The Adipose Stem Cell as a Novel Metabolic Actor in Adrenocortical Carcinoma Progression: Evidence from an In Vitro Tumor Microenvironment Crosstalk Model


 ,
, 
Abstract
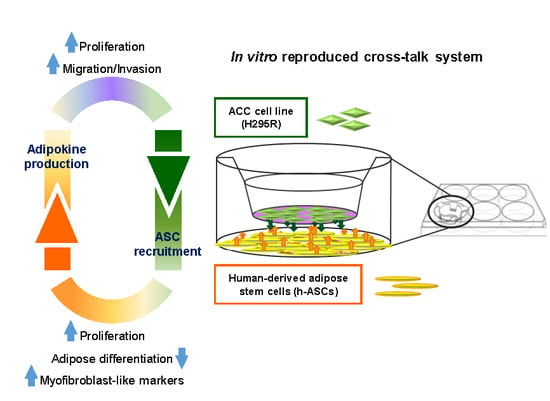
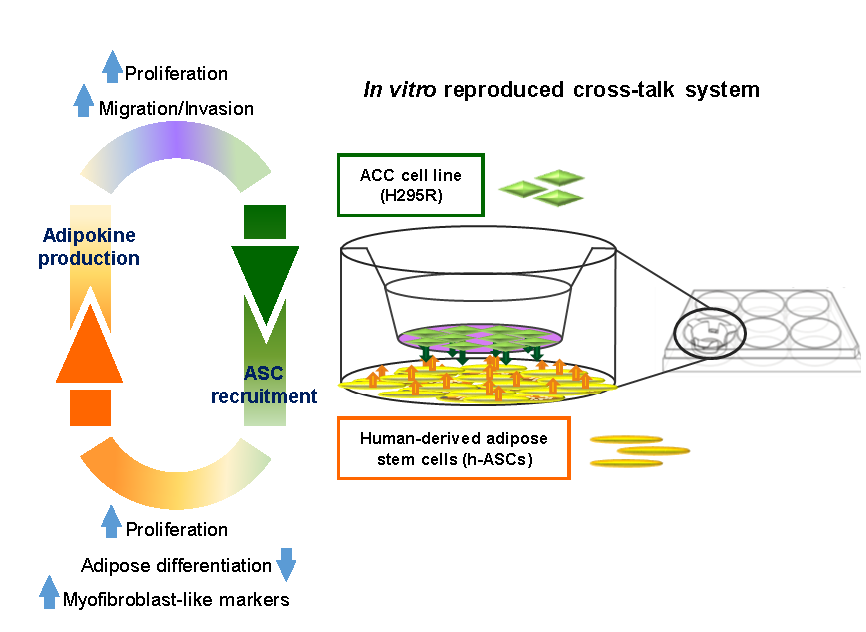
:
1. Introduction
2. Results
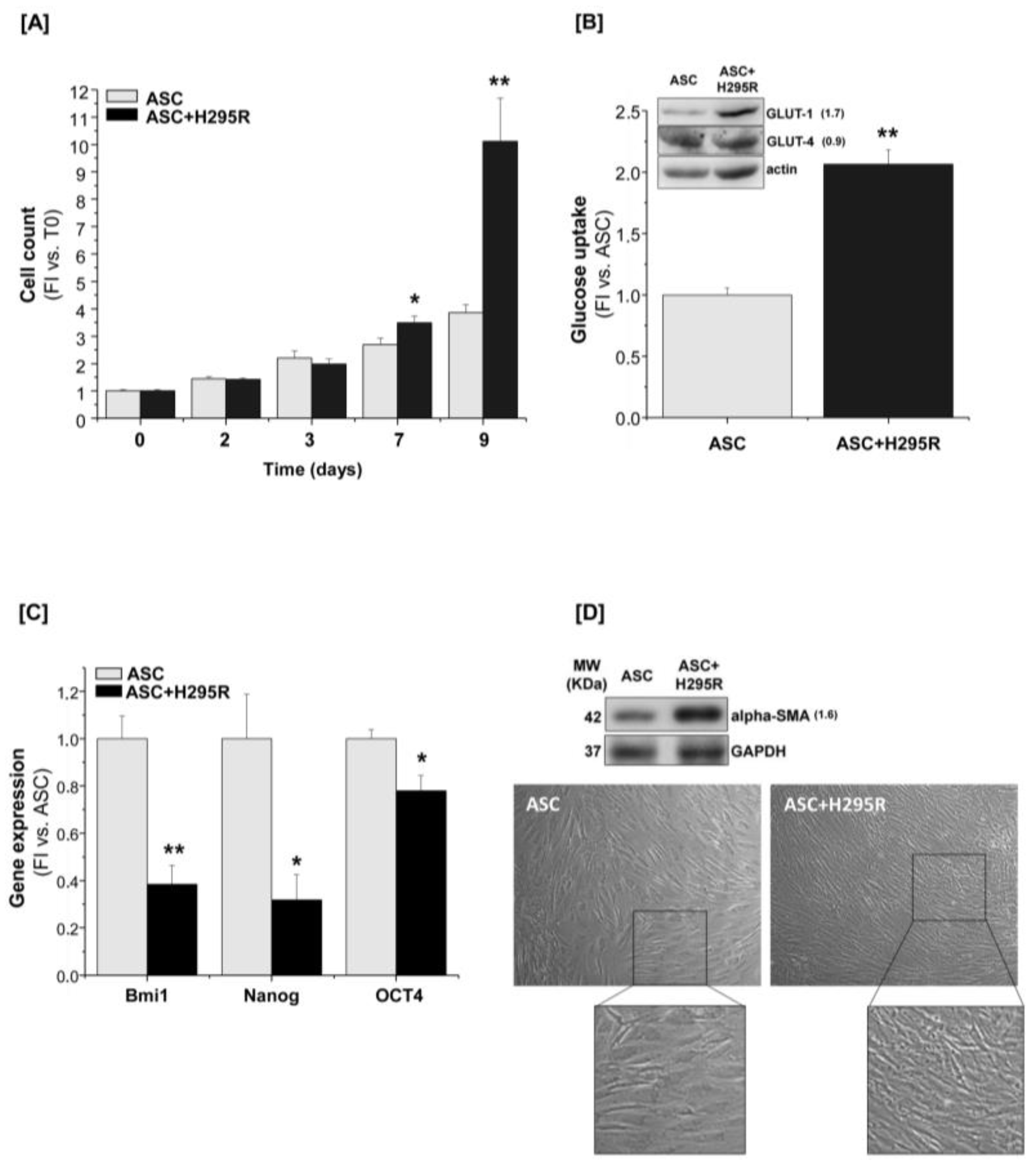
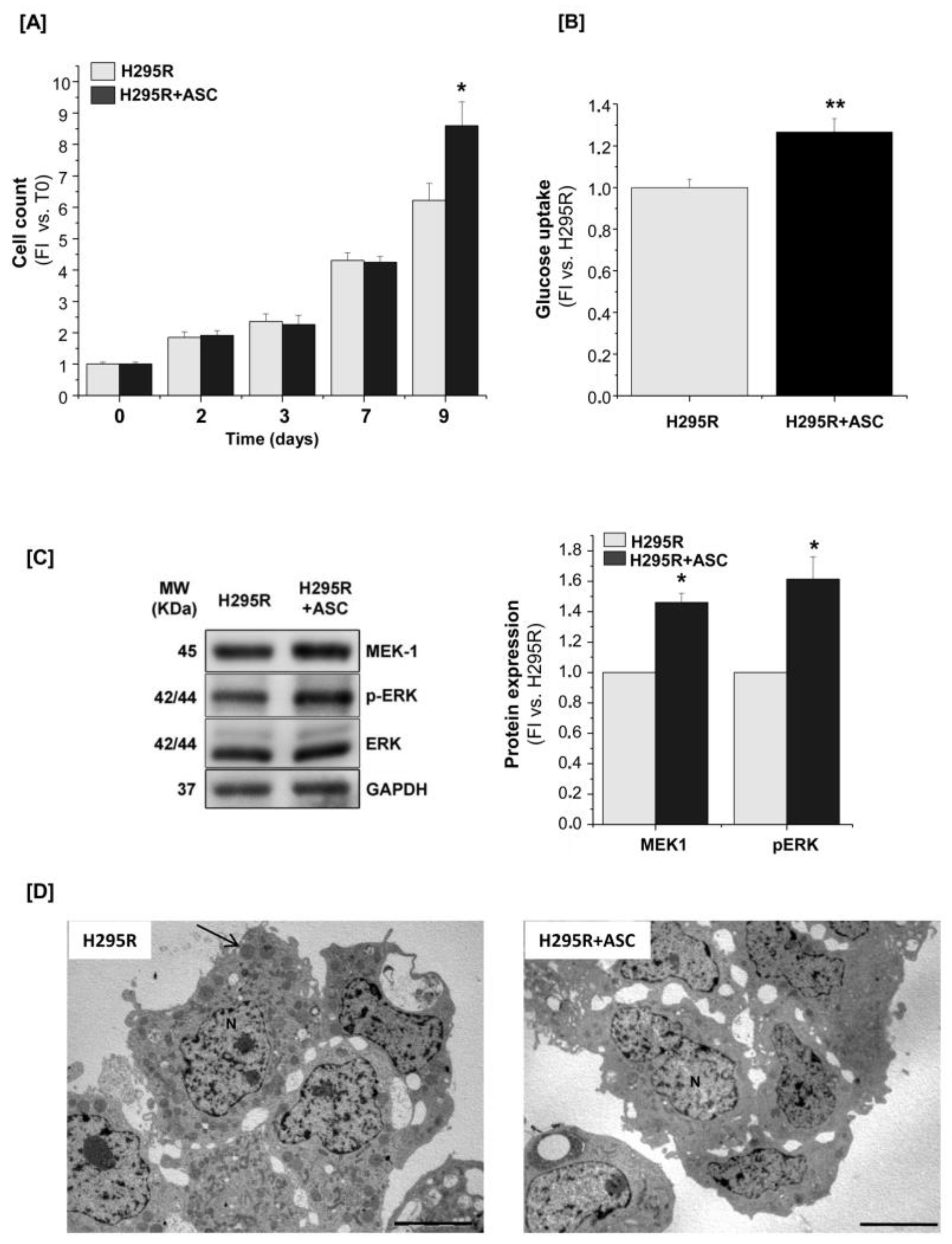
2.1. Proliferative, Metabolic and Morphological Changes in ASCs Co-Cultured with H295R Cells
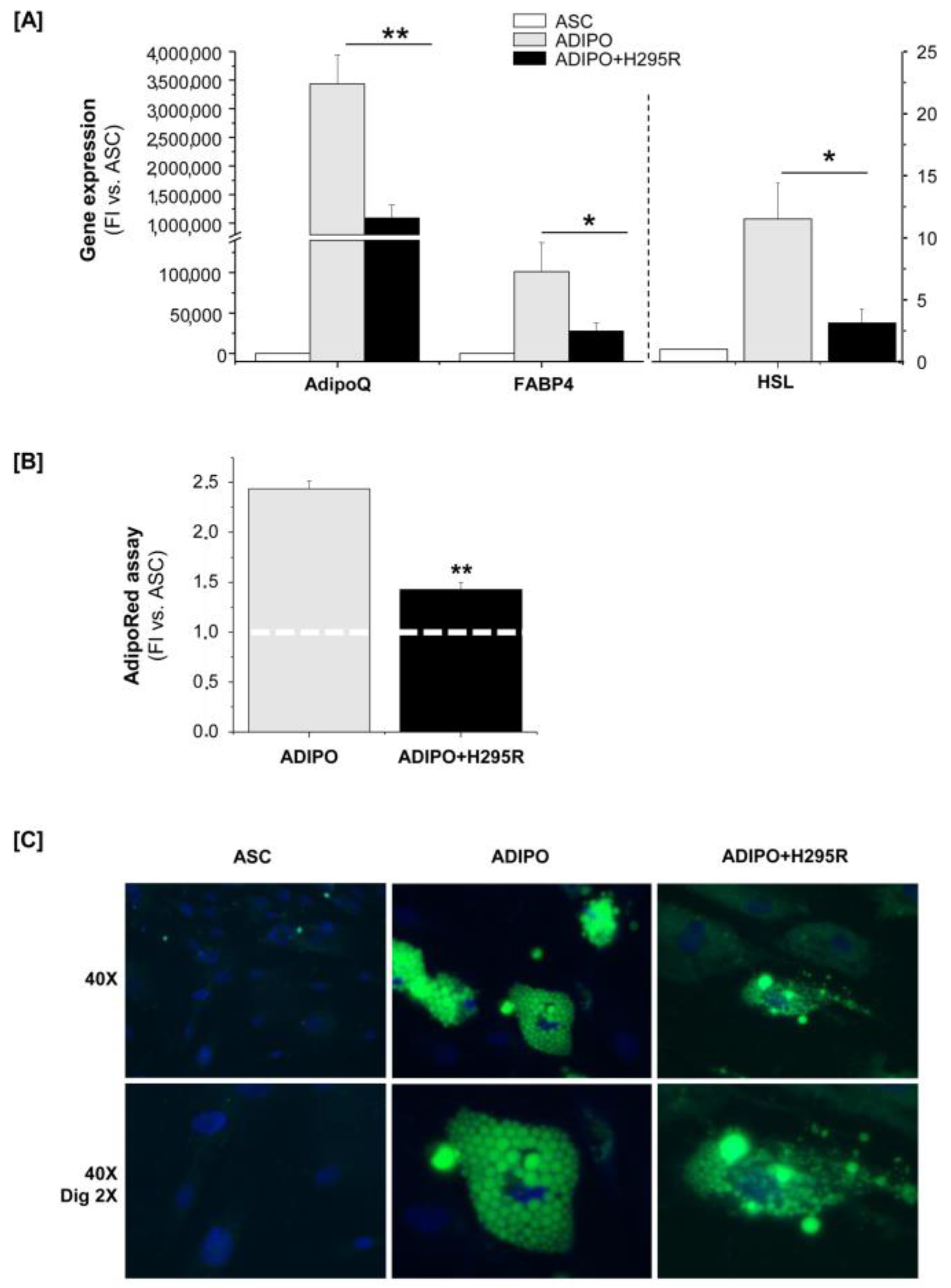
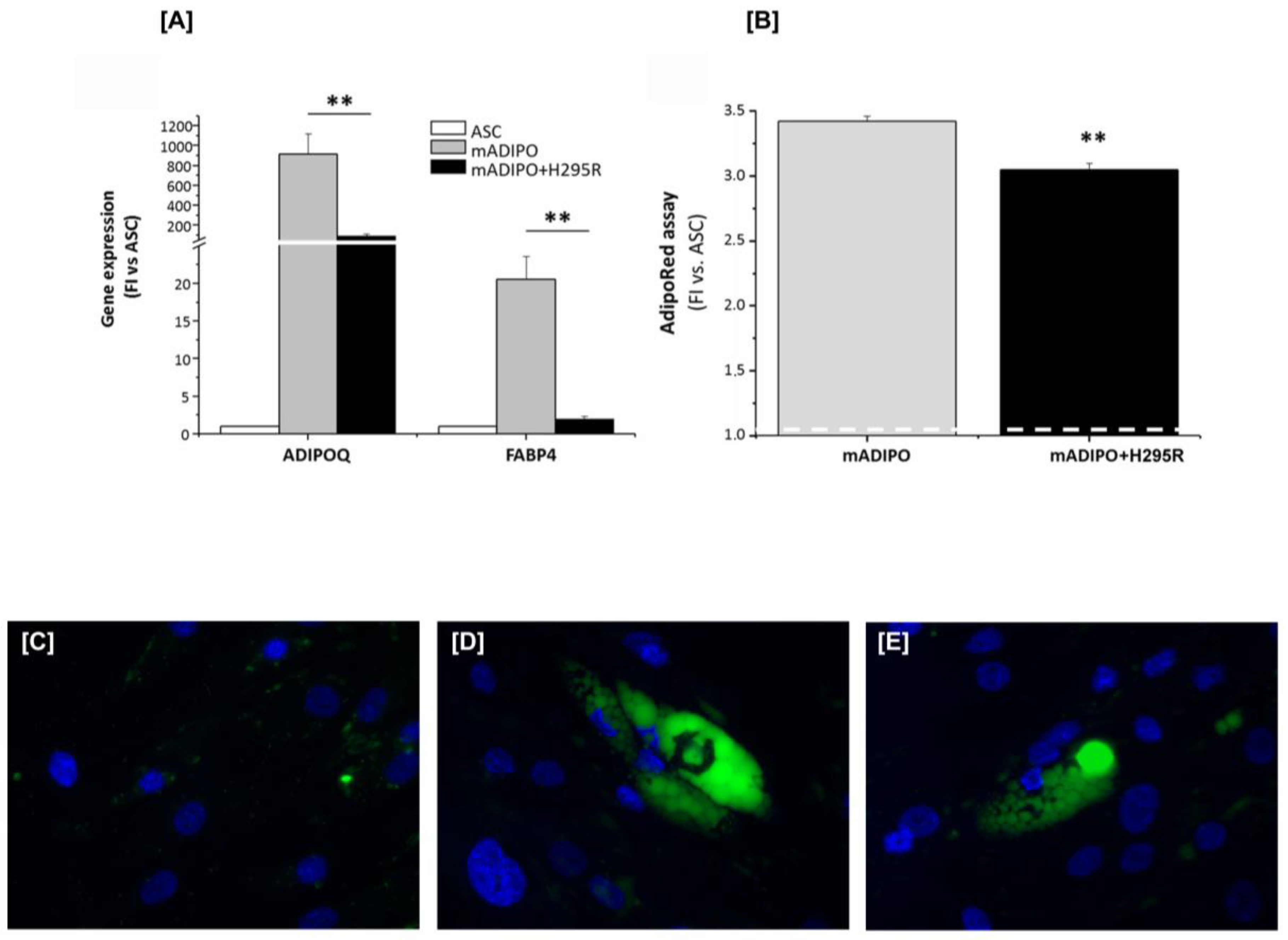
2.2. H295R Cells Hamper ASC Ability to Differentiate toward Mature White Adipocytes
2.3. H295R Cells Increase Proliferation and Glucose Metabolism When Co-Cultured with ASCs
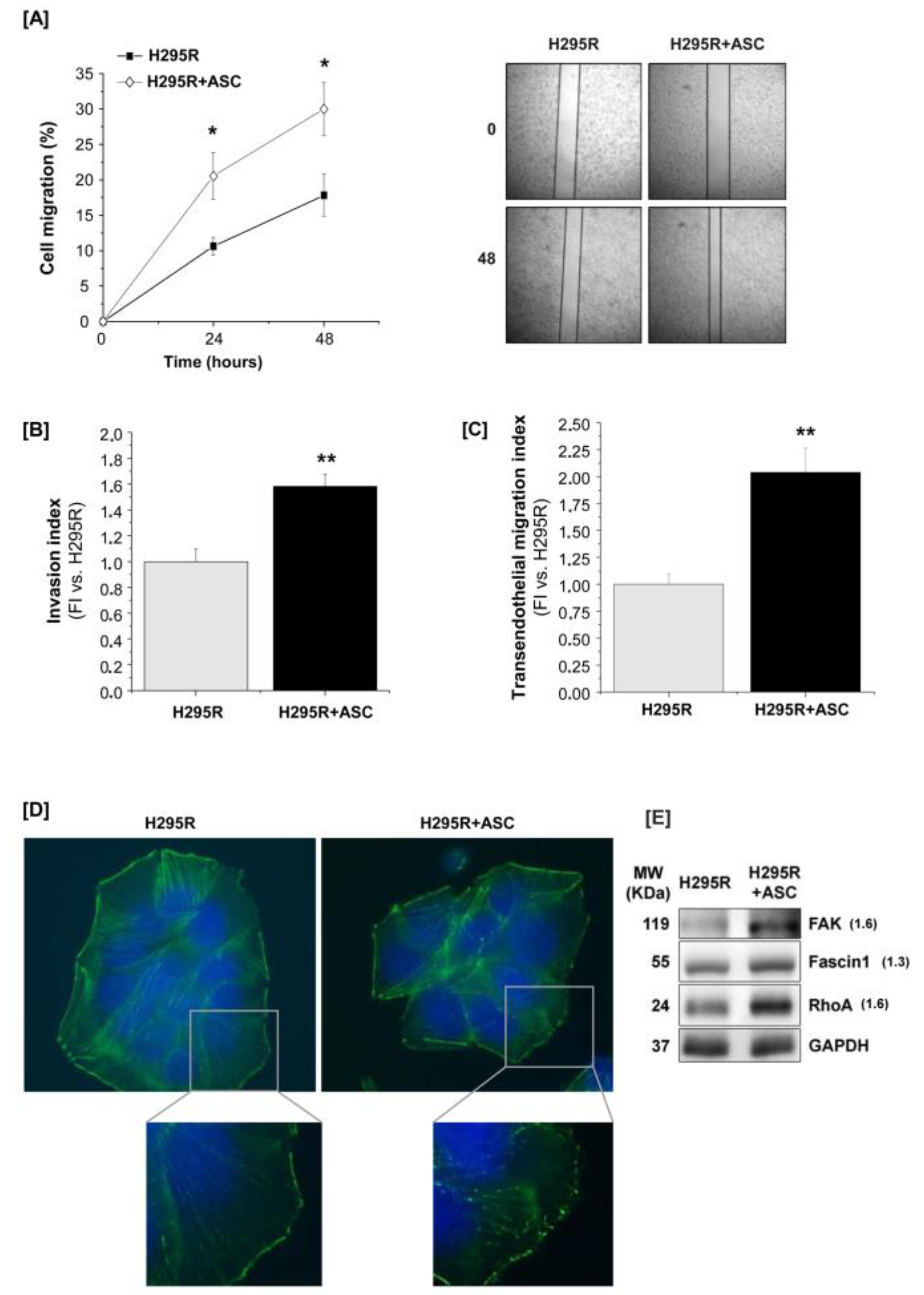
2.4. H295R Increase Migration and Invasive Ability after Co-Culture with ASCs
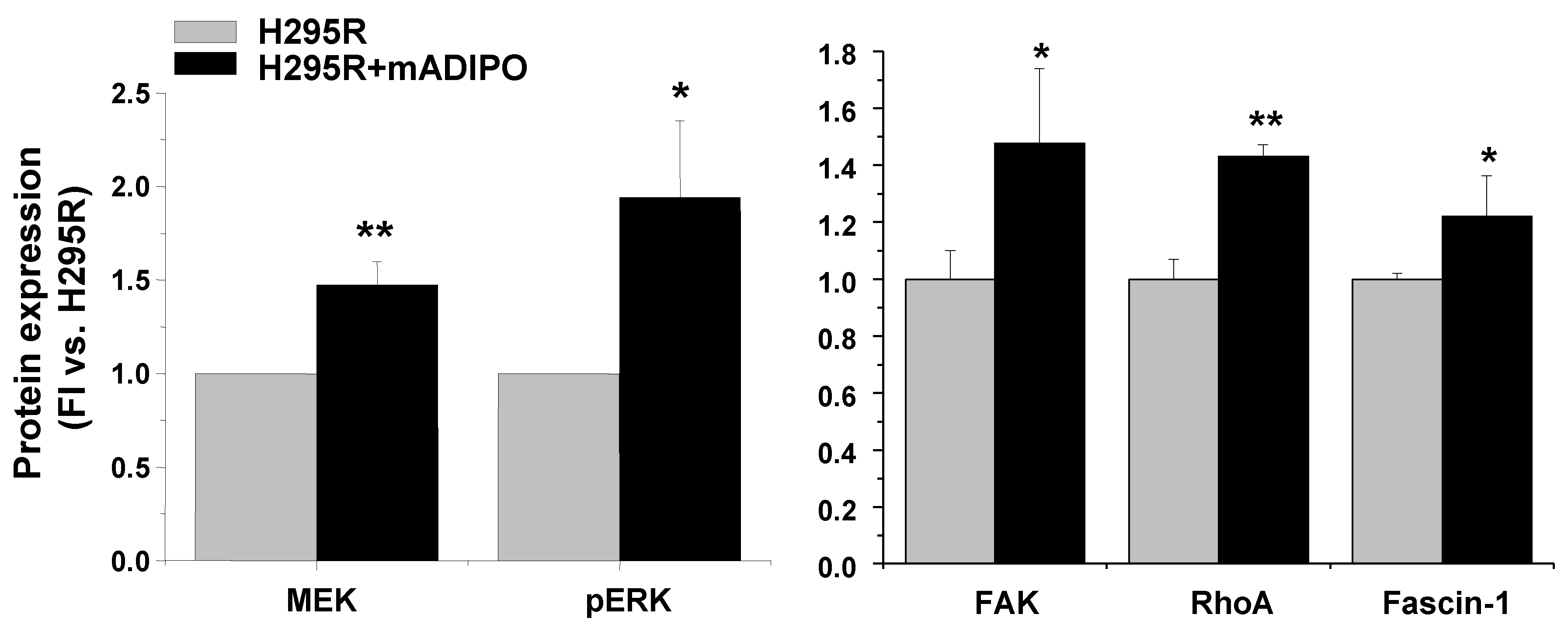
2.5. Molecular Mechanisms Activated by ASCs/H295R Cell Crosstalk
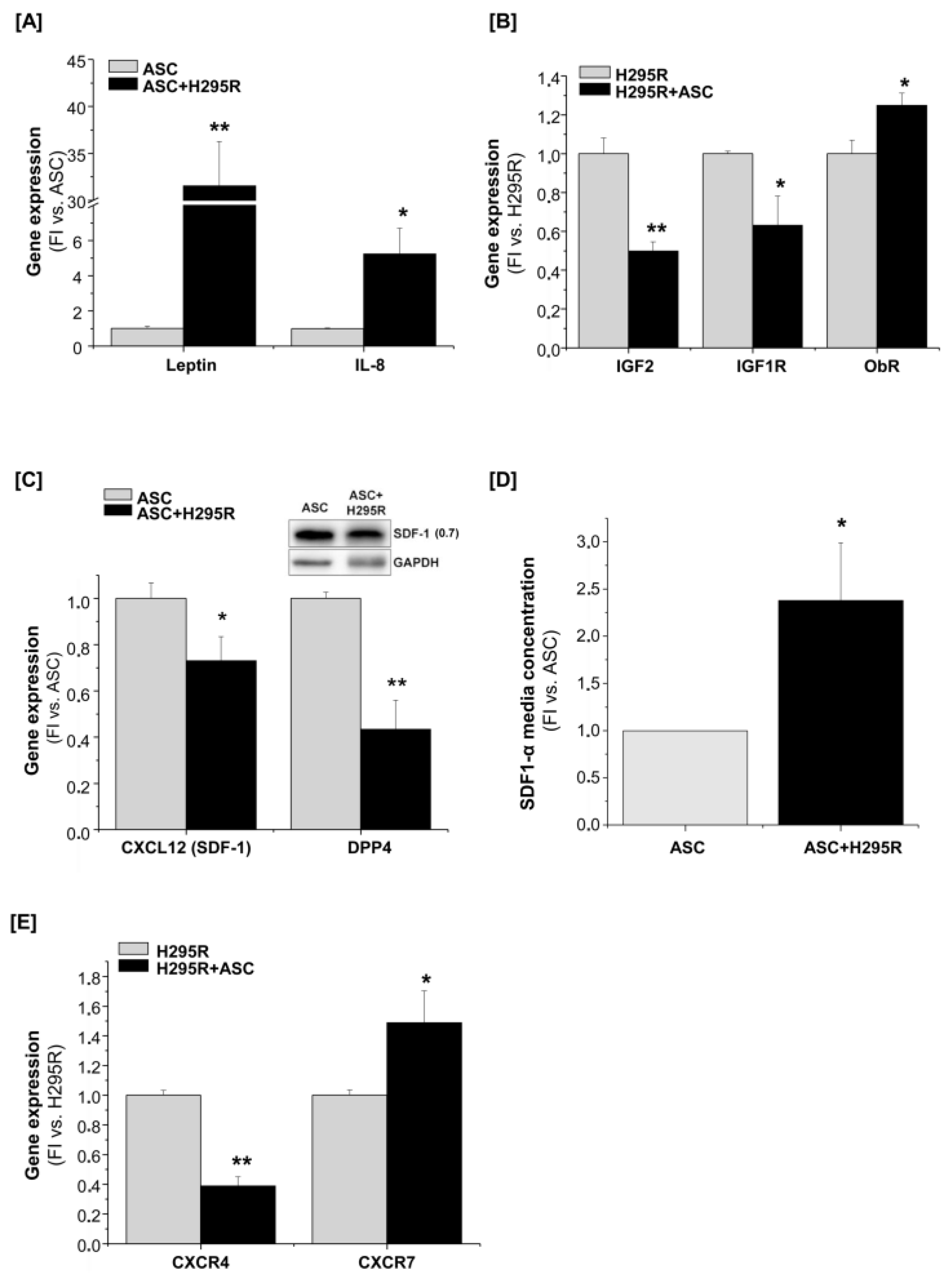
2.6. A Reciprocal Interaction Is Also Established between H295R Cells and Mature Adipocytes
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Histology of ACC
4.3. Cell Culture
4.4. Co-Culture Experiments
4.4.1. H295R-ASCs Co-Culture
4.4.2. H295R Co-Culture with ASCs during In-Vitro Induced Adipogenesis
4.4.3. H295R-mADIPO Co-Culture
4.5. Cell Count
4.6. Glucose Uptake Measurement
4.7. SDS-PAGE and Western Blot Analysis
4.8. mRNA Isolation and Quantitative Real-Time RT-PCR (RT-qPCR)
4.9. Glucose and Lactate Measurements
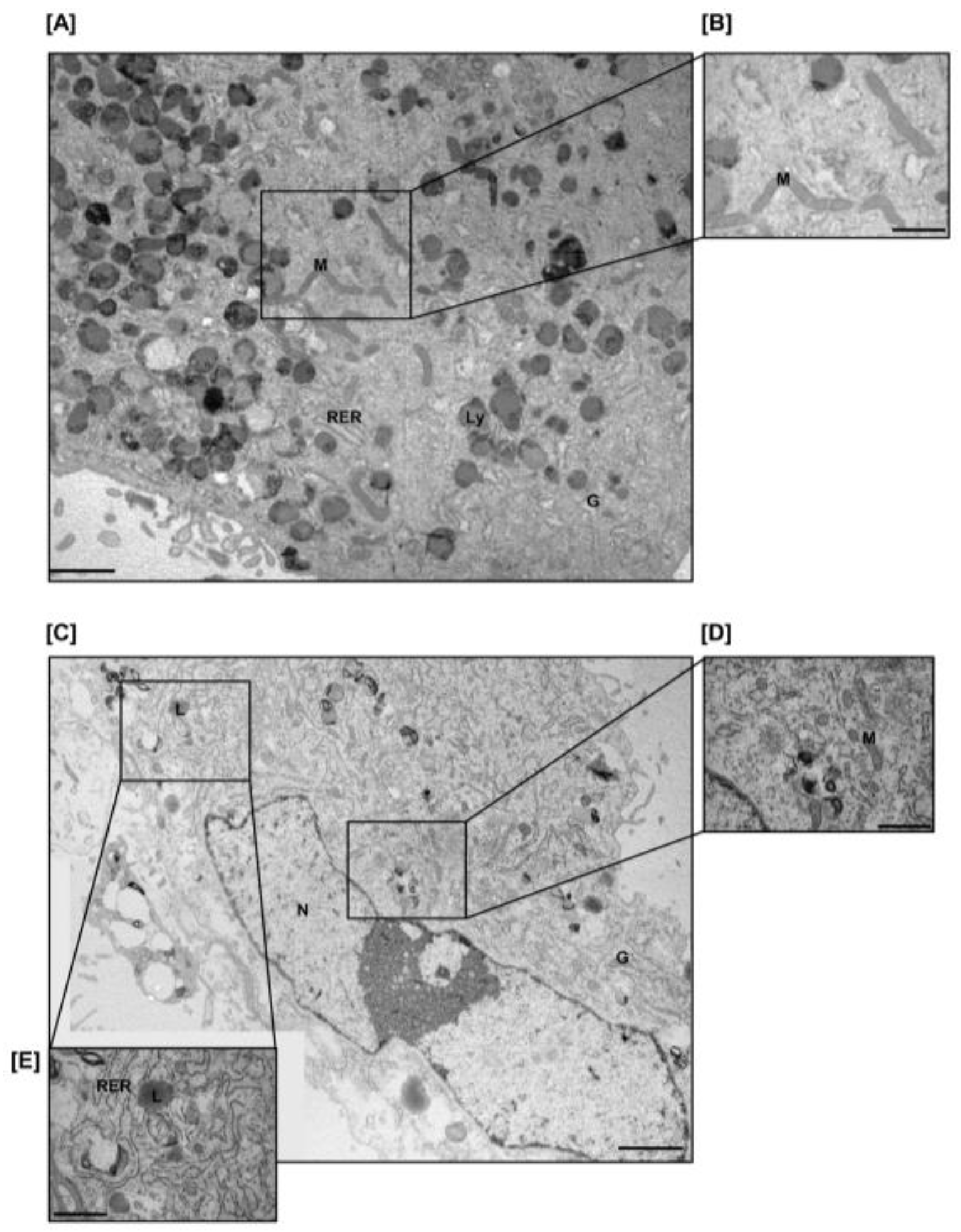
4.10. Transmission Electron Microscopy
4.11. Intracellular Lipid Content Quantification
4.12. Scratch Test
4.13. Invasion Assay
4.14. Trans-Endothelial Migration Assay
4.15. F-actin Cytoskeleton Fluorescence Stain
4.16. ELISA Assay
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Romero, I.L.; Mukherjee, A.; Kenny, H.A.; Litchfield, L.M.; Lengyel, E. Molecular pathways: Trafficking of metabolic resources in the tumor microenvironment. Clin. Cancer Res. 2015, 21, 680–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Du, L.; Lin, L.; Wang, Y. Tumour-associated mesenchymal stem/stromal cells: Emerging therapeutic targets. Nat. Rev. Drug Discov. 2017, 16, 35–52. [Google Scholar] [CrossRef]
