Assessing the Concordance of Genomic Alterations between Circulating-Free DNA and Tumour Tissue in Cancer Patients


Abstract
:1. Introduction
2. The Concordance Rate of SGAs between cfDNA and Tumour Tissue across Solid Tumours
3. The Underlying Factors Contributing to Perceived Discordance between SGAs Detected in Solid Tumours and cfDNA
3.1. Tumour Fraction and Mutation Allele Frequency (MAF)
3.2. Gene Type and the Effect of Drug Therapy
3.3. Sampling and Processing of Tumour Tissue
3.4. Detection Method
3.5. Heterogeneity
4. The future of cfDNA in precision oncology
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Jang, H.; Tsai, C.J.; Cheng, F. Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers. PLoS Comput. Biol. 2019, 15, e1006658. [Google Scholar]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiley, C.; de Bruin, E.C.; McGranahan, N.; Swanton, C. Deciphering intratumor heterogeneity and temporal acquisition of driver events to refine precision medicine. Genome Biol. 2014, 15, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, B.; Chakrabarty, M.; Cohn, E.M.; Leon, S.A. Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer 1983, 51, 2116–2120. [Google Scholar] [CrossRef]
- Chen, X.; Bonnefoi, H.; Diebold-Berger, S.; Lyautey, J.; Lederrey, C.; Faltin-Traub, E.; Stroun, M.; Anker, P. Detecting tumor-related alterations in plasma or serum DNA of patients diagnosed with breast cancer. Clin. Cancer Res. 1999, 5, 2297–2303. [Google Scholar] [PubMed]
- Riviere, P.; Fanta, P.T.; Ikeda, S.; Baumgartner, J.; Heestand, G.M.; Kurzrock, R. The Mutational Landscape of Gastrointestinal Malignancies as Reflected by Circulating Tumor DNA. Mol. Cancer Ther. 2018, 17, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, A.W.; Annala, M.; Aggarwal, R.; Beja, K.; Feng, F.; Youngren, J.; Foye, A.; Lloyd, P.; Nykter, M.; Beer, T.M.; et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Muluhngwi, P.; Valdes, R., Jr.; Fernandez-Botran, R.; Burton, E.; Williams, B.; Linder, M.W. Cell-free DNA diagnostics: Current and emerging applications in oncology. Pharmacogenomics 2019, 20, 357–380. [Google Scholar] [CrossRef] [PubMed]
- Sozzi, G.; Conte, D.; Mariani, L.; Lo Vullo, S.; Roz, L.; Lombardo, C.; Pierotti, M.A.; Tavecchio, L. Analysis of circulating tumor DNA in plasma at diagnosis and during follow-up of lung cancer patients. Cancer Res. 2001, 61, 4675–4678. [Google Scholar] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1997, 37, 646–650. [Google Scholar]
- Thompson, J.C.; Yee, S.S.; Troxel, A.B.; Savitch, S.L.; Fan, R.; Balli, D.; Lieberman, D.B.; Morrissette, J.D.; Evans, T.L.; Bauml, J.; et al. Detection of Therapeutically Targetable Driver and Resistance Mutations in Lung Cancer Patients by Next-Generation Sequencing of Cell-Free Circulating Tumor DNA. Clin. Cancer Res. 2016, 22, 5772–5782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combaret, V.; Audoynaud, C.; Iacono, I.; Favrot, M.C.; Schell, M.; Bergeron, C.; Puisieux, A. Circulating MYCN DNA as a tumor-specific marker in neuroblastoma patients. Cancer Res. 2002, 62, 3646–3648. [Google Scholar]
- Cristofanilli, M.; Hayes, D.F.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Reuben, J.M.; Doyle, G.V.; Matera, J.; Allard, W.J.; Miller, M.C.; et al. Circulating tumor cells: A novel prognostic factor for newly diagnosed metastatic breast cancer. J. Clin. Oncol. 2005, 23, 1420–1430. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Belic, J.; Gutschi, S.; Quehenberger, F.; Fischereder, K.; Benezeder, T.; Auer, M.; Pischler, C.; Mannweiler, S.; et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A. Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 54–548. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Caldas, C. Cell-free circulating tumour DNA as a liquid biopsy in breast cancer. Mol. Oncol. 2016, 10, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, M.; Schmidt, B. Circulating nucleic acids (CNAs) and cancer—a survey. Biochim. Biophys. Acta 2007, 1775, 123–181. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating tumor DNA as a liquid biopasy for cancer. Clin. Chem. 2015, 61, 23–112. [Google Scholar] [CrossRef]
- Montagut, C.; Siravegna, G.; Bardelli, A. Liquid biopsies to evaluate early therapeutic response in colorectal cancer. Ann. Oncol. 2015, 26, 1525–1527. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Luke, J.J.; Oxnard, G.R.; Paweletz, C.P.; Camidge, D.R.; Heymach, J.V.; Solit, D.B.; Johnson, B.E.; Cell Free DNA Working Group. Realizing the potential of plasma genotyping in an age of genotypedirected therapies. J. Natl. Cancer Inst. 2014, 106, dju214. [Google Scholar] [CrossRef] [Green Version]
- Bokemeyer, C.; Köhne, C.H.; Ciardiello, F.; Lenz, H.J.; Heinemann, V.; Klinkhardt, U.; Beier, F.; Duecker, K.; Krieken van, J.H.; Tejpar, S. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur. J. Cancer 2015, 51, 1243–1252. [Google Scholar] [CrossRef]
