S.; Nanus, D.M.; Glode, L.M.; Trump, D.L.; Chen, H.; et al. Trastuzumab, Paclitaxel, Carboplatin, and Gemcitabine in Advanced Human Epidermal Growth Factor Receptor-2/neu-Positive Urothelial Carcinoma: Results of a Multicenter Phase II National Cancer Institute Trial. J. Clin. Oncol. 2007, 25, 2218–2224. [Google Scholar]
- Ferre, F.; Colantoni, A.; Helmer-Citterich, M. Revealing Protein-lncRNA Interaction. Brief. Bioinform. 2016, 17, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Kraus, W.L. From discovery to function: The expanding roles of long noncoding RNAs in Physiology and Disease. Endocr. Rev. 2015, 36, 25–64. [Google Scholar] [CrossRef]
- Taheri, M.; Omrani, M.D.; Ghafouri-Fard, S. Long Non-Coding RNA Expression in Bladder Cancer. Biophys. Rev. 2018, 10, 1205–1213. [Google Scholar]
- Zur Bruegge, J.; Einspanier, R.; Sharbati, S. A Long Journey Ahead: Long Non-coding RNAs in Bacterial Infections. Front. Cell. Infect. Microbiol. 2017, 7, 95. [Google Scholar] [CrossRef]
- Atala, A. Re: Profiling of Long Non-Coding RNAs Identifies LINC00958 and LINC01296 as Candidate Oncogenes in Bladder Cancer. J. Urol. 2017, 198, 983–985. [Google Scholar] [CrossRef]
- Cao, X.; Xu, J.; Yue, D. LncRNA-SNHG16 Predicts Poor Prognosis and Promotes Tumor Proliferation through Epigenetically Silencing p21 in Bladder Cancer. Cancer Gene Ther. 2018, 25, 10–17. [Google Scholar] [CrossRef]
- Liao, X.; Chen, J.; Liu, Y.; He, A.; Chen, J.; Zhang, X.; Lv, Z.; Wang, F.; Mei, H. Knockdown of Long Noncoding RNA FGFR3- AS1 Induces Cell Proliferation Inhibition, Apoptosis and Motility Reduction in Bladder Cancer. Cancer Biomark. 2018, 21, 277–285. [Google Scholar] [CrossRef]
- Wang, H.; Niu, L.; Jiang, S.; Zhai, J.; Wang, P.; Kong, F.; Jin, X. Comprehensive Analysis of Aberrantly Expressed Profiles of lncRNAs and miRNAs with Associated ceRNA Network in Muscle-Invasive Bladder Cancer. Oncotarget 2016, 7, 86174–86185. [Google Scholar] [CrossRef]
- Seitz, A.K.; Christensen, L.L.; Christensen, E.; Faarkrog, K.; Ostenfeld, M.S.; Hedegaard, J.; Nordentoft, I.; Nielsen, M.M.; Palmfeldt, J.; Thomson, M.; et al. Profiling of Long Non-Coding RNAs Identifies LINC00958 and LINC01296 as Candidate Oncogenes in Bladder Cancer. Sci. Rep. 2017, 7, 395. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, M.D.; Vlachos, I.S.; Karagkouni, D.; Georgakilas, G.; Kanellos, I.; Vergoulis, T.; Zagganas, K.; Tsanakas, P.; Floros, E.; Dalamagas, T.; et al. DIANA-LncBase v2: Indexing microRNA Targets on Non-Coding Transcripts. Nucleic. Acids Res. 2016, 44, D231–D238. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Botvinnik, O.B.; Abudayyeh, O.; Birger, C.; Rosenbluh, J.; Shrestha, Y.; Abazeed, M.E.; Hammerman, P.S.; DiCara, D.; Konieczkowski, D.J.; et al. Characterizing Genomic Alterations in Cancer by Complementary Functional Associations. Nat. Biotechnol. 2016, 34, 539–546. [Google Scholar]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic Dysfunction across all Human Cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Anaya, J. OncoLnc: Linking TCGA Survival Data to mRNAs, miRNAs, and lncRNAs. PeerJ. Computer Science 2016. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Hatzigeorgiou, A.G. Analyzing MiRNA-LncRNA Interactions. Methods Mol. Biol 2016, 1402, 271–286. [Google Scholar]
- Lin, C.; Yang, L. Long Noncoding RNA in Cancer: Wiring Signaling Circuitry. Trends Cell Biol. 2018, 28, 287–301. [Google Scholar] [CrossRef]