- Duong, M.N.; Geneste, A.; Fallone, F.; Li, X.; Dumontet, C.; Muller, C. The fat and the bad: Mature adipocytes, key actors in tumor progression and resistance. Oncotarget 2017, 8, 57622–57641. [Google Scholar] [CrossRef] [Green Version]
- van Kruijsdijk, R.C.M.; van der Wall, E.; Visseren, F.L.J. Obesity and cancer: The role of dysfunctional adipose tissue. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 2569–2578. [Google Scholar] [CrossRef] [Green Version]
- Font-Burgada, J.; Sun, B.; Karin, M. Obesity and Cancer: The Oil that Feeds the Flame. Cell Metab. 2016, 23, 48–62. [Google Scholar] [CrossRef] [Green Version]
- Obesity and Cancer. Available online: https://www.cancer.gov/about-cancer/causes-prevention/risk/obesity/obesity-fact-sheet (accessed on 30 May 2019).
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [Green Version]
- Andarawewa, K.L.; Motrescu, E.R.; Chenard, M.-P.; Gansmuller, A.; Stoll, I.; Tomasetto, C.; Rio, M.-C. Stromelysin-3 is a potent negative regulator of adipogenesis participating to cancer cell-adipocyte interaction/crosstalk at the tumor invasive front. Cancer Res. 2005, 65, 10862–10871. [Google Scholar] [CrossRef] [Green Version]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Laurent, V.; Toulet, A.; Attané, C.; Milhas, D.; Dauvillier, S.; Zaidi, F.; Clement, E.; Cinato, M.; Le Gonidec, S.; Guérard, A.; et al. Periprostatic Adipose Tissue Favors Prostate Cancer Cell Invasion in an Obesity-Dependent Manner: Role of Oxidative Stress. Mol. Cancer Res. 2019, 17, 821–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 2013, 1831, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Morley, T.S.; Kim, M.; Clegg, D.J.; Scherer, P.E. Obesity and cancer--mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 2014, 10, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.-A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef]