- Grasselli, J.; Elez, E.; Caratù, G.; Matito, J.; Santos, C.; Macarulla, T.; Vidal, J.; Garcia, M.; Viéitez, J.M.; Paéz, D.; et al. Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2017, 28, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Bando, H.; Kagawa, Y.; Kato, T.; Akagi, K.; Denda, T.; Nishina, T.; Komatsu, Y.; Oki, E.; Kudo, T.; Kumamoto, H.; et al. A multicentre, prospective study of plasma circulating tumour DNA test for detecting RAS mutation in patients with metastatic colorectal cancer. Br. J. Cancer 2019, 120, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Schmiegel, W.; Scott, R.J.; Dooley, S.; Lewis, W.; Meldrum, C.J.; Pockney, P.; Draganic, B.; Smith, S.; Hewitt, C.; Philimore, H.; et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: Concordance of results from circulating tumor DNA and tissuebased RAS testing. Mol. Oncol. 2017, 11, 208–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettinger, D.S.; Akerley, W.; Borghaei, H.; Chang, A.C.; Cheney, R.T.; Chirieac, L.R.; D’Amico, T.A.; Demmy, T.L.; Govindan, R.; Grannis, F.W.; et al. Nonsmall cell lung cancer, version 2.2013. J. Natl. Compr. Cancer Netw. 2013, 11, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Oxnard, G.R.; Binder, A.J.P.A. New targetable oncogenes in non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 1097–1104. [Google Scholar] [CrossRef] [Green Version]
- Sung, J.S.; Chong, H.Y.; Kwon, N.J.; Kim, H.M.; Lee, J.W.; Kim, B.; Lee, S.B.; Park, C.W.; Choi, J.Y.; Chang, W.J.; et al. Detection of somatic variants and EGFR mutations in cell-free DNA from non-small cell lung cancer patients by ultra-deep sequencing using the ion ampliseq cancer hotspot panel and droplet digital polymerase chain reaction. Oncotarget 2017, 8, 106901. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Qing, X.; Xiumin, W.; Yali, B.; Chi, S.; Bak, S.H.; Lee, H.Y.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; et al. Longitudinal monitoring of EGFR mutations in plasma predicts outcomes of NSCLC patients treated with EGFR TKIs: Korean Lung Cancer Consortium (KLCC-12-02). Oncotarget 2016, 7, 6984–6993. [Google Scholar] [CrossRef] [Green Version]
- Stewart, E.L.; Tan, S.Z.; Liu, G.T.M.S. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations-a review. Transl. Lung Cancer Res. 2015, 4, 67–81. [Google Scholar]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.J.D.M. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin. Cancer Res. Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [Green Version]
- Yung, T.K.; Chan, K.C.; Mok, T.S.; Tong, J.; To, K.F.; Lo, Y.M. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin. Cancer Res. 2009, 15, 2076–2084. [Google Scholar] [CrossRef] [Green Version]
- Sakai, K.; Horiike, A.; Irwin, D.L.; Kudo, K.; Fujita, Y.; Tanimoto, A.; Sakatani, T.; Saito, R.; Kaburaki, K.; Yanagitani, N.; et al. Detection of epidermal growth factor receptor T790M mutation in plasma DNA from patients refractory to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci. 2013, 104, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.H.; Li, L.H.; Hua, D. Quantitative analysis of plasma circulating DNA at diagnosis and during follow-up of breast cancer patients. Cancer Lett. 2006, 243, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, T.; Yamamoto, Y.; Yamamoto-Ibusuki, M.; Tomiguchi, M.; Sueta, A.; Murakami, K.; Omoto, Y.; Iwase, H. Comparison of ESR1 Mutations in Tumor Tissue and Matched Plasma Samples from Metastatic Breast Cancer Patients. Transl. Oncol. 2017, 10, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Higgins, M.J.; Jelovac, D.; Barnathan, E.; Blair, B.; Slater, S.; Powers, P.; Zorzi, J.; Jeter, S.C.; Oliver, G.R.; Fetting, J.; et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin. Cancer Res. 2012, 18, 3462–3469. [Google Scholar] [CrossRef] [Green Version]
- Board, R.E.; Wardley, A.M.; Dixon, J.M.; Armstrong, A.C.; Howell, S.; Renshaw, L.; Donald, E.; Greystoke, A.; Ranson, M.; Hughes, A.; et al. Detection of PIK3CA mutations in circulating free DNA in patients with breast cancer. Breast Cancer Res. Treat. 2010, 120, 461–467. [Google Scholar] [CrossRef]
- Garcia-Saenz, J.A.; Ayllon, P.; Laig, M.; Acosta-Eyzaguirre, D.; Garcia-Esquinas, M.; Montes, M.; Sanz, J.; Barquín, M.; Moreno, F.; Garcia-Barberan, V.; et al. Tumor burden monitoring using cell-free tumor DNA could be limited by tumor heterogeneity in advanced breast cancer and should be evaluated together with radiographic imaging. BMC Cancer 2017, 17, 210. [Google Scholar] [CrossRef] [Green Version]
- Beaver, J.A.; Jelovac, D.; Balukrishna, S.; Cochran, R.; Croessmann, S.; Zabransky, D.J.; Wong, H.Y.; Toro, P.V.; Cidado, J.; Blair, B.G.; et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin. Cancer Res. 2014, 20, 2643–2650. [Google Scholar] [CrossRef] [Green Version]
- Kodahl, A.R.; Ehmsen, S.; Pallisgaard, N.; Jylling, A.M.B.; Jensen, J.D.; Laenkholm, A.V.; Knoop, A.S.; Ditzel, H.J. Correlation between circulating cell-free PIK3CA tumor DNA levels and treatment response in patients with PIK3CA-mutated metastatic breast cancer. Mol. Oncol. 2018, 12, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Di Cataldo, A.; Dau, D.; Conte, M.; Parodi, S.; De Bernardi, B.; Giuliano, M.; Pession, A.; Viscardi, E.; Luksch, R.; Castellano, A.; et al. Diagnostic and prognostic markers in infants with disseminated neuroblastoma: A retrospective analysis from the Italian Cooperative Group for Neuroblastoma. Med. Sci. Monit. 2009, 1, MT11–MT18. [Google Scholar]