- Wen, L.; Zhang, X.; Bian, J.; Han, L.; Huang, H.; He, M.; Wei, M.; Wang, P. The Long Non-Coding RNA LINC00460 Predicts the Prognosis and Promotes the Proliferation and Migration of Cells in Bladder Urothelial Carcinoma. Oncol. Lett. 2019, 17, 3874–3880. [Google Scholar] [CrossRef]
- Volders, P.-J.; Anckaert, J.; Verheggen, K.; Nuytens, K.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a Reference Set of Human Long Non-Coding RNAs. Nucleic. Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef]
- Avgeris, M.; Tsilimantou, A.; Levis, P.K.; Rampias, T.; Papadimitriou, M.-A.; Panoutsopoulou, K.; Stravodimos, K.; Scorilas, A. Unraveling UCA1 lncRNA Prognostic Utility in Urothelial Bladder Cancer. Carcinogenesis 2019. [Google Scholar] [CrossRef]
- Jiang, B.; Su, H.; Yuan, J.; Zhao, H.; Xia, W.; Zha, Z.; Wu, B.; Liu, Z. Identification of Oncogenic Long Noncoding RNA SNHG12 and DUXAP8 in Human Bladder Cancer through a Comprehensive Profiling Analysis. Biomed. Pharmacother. 2018, 108, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Jiang, F.; Görög, J.-P.; Sartorius, G.; Egenter, C.; Gasser, T.C.; Moch, H.; Mihatsch, M.j.; Sauter, g. Marked Genetic Differences between Stage pTa and Stage pT1 Papillary Bladder Cancer Detected by Comparative Genomic Hybridization. Cancer Res. 1997, 57, 2860–2864. [Google Scholar] [PubMed]
- Qin, S.-L.; Chen, X.-J.; Xu, X.; Shou, J.-Z.; Bi, X.-G.; Ji, L.; Han, Y.-L.; Cai, Y.; Wei, F.; Ma, J.-H.; et al. Detection of Chromosomal Alterations in Bladder Transitional Cell Carcinomas from Northern China by Comparative Genomic Hybridization. Cancer Lett. 2006, 238, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.Y.-H.; Cairns, P.; Schoenberg, M.P.; Polascik, T.J.; Sidransky, D. Novel Suppressor Loci on Chromosome 14q in Primary Bladder Cancer. Cancer Res. 1995, 55, 3246–3249. [Google Scholar] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Song, C.; Jin, S.; Chen, Z.; Ni, M.; Han, L.; Wu, J.; Jin, Y. Gene Set Enrichment Analysis: A Genome-Wide Expression Profile-Based Strategy for Discovering Functional microRNA-Disease Relationships. J. Int. Med. Res. 2018, 46, 596–611. [Google Scholar] [CrossRef]
- Li, W.T.; Zheng, H.; Nguyen, V.; Wang-Rodriguez, J.; Ongkeko, W.M. Functional Genomics Profiling of Bladder Urothelial Carcinoma MicroRNAome as a Potential Biomarker. Neoplasia 2018, 20, 364–373. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, H.; Wong, L.M.; Li, W.T.; Chu, M.; High, R.A.; Chang, E.Y.; Wang-Rodriguez, J.; Ongkeko, W.M. The Landscape of Long Non-Coding RNA Dysregulation and Clinical Relevance in Muscle Invasive Bladder Urothelial Carcinoma. Cancers 2019, 11, 1919. https://doi.org/10.3390/cancers11121919
Shen H, Wong LM, Li WT, Chu M, High RA, Chang EY, Wang-Rodriguez J, Ongkeko WM. The Landscape of Long Non-Coding RNA Dysregulation and Clinical Relevance in Muscle Invasive Bladder Urothelial Carcinoma. Cancers. 2019; 11(12):1919. https://doi.org/10.3390/cancers11121919
Chicago/Turabian StyleShen, Haotian, Lindsay M. Wong, Wei Tse Li, Megan Chu, Rachel A. High, Eric Y. Chang, Jessica Wang-Rodriguez, and Weg M. Ongkeko. 2019. "The Landscape of Long Non-Coding RNA Dysregulation and Clinical Relevance in Muscle Invasive Bladder Urothelial Carcinoma" Cancers 11, no. 12: 1919. https://doi.org/10.3390/cancers11121919
APA StyleShen, H., Wong, L. M., Li, W. T., Chu, M., High, R. A., Chang, E. Y., Wang-Rodriguez, J., & Ongkeko, W. M. (2019). The Landscape of Long Non-Coding RNA Dysregulation and Clinical Relevance in Muscle Invasive Bladder Urothelial Carcinoma. Cancers, 11(12), 1919. https://doi.org/10.3390/cancers11121919