- Freese, K.E.; Kokai, L.; Edwards, R.P.; Philips, B.J.; Sheikh, M.A.; Kelley, J.; Comerci, J.; Marra, K.G.; Rubin, J.P.; Linkov, F. Adipose-derived stems cells and their role in human cancer development, growth, progression, and metastasis: A systematic review. Cancer Res. 2015, 75, 1161–1168. [Google Scholar] [CrossRef] [Green Version]
- Maj, M.; Kokocha, A.; Bajek, A.; Drewa, T. The interplay between adipose-derived stem cells and bladder cancer cells. Sci. Rep. 2018, 8, 15118. [Google Scholar] [CrossRef] [Green Version]
- Preisner, F.; Leimer, U.; Sandmann, S.; Zoernig, I.; Germann, G.; Koellensperger, E. Impact of Human Adipose Tissue-Derived Stem Cells on Malignant Melanoma Cells in An in vitro Co-culture Model. Stem Cell Rev. 2018, 14, 125–140. [Google Scholar] [CrossRef]
- Su, F.; Ahn, S.; Saha, A.; DiGiovanni, J.; Kolonin, M.G. Adipose stromal cell targeting suppresses prostate cancer epithelial-mesenchymal transition and chemoresistance. Oncogene 2019, 38, 1979–1988. [Google Scholar] [CrossRef]
- Mohan, D.R.; Lerario, A.M.; Hammer, G.D. Therapeutic Targets for Adrenocortical Carcinoma in the Genomics Era. J. Endocr. Soc. 2018, 2, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Baglioni, S.; Francalanci, M.; Squecco, R.; Lombardi, A.; Cantini, G.; Angeli, R.; Gelmini, S.; Guasti, D.; Benvenuti, S.; Annunziato, F.; et al. Characterization of human adult stem-cell populations isolated from visceral and subcutaneous adipose tissue. FASEB J. 2009, 23, 3494–3505. [Google Scholar] [CrossRef] [PubMed]
- Baglioni, S.; Cantini, G.; Poli, G.; Francalanci, M.; Squecco, R.; Di Franco, A.; Borgogni, E.; Frontera, S.; Nesi, G.; Liotta, F.; et al. Functional differences in visceral and subcutaneous fat pads originate from differences in the adipose stem cell. PLoS ONE 2012, 7, e36569. [Google Scholar] [CrossRef] [PubMed]