- Pession, A.; De Bernardi, B.; Perri, P.; Mazzocco, K.; Rondelli, R.; Nigro, M.; Lolascon, A.; Forni, M.; Basso, G.; Conte, M.; et al. The prognostic effect of amplification of MYCN oncogene in neuroblstoma. The preliminary results of the Italian Copperative Group for Neuroblastoma (GCINB). Pediatr. Med. Chir. 1994, 16, 211–218. [Google Scholar] [PubMed]
- Combaret, V.; Iacono, I.; Bellini, A.; Bréjon, S.; Bernard, V.; Marabelle, A.; Coze, C.; Pierron, G.; Lapouble, E.; Schleiermacher, G.; et al. Detection of tumor ALK status in neuroblastoma patients using peripheral blood. Cancer Med. 2015, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Chicard, M.; Boyault, S.; Colmet Daage, L.; Richer, W.; Gentien, D.; Pierron, G.; Lapouble, E.; Bellini, A.; Clement, N.; Iacono, I.; et al. Genomic Copy Number Profiling Using Circulating Free Tumor DNA Highlights Heterogeneity in Neuroblastoma. Clin. Cancer Res. 2016, 22, 5564–5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chicard, M.; Colmet-Daage, L.; Clement, N.; Danzon, A.; Bohec, M.; Bernard, V.; Baulande, S.; Bellini, A.; Deveau, P.; Pierron, G.; et al. Whole-Exome Sequencing of Cell-Free DNA Reveals Temporo-spatial Heterogeneity and Identifies Treatment-Resistant Clones in Neuroblastoma. Clin. Cancer Res. 2018, 24, 939–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Roy, N.; Van Der Linden, M.; Menten, B.; Dheedene, A.; Vandeputte, C.; Van Dorpe, J.; Laureys, G.; Renard, M.; Sante, T.; Lammens, T.; et al. Shallow Whole Genome Sequencing on Circulating Cell-Free DNA Allows Reliable Noninvasive Copy-Number Profiling in Neuroblastoma Patients. Clin. Cancer Res. 2017, 23, 6305–6314. [Google Scholar] [CrossRef] [Green Version]
- Leary, R.J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J.D.; Craig, D.; O’Shaughnessy, J.; Kinzler, K.W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci. Transl. Med. 2012, 1, 162ra154. [Google Scholar] [CrossRef] [Green Version]
- Lavon, I.; Refael, M.; Zelikovitch, B.; Shalom, E.S. Serum DNA can define tumor-specific genetic and epigenetic markers in gliomas of various grades. Neuro. Oncol. 2010, 12, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Molparia, B.; Oliveira, G.; Wagner, J.L.; Spencer, E.G.; Torkamani, A. A feasibility study of colorectal cancer diagnosis via circulating tumor DNA derived CNV detection. PLoS. ONE 2018, 13, e0196826. [Google Scholar] [CrossRef]
- Campbell, P.J.; Stephens, P.J.; Pleasance, E.D.; O’Meara, S.; Li, H.; Santarius, T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Hardy, C.; et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008, 40, 722–729. [Google Scholar] [CrossRef]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Ignatiadis, M.; Dawson, S.J. Circulating tumor cells and circulating tumor DNA for precision medicine: Dream or reality? Ann. Oncol. 2014, 25, 2304–2313. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef] [Green Version]
- García-Foncillas, J.; Tabernero, J.; Élez, E.; Aranda, E.; Benavides, M.; Camps, C.; Jantus-Lewintre, E.; López, R.; Muinelo-Romay, L.; Montagut, C.; et al. Prospective multicenter real-world RAS mutation comparison between OncoBEAM-based liquid biopsy and tissue analysis in metastatic colorectal cancer. Br. J. Cancer 2018, 119, 1464–1470. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Mollevi, C.; Raoul, J.L.; Guimbaud, R.; Pezet, D.; Artru, P.; Assenat, E.; Borg, C.; Mathonnet, M.; et al. Clinical utility of circulating DNA analysis for rapid detection of actionable mutations to select metastatic colorectal patients for anti-EGFR treatment. Ann. Oncol. 2017, 28, 2149–2159. [Google Scholar] [CrossRef]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhuo, M.; Ye, X.; Bai, H.; Wang, Z.; Sun, Y.; Zhao, J.; An, T.; Duan, J.; Wu, M.; et al. Quantification of mutant alleles in circulating tumor DNA can predict survival in lung cancer. Oncotarget 2016, 7, 20810. [Google Scholar] [CrossRef] [Green Version]
- Bartels, S.; Persing, S.; Hasemeier, B.; Schipper, E.; Kreipe, H.; Lehmann, U. Molecular Analysis of Circulating Cell-Free DNA from Lung Cancer Patients in Routine Laboratory Practice: A Cross-Platform Comparison of Three Different Molecular Methods for Mutation Detection. J. Mol. Diagn. 2017, 19, 722–723. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.K.; Davis, A.A.; Jain, S.; Santa-Maria, C.; Flaum, L.; Beaubier, N.; Platanias, L.C.; Gradishar, W.; Giles, F.J.; Cristofanilli, M. Concordance of genomic alterations by next-generation sequencing in tumor tissue versus circulating tumor DNA in breast cancer. Mol. Cancer Ther. 2017, 16, 1412–1420. [Google Scholar] [CrossRef] [Green Version]
- Jen, J.; Wu, L.; Sidransky, D. An overview on the isolation and analysis of circulating tumor DNA in plasma and serum. Ann. N. Y. Acad. Sci. 2000, 906, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, J.; Xiao, J.; Wang, L.; Hu, X.; Yu, W.; Song, G.; Lou, J.; Chen, J. Heterogeneous mutation pattern in tumor tissue and circulating tumor DNA warrants parallel NGS panel testing. Mol. Cancer 2018, 17, 131. [Google Scholar] [CrossRef] [PubMed]