- Cantini, G.; Lombardi, A.; Piscitelli, E.; Poli, G.; Ceni, E.; Marchiani, S.; Ercolino, T.; Galli, A.; Serio, M.; Mannelli, M.; et al. Rosiglitazone inhibits adrenocortical cancer cell proliferation by interfering with the IGF-IR intracellular signaling. PPAR Res. 2008, 2008, 904041. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Chemi, F.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Cordella, A.; Campana, A.; Hashim, A.; Rizza, P.; et al. Leptin as a mediator of tumor-stromal interactions promotes breast cancer stem cell activity. Oncotarget 2016, 7, 1262–1275. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Liu, Y.; Wei, D.; Wang, T.; Wang, K.; Huang, S.; Liu, L.; Li, Y.; Ge, J.; Li, X.; et al. Leptin promotes proliferation and metastasis of human gallbladder cancer through OB-Rb leptin receptor. Int. J. Oncol. 2016, 49, 197–206. [Google Scholar] [CrossRef]
- Wei, L.; Li, K.; Pang, X.; Guo, B.; Su, M.; Huang, Y.; Wang, N.; Ji, F.; Zhong, C.; Yang, J.; et al. Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J. Exp. Clin. Cancer Res. 2016, 35, 166. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, A.; Saeidi, J.; Azimi-Nejad, M.; Hashemy, S.I. Leptin-induced signaling pathways in cancer cell migration and invasion. Cell Oncol. 2019, 42, 243–260. [Google Scholar] [CrossRef]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef]
- Lalli, E.; Luconi, M. The next step: Mechanisms driving adrenocortical carcinoma metastasis. Endocr. Relat. Cancer 2018, 25, R31–R48. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y. Adipocyte and lipid metabolism in cancer drug resistance. J. Clin. Investig. 2019, 129, 3006–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, E.S.; Moon, H.J.; Lee, M.J.; Song, H.Y.; Kim, Y.M.; Cho, M.; Suh, D.-S.; Yoon, M.-S.; Chang, C.L.; Jung, J.S.; et al. Cancer-derived lysophosphatidic acid stimulates differentiation of human mesenchymal stem cells to myofibroblast-like cells. Stem Cells 2008, 26, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.A.; Park, H.; Lim, E.H.; Kim, K.H.; Choi, J.S.; Lee, J.H.; Shin, J.W.; Lee, K.W. Exosomes from ovarian cancer cells induce adipose tissue-derived mesenchymal stem cells to acquire the physical and functional characteristics of tumor-supporting myofibroblasts. Gynecol. Oncol. 2011, 123, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.A.; Park, H.; Lim, E.H.; Lee, K.W. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int. J. Oncol. 2012, 40, 130–138. [Google Scholar] [PubMed] [Green Version]
- Park, Y.M.; Yoo, S.H.; Kim, S.-H. Adipose-derived stem cells induced EMT-like changes in H358 lung cancer cells. Anticancer Res. 2013, 33, 4421–4430. [Google Scholar] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Barreau, O.; Assié, G.; Wilmot-Roussel, H.; Ragazzon, B.; Baudry, C.; Perlemoine, K.; René-Corail, F.; Bertagna, X.; Dousset, B.; Hamzaoui, N.; et al. Identification of a CpG island methylator phenotype in adrenocortical carcinomas. J. Clin. Endocrinol. Metab. 2013, 98, E174–E184. [Google Scholar] [CrossRef] [Green Version]
- Mohan, D.R.; Lerario, A.M.; Else, T.; Mukherjee, B.; Almeida, M.Q.; Vinco, M.; Rege, J.; Mariani, B.M.P.; Zerbini, M.C.N.; Mendonca, B.B.; et al. Targeted Assessment of G0S2 Methylation Identifies a Rapidly Recurrent, Routinely Fatal Molecular Subtype of Adrenocortical Carcinoma. Clin. Cancer Res. 2019, 25, 3276–3288. [Google Scholar] [CrossRef] [Green Version]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Ray, A.; Cleary, M.P. The potential role of leptin in tumor invasion and metastasis. Cytokine Growth Factor Rev. 2017, 38, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Barone, I.; Catalano, S.; Gelsomino, L.; Marsico, S.; Giordano, C.; Panza, S.; Bonofiglio, D.; Bossi, G.; Covington, K.R.; Fuqua, S.A.W.; et al. Leptin mediates tumor-stromal interactions that promote the invasive growth of breast cancer cells. Cancer Res. 2012, 72, 1416–1427. [Google Scholar] [PubMed] [Green Version]
- Wang, L.; Tang, C.; Cao, H.; Li, K.; Pang, X.; Zhong, L.; Dang, W.; Tang, H.; Huang, Y.; Wei, L.; et al. Activation of IL-8 via PI3K/Akt-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol. Ther. 2015, 16, 1220–1230. [Google Scholar] [PubMed] [Green Version]
- Cao, H.; Huang, Y.; Wang, L.; Wang, H.; Pang, X.; Li, K.; Dang, W.; Tang, H.; Wei, L.; Su, M.; et al. Leptin promotes migration and invasion of breast cancer cells by stimulating IL-8 production in M2 macrophages. Oncotarget 2016, 7, 65441–65453. [Google Scholar]