- Li, B.T.; Janku, F.; Jung, B.; Hou, C.; Madwani, K.; Alden, R.; Razavi, P.; Reis-Filho, J.S.; Shen, R.; Isbell, J.M.; et al. Ultra-deep next-generation sequencing of plasma cell-free DNA in patients with advanced lung cancers: Results from the Actionable Genome Consortium. Ann. Oncol. 2019, 30, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Sefrioui, D.; Beaussire, L.; Perdrix, A.; Clatot, F.; Michel, P.; Frebourg, T.; Di Fiore, F.; Sarafan-Vasseur, N. Direct circulating tumor DNA detection from unpurified plasma using a digital PCR platform. Clin. BioChem. 2017, 50, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Demuth, C.; Spindler, K.G.; Johansen, J.S.; Pallisgaard, N.; Nielsen, D.; Hogdall, E.; Vittrup, B.; Sorensen, B.S. Measuring KRAS Mutations in Circulating Tumor DNA by Droplet Digital PCR and Next-Generation Sequencing. Transl. Oncol. 2018, 11, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Jovelet, C.; Ileana, E.; Le Deley, M.C.; Motté, N.; Rosellini, S.; Romero, A.; Lefebvre, C.; Pedrero, M.; Pata-Merci, N.; Droin, N.; et al. Circulating Cell-Free Tumor DNA Analysis of 50 Genes by Next-Generation Sequencing in the Prospective MOSCATO Trial. Clin. Cancer Res. 2016, 22, 2960. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Huang, H.J.; Fujii, T.; Shelton, D.N.; Madwani, K.; Fu, S.; Tsimberidou, A.M.; Piha-Paul, S.A.; Wheler, J.J.; Zinner, R.G.; et al. Multiplex KRASG12/G13 mutation testing of unamplified cell-free DNA from the plasma of patients with advanced cancers using droplet digital polymerase chain reaction. Ann. Oncol. 2017, 28, 642–650. [Google Scholar]
- Klega, K.; Imamovic-Tuco, A.; Ha, G.; Clapp, A.N.; Meyer, S.; Ward, A.; Clinton, C.; Nag, A.; Van Allen, E.; Mullen, E.; et al. Detection of Somatic Structural Variants Enables Quantification and Characterization of Circulating Tumor DNA in Children With Solid Tumors. JCO Precis. Oncol. 2018, 2, 1–3. [Google Scholar] [CrossRef]
- Hemming, M.L.; Klega, K.S.; Rhoades, J.; Ha, G.; Acker, K.E.; Andersen, J.L.; Thai, E.; Nag, A.; Thorner, A.R.; Raut, C.P.; et al. Detection of Circulating Tumor DNA in Patients With Leiomyosarcoma With Progressive Disease. JCO Precis. Oncol. 2019, 3, 1–11. [Google Scholar] [CrossRef]
- Namløs, H.M.; Boye, K.; Mishkin, S.J.; Barøy, T.; Lorenz, S.; Bjerkehagen, B.; Stratford, E.W.; Munthe, E.; Kudlow, B.A.; Myklebost, O.; et al. Noninvasive Detection of ctDNA Reveals Intratumor Heterogeneity and Is Associated with Tumor Burden in Gastrointestinal Stromal Tumor. Mol. Cancer Ther. 2018, 17, 2473–2480. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Liu, H.; Shao, D.; Liu, Z.; Wang, J.; Deng, Q.; Tang, H.; Yang, H.; Zhang, Y.; Qiu, Y.; et al. Development and clinical validation of a circulating tumor DNA test for the identification of clinically actionable mutations in nonsmall cell lung cancer. Genes Chromosom. Cancer 2018, 57, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Guleria, R.; Singh, V.; Bharti, A.C.; Mohan, A.; Das, B.C. Plasma DNA level in predicting therapeutic efficacy in advanced nonsmall cell lung cancer. Eur. Respir. J. 2010, 36, 885–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Zhang, Y.; Mao, X.; Zheng, X.; Han-Zhang, H.; Ye, J.; Zhao, R.; Zhang, X.; Sun, J. Comparison of genetic profiles among primary lung tumor, metastatic lymph nodes and circulating tumor DNA in treatment-naïve advanced nonsquamous non-small cell lung cancer patients. Lung Cancer 2018, 121, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Tzanikou, E.; Markou, A.; Politaki, E.; Koutsopoulos, A.; Psyrri, A.; Mavroudis, D.; Georgoulias, V.; Lianidou, E. PIK3CA hotspot mutations in circulating tumor cells and paired circulating tumor DNA in breast cancer: A direct comparison study. Mol. Oncol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Okamura, R.; Baumgartner, J.M.; Patel, H.; Leichman, L.; Kelly, K.; Sicklick, J.K.; Fanta, P.T.; Lippman, S.M.; Kurzrock, R. Analysis of Circulating Tumor DNA and Clinical Correlates in Patients with Esophageal, Gastroesophageal Junction, and Gastric Adenocarcinoma. Clin. Cancer Res. 2018, 24, 6248–6256. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra54. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Gerlinger, M.; Futreal, P.A.; Pusztai, L.; Swanton, C. Intratumor heterogeneity: Seeing the wood for the trees. Sci. Transl. Med. 2012, 4, 127ps10. [Google Scholar] [CrossRef] [Green Version]
- Stratton, M.; Campbell, P.; Futreal, P. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Toor, O.M.; Ahmed, Z.; Bahaj, W.; Boda, U.; Cummings, L.S.; McNally, M.E.; Kennedy, K.F.; Pluard, T.J.; Hussain, A.; Subramanian, J.; et al. Correlation of Somatic Genomic Alterations Between Tissue Genomics and ctDNA Employing Next-Generation Sequencing: Analysis of Lung and Gastrointestinal Cancers. Mol. Cancer Ther. 2018, 17, 1123–1132. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.W.; Gill, D.M.; Maughan, B.; Agarwal, A.; Arjyal, L.; Gupta, S.; Streeter, J.; Bailey, E.; Pal, S.K.; Agarwal, N. Correlation of genomic alterations assessed by next-generation sequencing (NGS) of tumor tissue DNA and circulating tumor DNA (ctDNA) in metastatic renal cell carcinoma (mRCC): Potential clinical implications. Oncotarget 2017, 8, 33614–33620. [Google Scholar] [CrossRef] [Green Version]
- Kuderer, N.M.; Burton, K.A.; Blau, S.; Rose, A.L.; Parker, S.; Lyman, G.H.; Blau, C.A. Comparison of 2 commercially available next-generation sequencing platforms in oncology. JAMA Oncol. 2017, 3, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.C.; Koshkin, V.S.; Funchain, P.; Sohal, D.; Pritchard, A.; Klek, S.; Adamowicz, T.; Gopalakrishnan, D.; Garcia, J.; Rini, B.; et al. Next-generation sequencing (NGS) of cell-free circulating tumor DNA and tumor tissue in patients with advanced urothelial cancer: A pilot assessment of concordance. Ann. Oncol. 2017, 28, 2458–2463. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202–11020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pishvaian, M.J.; Joseph Bender, R.; Matrisian, L.M.; Rahib, L.; Hendifar, A.; Hoos, W.A.; Mikhail, S.; Chung, V.; Picozzi, V.; Heartwell, C.; et al. A pilot study evaluating concordance between blood-based and patient-matched tumor molecular testing within pancreatic cancer patients participating in the Know Your Tumor (KYT) initiative. Oncotarget 2016, 8, 83446–83456. [Google Scholar] [CrossRef] [Green Version]
- Vandekerkhove, G.; Struss, W.J.; Annala, M.; Kallio, H.M.L.; Khalaf, D.; Warner, E.W.; Herberts, C.; Ritch, E.; Beja, K.; Loktionova, Y.; et al. Circulating Tumor DNA Abundance and Potential Utility in De Novo Metastatic Prostate Cancer. Eur. Urol. 2019, 75, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Spindler, K.L.; Pallisgaard, N.; Andersen, R.F.; Brandslund, I.; Jakobsen, A. Circulating free DNA as biomarker and source for mutation detection in metastatic colorectal cancer. PLoS ONE 2015, 10, e0108247. [Google Scholar] [CrossRef]
- Bachet, J.B.; Bouché, O.; Taieb, J.; Dubreuil, O.; Garcia, M.L.; Meurisse, A.; Normand, C.; Gornet, J.M.; Artru, P.; Louafi, S.; et al. RAS mutation analysis in circulating tumor DNA from patients with metastatic colorectal cancer: The AGEO RASANC prospective multicenter study. Ann. Oncol. Oncol. 2018, 29, 1211–1219. [Google Scholar] [CrossRef]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef]
- Buim, M.E.; Fanelli, M.F.; Souza, V.S.; Romero, J.; Abdallah, E.A.; Mello, C.A.; Alves, V.; Ocea, L.M.; Mingues, N.B.; Barbosa, P.N.; et al. Detection of KRAS mutations in circulating tumor cells from patients with metastatic colorectal cancer. Cancer Biol. Ther. 2015, 16, 1289–1295. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wu, S.; Huang, F.; Shen, M.; Jiang, H.; Yu, Y.; Yu, Q.; Yang, Y.; Zhao, Y.; Zhou, Y.; et al. Analytical and clinical validation of a novel amplicon-based NGS assay for the evaluation of circulating tumor DNA in metastatic colorectal cancer patients. Clin. Chem. Lab. Med. 2019, 57, 1501–1510. [Google Scholar] [CrossRef]
- Osumi, H.; Shinozaki, E.; Takeda, Y.; Wakatsuki, T.; Ichimura, T.; Saiura, A.; Yamaguchi, K.; Takahashi, S.; Noda, T.; Zembutsu, H. Clinical relevance of circulating tumor DNA assessed through deep sequencing in patients with metastatic colorectal cancer. Cancer Med. 2019, 8, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Mauri, G.; Siravegna, G.; Dive, C.; Pierce, J.; Di Nicolantonio, F.; D’Incalci, M.; Bardelli, A.; Siena, S.; Sartore-Bianchi, A. Parallel Evaluation of Circulating Tumor DNA and Circulating Tumor Cells in Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2018, 17, 80–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beije, N.; Helmijr, J.C.; Weerts, M.J.A.; Beaufort, C.M.; Wiggin, M.; Marziali, A.; Verhoef, C.; Sleijfer, S.; Jansen, M.P.H.M.; Martens, J.W.M. Somatic mutation detection using various targeted detection assays in paired samples of circulating tumor DNA, primary tumor and metastases from patients undergoing resection of colorectal liver metastases. Mol. Oncol. 2016, 10, 1575–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Schwaederlé, M.C.; Fanta, P.T.; Okamura, R.; Leichman, L.; Lippman, S.M.; Lanman, R.B.; Raymond, V.M.; Talasaz, A.; Kurzrock, R. Genomic Assessment of Blood-Derived Circulating Tumor DNA in Patients With Colorectal Cancers: Correlation With Tissue Sequencing, Therapeutic Response, and Survival. JCO Precis. Oncol. 2019, 3, 1–16. [Google Scholar] [CrossRef]
- Mohamed Suhaimi, N.A.; Foong, Y.M.; Lee, D.Y.; Phyo, W.M.; Cima, I.; Lee, E.X.; Goh, W.L.; Lim, W.Y.; Chia, K.S.; Kong, S.L.; et al. Non-invasive sensitive detection of KRAS and BRAF mutation in circulating tumor cells of colorectal cancer patients. Mol. Oncol. 2015, 9, 850–860. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, S.; Ueda, Y.; Onitake, Y.; Sueda, T.; Ohta, E.; Morihara, N.; Hirano, S.; Irisuna, F.; Hiyama, E. Circulating free DNA as non-invasive diagnostic biomarker for childhood solid tumors. J. Pediatr. Surg. 2015, 50, 2094–2097. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.Z.; Lou, F.; Yang, F.; Zhang, J.B.; Ye, H.; Chen, W.; Guan, T.; Zhao, M.Y.; Su, X.X.; Shi, R.; et al. Circulating Tumor DNA Detection in Early-Stage Non-Small Cell Lung Cancer Patients by Targeted Sequencing. Sci. Rep. 2016, 6, 31985. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ren, R.; Ren, S.; Chen, X.; Cai, W.; Zhou, F.; Zhang, Y.; Su, C.; Zhao, C.; Li, J.; et al. Peripheral blood for epidermal growth factor receptor mutation detection in non-small cell lung cancer patients. Transl. Oncol. 2014, 7, 341. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Shi, X.; Zhao, J.; He, Q.; Chen, M.; Yan, J.; Ou, Q.; Wu, X.; Shao, Y.W.; Yu, X. Mechanisms of primary resistance to EGFR targeted therapy in advanced lung adenocarcinomas. Lung Cancer 2018, 124, 110–116. [Google Scholar] [CrossRef]