- Poli, G.; Ruggiero, C.; Cantini, G.; Canu, L.; Baroni, G.; Armignacco, R.; Jouinot, A.; Santi, R.; Ercolino, T.; Ragazzon, B.; et al. Fascin-1 Is a Novel Prognostic Biomarker Associated with Tumor Invasiveness in Adrenocortical Carcinoma. J. Clin. Endocrinol. Metab. 2019, 104, 1712–1724. [Google Scholar]
- Hattermann, K.; Held-Feindt, J.; Lucius, R.; Müerköster, S.S.; Penfold, M.E.T.; Schall, T.J.; Mentlein, R. The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res. 2010, 70, 3299–3308. [Google Scholar]
- Deutsch, A.J.A.; Steinbauer, E.; Hofmann, N.A.; Strunk, D.; Gerlza, T.; Beham-Schmid, C.; Schaider, H.; Neumeister, P. Chemokine receptors in gastric MALT lymphoma: Loss of CXCR4 and upregulation of CXCR7 is associated with progression to diffuse large B-cell lymphoma. Mod. Pathol. 2013, 26, 182–194. [Google Scholar]
- Wu, Y.-C.; Tang, S.-J.; Sun, G.-H.; Sun, K.-H. CXCR7 mediates TGFβ1-promoted EMT and tumor-initiating features in lung cancer. Oncogene 2016, 35, 2123–2132. [Google Scholar] [CrossRef]
- Ma, D.-M.; Luo, D.-X.; Zhang, J. SDF-1/CXCR7 axis regulates the proliferation, invasion, adhesion, and angiogenesis of gastric cancer cells. World J. Surg. Oncol. 2016, 14, 256. [Google Scholar]
- Shi, A.; Shi, H.; Dong, L.; Xu, S.; Jia, M.; Guo, X.; Wang, T. CXCR7 as a chemokine receptor for SDF-1 promotes gastric cancer progression via MAPK pathways. Scand. J. Gastroenterol. 2017, 52, 745–753. [Google Scholar]
- Qian, T.; Liu, Y.; Dong, Y.; Zhang, L.; Dong, Y.; Sun, Y.; Sun, D. CXCR7 regulates breast tumor metastasis and angiogenesis in vivo and in vitro. Mol. Med. Rep. 2018, 17, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Al-Toub, M.; Almohawes, M.; Vishnubalaji, R.; Alfayez, M.; Aldahmash, A.; Kassem, M.; Alajez, N.M. CXCR7 signaling promotes breast cancer survival in response to mesenchymal stromal stem cell-derived factors. Cell Death Discov. 2019, 5, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.C.