- Guo, N.; Lou, F.; Ma, Y.; Li, J.; Yang, B.; Chen, W.; Ye, H.; Zhang, J.B.; Zhao, M.Y.; Wu, W.J.; et al. Circulating tumor DNA detection in lung cancer patients before and after surgery. Sci. Rep. 2016, 6, 33519. [Google Scholar] [CrossRef] [Green Version]
- Villaflor, V.; Won, B.; Nagy, R.; Banks, K.; Lanman, R.B.; Talasaz, A.; Salgia, R. Biopsy-free circulating tumor DNA assay identifies actionable mutations in lung cancer. Oncotarget 2016, 7, 66880–66891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaederlé, M.C.; Patel, S.P.; Husain, H.; Ikeda, M.; Lanman, R.B.; Banks, K.C.; Talasaz, A.; Bazhenova, L.; Kurzrock, R. Utility of Genomic Assessment of Blood-Derived Circulating Tumor DNA (ctDNA) in Patients with Advanced Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 5101–5111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Shen, X.; Li, R.; Shen, J.; Zhang, H.; Yu, L.; Liu, B.; Wang, L. The detection and significance of EGFR and BRAF in cell-free DNA of peripheral blood in NSCLC. Oncotarget 2017, 8, 49773–49782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria-Comes, T.; Palomar-Abril, V.; Ureste, M.M.; Guerola, M.T.; Maiques, I.C.M. Real-World Data of the Correlation between EGFR Determination by Liquid Biopsy in Non-squamous Non-small Cell Lung Cancer (NSCLC) and the EGFR Profile in Tumor Biopsy. Pathol. Oncol. Res. 2019, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Huang, F.; Zhang, M.; Ji, H.; Wu, S.; Zhao, Y.; Zhang, C.; Wu, J.; Wang, B.; Pan, B.; et al. Multiplex picoliter-droplet digital PCR for quantitative assessment of EGFR mutations in circulating cell-free DNA derived from advanced non-small cell lung cancer patients. Mol. Med. Rep. 2017, 16, 1157–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.; Wu, Y.L.; Lee, J.S.; Yu, C.J.; Sriuranpong, V.; Sandoval-Tan, J.; Ladrera, G.; Thongprasert, S.; Srimuninnimit, V.; Liao, M.; et al. Detection and dynamic changes of EGFR mutations from circulating tumor DNA as a predictor of survival outcomes in NSCLC patients treated with first-line intercalated erlotinib and chemotherapy. Clin. Cancer Res. 2015, 21, 3196–3203. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.J.; Zhang, H.B.; Liu, Y.H.; Zhang, F.L.; Zhu, Y.Z.; Li, Y.; Bai, J.P.; Liu, L.R.; Qu, Y.C.; Qu, X.; et al. Quantitative cell-free circulating EGFR mutation concentration is correlated with tumor burden in advanced NSCLC patients. Lung Cancer 2017, 109, 124–127. [Google Scholar] [CrossRef]
- Yao, Y.; Liu, J.; Li, L.; Yuan, Y.; Nan, K.; Wu, X.; Zhang, Z.; Wu, Y.; Li, X.; Zhu, J.; et al. Detection of circulating tumor DNA in patients with advanced non-small cell lung cancer. Oncotarget 2017, 8, 2130–2140. [Google Scholar] [CrossRef] [Green Version]
- Cui, S.; Ye, L.; Wang, H.; Chu, T.; Zhao, Y.; Gu, A.; Xiong, L.; Shi, C.; Jiang, L. Use of SuperARMS EGFR Mutation Detection Kit to Detect EGFR in Plasma Cell-free DNA of Patients With Lung Adenocarcinoma. Clin. Lung Cancer 2018, 19, 313–322. [Google Scholar] [CrossRef]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non-small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Yang, Z.; Li, C.S.; Zhao, W.; Liang, Z.X.; Dai, Y.; Zhu, Q.; Miao, K.L.; Cui, D.H.; Chen, L.A. Differences in the genomic profiles of cell-free DNA between plasma, sputum, urine, and tumor tissue in advanced NSCLC. Cancer Med. 2019, 8, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Sim, W.C.; Loh, C.H.; Toh, G.L.; Lim, C.W.; Chopra, A.; Chang, A.Y.C.; Goh, L.L. Non-invasive detection of actionable mutations in advanced non-small-cell lung cancer using targeted sequencing of circulating tumor DNA. Lung Cancer 2018, 124, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lou, F.; Wu, Y.; Sun, D.Q.; Zhang, J.B.; Chen, W.; Ye, H.; Liu, J.H.; Wei, S.; Zhao, M.Y.; et al. Circulating tumor DNA identified by targeted sequencing in advanced-stage non-small cell lung cancer patients. Cancer Lett. 2016, 370, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Hagiwara, K.; Han, B.; Tjulandin, S.; Grohé, C.; Yokoi, T.; Morabito, A.; Novello, S.; Arriola, E.; Molinier, O.; et al. ctDNA Determination of EGFR Mutation Status in European and Japanese Patients with Advanced NSCLC: The ASSESS Study. J. Thorac. Oncol. Oncol. 2016, 11, 1682–1689. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Huang, B.; Zhuang, Z.; Chen, S. Circulating tumor DNA as prognostic markers for late stage NSCLC with bone metastasis. Int. J. Biol. Markers 2018, 33, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Veldore, V.H.; Choughule, A.; Routhu, T.; Mandloi, N.; Noronha, V.; Joshi, A.; Dutt, A.; Gupta, R.; Vedam, R.; Prabhash, K. Validation of liquid biopsy: Plasma cell-free DNA testing in clinical management of advanced non-small cell lung cancer. Lung Cancer 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Shi, C.; Qian, J.; Teng, J.; Zhong, H.; Han, B. Comparison of plasma and tissue samples in epidermal growth factor receptor mutation by ARMS in advanced non-small cell lung cancer. Gene 2016, 591, 58–64. [Google Scholar] [CrossRef]
- Denis, M.G.; Lafourcade, M.P.; Le Garff, G.; Dayen, C.; Falchero, L.; Thomas, P.; Locher, C.; Oliviero, G.; Licour, M.; Reck, M.; et al. Circulating free tumor-derived DNA to detect EGFR mutations in patients with advanced NSCLC: French subset analysis of the ASSESS study. J. Thorac. Dis. 2019, 11, 1370–1378. [Google Scholar] [CrossRef]
- Guibert, N.; Hu, Y.; Feeney, N.; Kuang, Y.; Plagnol, V.; Jones, G.; Howarth, K.; Beeler, J.F.; Paweletz, C.P.; Oxnard, G.R. Amplicon-based next-generation sequencing of plasma cell-free DNA for detection of driver and resistance mutations in advanced non-small cell lung cancer. Ann. Oncol. 2018, 29, 49–1055. [Google Scholar] [CrossRef]
- Howell, J.; Atkinson, S.R.; Pinato, D.J.; Knapp, S.; Ward, C.; Minisini, R.; Burlone, M.E.; Leutner, M.; Pirisi, M.; Büttner, R.; et al. Identification of mutations in circulating cell-free tumour DNA as a biomarker in hepatocellular carcinoma. Eur. J. Cancer 2019, 116, 56–66. [Google Scholar] [CrossRef]
- Bernard, V.; Kim, D.U.; San Lucas, F.A.; Castillo, J.; Allenson, K.; Mulu, F.C.; Stephens, B.M.; Huang, J.; Semaan, A.; Guerrero, P.A.; et al. Circulating Nucleic Acids Are Associated With Outcomes of Patients With Pancreatic Cancer. Gastroenterology 2019, 156, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinugasa, H.; Nouso, K.; Miyahara, K.; Morimoto, Y.; Dohi, C.; Tsutsumi, K.; Kato, H.; Matsubara, T.; Okada, H.; Yamamoto, K. Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer 2015, 121, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Gangadhar, T.C.; Savitch, S.L.; Yee, S.S.; Xu, W.; Huang, A.C.; Harmon, S.; Lieberman, D.B.; Soucier, D.; Fan, R.; Black, T.A.; et al. Feasibility of monitoring advanced melanoma patients using cell-free DNA from plasma. Pigment. Cell Melanoma Res. 2018, 31, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haselmann, V.; Gebhardt, C.; Brechtel, I.; Duda, A.; Czerwinski, C.; Sucker, A.; Holland-Letz, T.; Utikal, J.; Schadendorf, D.; Neumaier, M. Liquid Profiling of Circulating Tumor DNA in Plasma of Melanoma Patients for Companion Diagnostics and Monitoring of BRAF Inhibitor Therapy. Clin. Chem. 2018, 64, 830–842. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Kong, Y.; Si, L.; Cui, C.; Sheng, X.; Chi, Z.; Dai, J.; Yu, S.; Ma, M.; Wu, X.; et al. Clinical significance of BRAFV600E mutation in circulating tumor DNA in Chinese patients with melanoma. Oncol. Lett. 2018, 15, 1839–1844. [Google Scholar] [CrossRef]
- Pinzani, P.; Salvianti, F.; Cascella, R.; Massi, D.; De Giorgi, V.; Pazzagli, M.; Orlando, C. Allele specific Taqman-based real-time PCR assay to quantify circulating BRAFV600E mutated DNA in plasma of melanoma patients. Clin. Chim. Acta 2010, 411, 1319–1324. [Google Scholar] [CrossRef]
- Calapre, L.; Giardina, T.; Robinson, C.; Reid, A.L.; Al-Ogaili, Z.; Pereira, M.R.; McEvoy, A.C.; Warburton, L.; Hayward, N.K.; Khattak, M.A.; et al. Locus-specific concordance of genomic alterations between tissue and plasma circulating tumor DNA in metastatic melanoma. Mol. Oncol. 2019, 13, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Sandulache, V.C.; Williams, M.D.; Lai, S.Y.; Lu, C.; William, W.N.; Busaidy, N.L.; Cote, G.J.; Singh, R.R.; Luthra, R.; Cabanillas, M.E. Real-Time Genomic Characterization Utilizing Circulating Cell-Free DNA in Patients with Anaplastic Thyroid Carcinoma. Thyroid 2017, 27, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Lee, W.S.; Lanman, R.B.; Mortimer, S.; Zill, O.A.; Kim, K.M.; Jang, K.T.; Kim, S.H.; Park, S.H.; Park, J.O.; et al. Prospective blinded study of somatic mutation detection in cell-free DNA utilizing a targeted 54-gene next generation sequencing panel in metastatic solid tumor patients. Oncotarget 2015, 6, 40360–40369. [Google Scholar] [CrossRef] [Green Version]
- Rachiglio, A.M.; Esposito Abate, R.; Sacco, A.; Pasquale, R.; Fenizia, F.; Lambiase, M.; Morabito, A.; Montanino, A.; Rocco, G.; Romano, C.; et al. Limits and potential of targeted sequencing analysis of liquid biopsy in patients with lung and colon carcinoma. Oncotarget 2016, 7, 66595–66605. [Google Scholar] [CrossRef] [Green Version]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgartner, J.M.; Raymond, V.M.; Lanman, R.B.; Tran, L.; Kelly, K.J.; Lowy, A.M.; Kurzrock, R. Preoperative Circulating Tumor DNA in Patients with Peritoneal Carcinomatosis is an Independent Predictor of Progression-Free Survival. Ann. Surg Oncol. 2018, 25, 2400–2408. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.; Yap, T.A.; Pope, L.; Cassidy, A.M.; Dukes, J.P.; Riisnaes, R.; Massard, C.; Cassier, P.A.; Miranda, S.; Clark, J.; et al. Multi-purpose utility of circulating plasma DNA testing in patients with advanced cancers. PLoS ONE 2012, 7, E47020. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, S.; Caldas, C. The implications of clonal genome evolution for cancer medicine. N. Engl. J. Med. 2013, 368, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Cleary, A.S.; Leonard, T.L.; Gestl, S.A.; Gunther, E.J. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature 2014, 508, 113–117. [Google Scholar] [CrossRef]