-S.; Yu, H.-S.; Yen, F.-L.; Chen, G.-S.; Lan, C.-C.E. CXCR7 expression correlates with tumor depth in cutaneous squamous cell carcinoma skin lesions and promotes tumor cell survival through ERK activation. Exp. Dermatol. 2014, 23, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Han, M.-M.; Wang, F.; Xu, L.-L.; Yu, H.-X.; Yang, P.-Y. CXCR7 stimulates MAPK signaling to regulate hepatocellular carcinoma progression. Cell Death Dis. 2014, 5, e1488. [Google Scholar] [CrossRef] [PubMed]
- Mazzinghi, B.; Ronconi, E.; Lazzeri, E.; Sagrinati, C.; Ballerini, L.; Angelotti, M.L.; Parente, E.; Mancina, R.; Netti, G.S.; Becherucci, F.; et al. Essential but differential role for CXCR4 and CXCR7 in the therapeutic homing of human renal progenitor cells. J. Exp. Med. 2008, 205, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Weiss, L.M. The Weiss system for evaluating adrenocortical neoplasms: 25 years later. Hum. Pathol. 2009, 40, 757–768. [Google Scholar] [CrossRef]
- Fassnacht, M.; Johanssen, S.; Quinkler, M.; Bucsky, P.; Willenberg, H.S.; Beuschlein, F.; Terzolo, M.; Mueller, H.H.; Hahner, S.; Allolio, B. German Adrenocortical Carcinoma Registry Group European Network for the Study of Adrenal Tumors. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: Proposal for a Revised TNM Classification. Cancer 2009, 115, 243–250. [Google Scholar]
- Poli, G.; Ceni, E.; Armignacco, R.; Ercolino, T.; Canu, L.; Baroni, G.; Nesi, G.; Galli, A.; Mannelli, M.; Luconi, M. 2D-DIGE proteomic analysis identifies new potential therapeutic targets for adrenocortical carcinoma. Oncotarget 2015, 6, 5695–5706. [Google Scholar] [CrossRef] [Green Version]
- Cantini, G.; Lombardi, A.; Borgogni, E.; Francalanci, M.; Ceni, E.; Degl’Innocenti, S.; Gelmini, S.; Poli, G.; Galli, A.; Serio, M.; et al. Peroxisome-proliferator-activated receptor gamma (PPARgamma) is required for modulating endothelial inflammatory response through a nongenomic mechanism. Eur. J. Cell. Biol. 2010, 89, 645–653. [Google Scholar] [CrossRef]
- Lombardi, A.; Cantini, G.; Piscitelli, E.; Gelmini, S.; Francalanci, M.; Mello, T.; Ceni, E.; Varano, G.; Forti, G.; Rotondi, M.; et al. A new mechanism involving ERK contributes to rosiglitazone inhibition of tumor necrosis factor-alpha and interferon-gamma inflammatory effects in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 718–724. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | ASC | ASC + H295R | H295R | H295R + ASC |
|---|---|---|---|---|
| Conditioned medium | ||||
| Glucose | 1.0 ± 0.02 | 0.67 ± 0.03 ** | 1.0 ± 0.05 | 0.83 ± 0.02 * |
| Lactic acid | 1.0 ± 0.10 | 1.96 ± 0.25 * | 1.0 ± 0.02 | 1.70 ± 0.05 ** |
| Intracellular lactate | ||||
| 1.0 ± 0.02 | 1.39 ± 0.03 ** | 1.0 ± 0.10 | 1.33 ± 0.11 * | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armignacco, R.; Cantini, G.; Poli, G.; Guasti, D.; Nesi, G.; Romagnoli, P.; Mannelli, M.; Luconi, M. The Adipose Stem Cell as a Novel Metabolic Actor in Adrenocortical Carcinoma Progression: Evidence from an In Vitro Tumor Microenvironment Crosstalk Model. Cancers 2019, 11, 1931. https://doi.org/10.3390/cancers11121931
Armignacco R, Cantini G, Poli G, Guasti D, Nesi G, Romagnoli P, Mannelli M, Luconi M. The Adipose Stem Cell as a Novel Metabolic Actor in Adrenocortical Carcinoma Progression: Evidence from an In Vitro Tumor Microenvironment Crosstalk Model. Cancers. 2019; 11(12):1931. https://doi.org/10.3390/cancers11121931
Chicago/Turabian StyleArmignacco, Roberta, Giulia Cantini, Giada Poli, Daniele Guasti, Gabriella Nesi, Paolo Romagnoli, Massimo Mannelli, and Michaela Luconi. 2019. "The Adipose Stem Cell as a Novel Metabolic Actor in Adrenocortical Carcinoma Progression: Evidence from an In Vitro Tumor Microenvironment Crosstalk Model" Cancers 11, no. 12: 1931. https://doi.org/10.3390/cancers11121931
APA StyleArmignacco, R., Cantini, G., Poli, G., Guasti, D., Nesi, G., Romagnoli, P., Mannelli, M., & Luconi, M. (2019). The Adipose Stem Cell as a Novel Metabolic Actor in Adrenocortical Carcinoma Progression: Evidence from an In Vitro Tumor Microenvironment Crosstalk Model. Cancers, 11(12), 1931. https://doi.org/10.3390/cancers11121931