- Ma, F.; Zhu, W.; Guan, Y.; Yang, L.; Xia, X.; Chen, S.; Li, Q.; Guan, X.; Yi, Z.; Qian, H.; et al. ctDNA dynamics: A novel indicator to track resistance in metastatic breast cancer treated with anti-HER2 therapy. Oncotarget 2016, 7, 66020–66031. [Google Scholar] [CrossRef]
- Shatsky, R.; Parker, B.A.; Bui, N.Q.; Helsten, T.; Schwab, R.B.; Boles, S.G.; Kurzrock, R. Next-Generation Sequencing of Tissue and Circulating Tumor DNA: The UC San Diego Moores Center for Personalized Cancer Therapy Experience with Breast Malignancies. Mol. Cancer Ther. 2019, 18, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Perdigones, N.; Murtaza, M. Capturing tumor heterogeneity and clonal evolution in solid cancers using circulating tumor DNA analysis. Pharmacol. Ther. 2017, 174, 22–26. [Google Scholar] [CrossRef]
- Molina, R.; Filella, X.; Augé, J.M.; Fuentes, R.; Bover, I.; Rifa, J.; Moreno, V.; Canals, E.; Viñolas, N.; Marquez, A.; et al. Tumor markers (CEA, CA 125, CYFRA 21-1, SCC and NSE) in patients with non-small cell lung cancer as an aid in histological diagnosis and prognosis. Comparison with the main clinical and pathological prognostic factors. Tumour Biol. 2003, 24, 209–218. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef]
- Hovelson, D.H.; Liu, C.J.; Wang, Y.; Kang, Q.; Henderson, J.; Gursky, A.; Brockman, S.; Ramnath, N.; Krauss, J.C.; Talpaz, M. Rapid, ultra low coverage copy number profiling of cell-free DNA as a precision oncology screening strategy. Oncotarget 2017, 8, 89848–89866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonavia, R.; Inda, M.M.; Cavenee, W.K.; Furnari, F.B. Heterogeneity maintenance in glioblastoma: A social network. Cancer Res. 2011, 71, 4055–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pectasides, E.; Stachler, M.D.; Derks, S.; Liu, Y.; Maron, S.; Islam, M.; Alpert, L.; Kwak, H.; Kindler, H.; Polite, B.; et al. Genomic Heterogeneity as a Barrier to Precision Medicine in Gastroesophageal Adenocarcinoma. Cancer Discov. 2018, 8, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, P.K.; Shen, R.; Won, H.; Rekhtman, N.; Wang, L.; Sima, C.S.; Arora, A.; Seshan, V.; Ladanyi, M.; Berger, M.F.; et al. NextGeneration Sequencing of Stage IV Squamous Cell Lung Cancers Reveals an Association of PI3K Aberrations and Evidence of Clonal Heterogeneity in Patients with Brain Metastases. Cancer Discov. 2015, 5, 610–621. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Lee, H.S.; Kim, J.H.; Kim, Y.J.; Kwon, J.H.; Lee, J.O.; Bang, S.M.; Park, K.U.; Kim, D.W.; Kang, S.B.; et al. Different metastatic pattern according to the KRAS mutational status and site-specific discordance of KRAS status in patients with colorectal cancer. BMC Cancer 2012, 12, 347. [Google Scholar] [CrossRef] [Green Version]
- Knijn, N.; Mekenkamp, L.J.; Klomp, M.; Vink-Borger, M.E.; Tol, J.; Teerenstra, S.; Meijer, J.W.; Tebar, M.; Riemersma, S.; van Krieken, J.H.; et al. KRAS mutation analysis: A comparison between primary tumours and matched liver metastases in 305 colorectal cancer patients. Br. J. Cancer 2011, 104, 1020–1026. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wang, H.; Zang, W.; Li, B.; Rao, G.; Li, L.; Yu, Y.; Li, Z.; Dong, B.; Lu, Z.; et al. Circulating tumor DNA functions as an alternative for tissue to overcome tumor heterogeneity in advanced gastric cancer. Cancer Sci. 2017, 108, 1881–1887. [Google Scholar] [CrossRef]
- Volik, S.; Alcaide, M.; Morin, R.D.; Collins, C. Cell-free DNA (cfDNA): Clinical Significance and Utility in Cancer Shaped by Emerging Technologies. Mol. Cancer Res. 2016, 14, 898–908. [Google Scholar] [CrossRef] [Green Version]
- Bosse, K.R.; Maris, J.M. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 2016, 122, 20–33. [Google Scholar] [CrossRef]
- Olsson, E.; Winter, C.; George, A.; Chen, Y.; Howlin, J.; Tang, M.H.; Dahlgren, M.; Schulz, R.; Grabau, D.; van Westen, D.; et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 2015, 7, 1034–1047. [Google Scholar] [CrossRef]
- Mondaca, S.; Offin, M.; Borsu, L.; Myers, M.; Josyula, S.; Makhnin, A.; Shen, R.; Riely, G.J.; Rudin, C.M.; Ladanyi, M.; et al. Lessons learned from routine, targeted assessment of liquid biopsies for EGFR T790M resistance mutation in patients with EGFR mutant lung cancers. Acta Oncol. 2019, 1634–1639. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Whisenant, J.G.; Wakelee, H.; Reckamp, K.L.; Qiao, H.; Leal, T.A.; Du, L.; Hernandez, J.; Huang, V.; Blumenschein, G.R.; et al. Monitoring therapeutic response and resistance: Analysis of circulating tumor DNA in patients with ALK+ lung cancer. J. Thorac. Oncol. 2019, 14, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
- Toledo, R.A.; Garralda, E.; Mitsi, M.; Pons, T.; Monsech, J.; Vega, E.; Otero, Á.; Albarran, M.I.; Baños, N.; Durán, Y.; et al. Exome Sequencing of Plasma DNA Portrays the Mutation Landscape of Colorectal Cancer and Discovers Mutated VEGFR2 Receptors as Modulators of Antiangiogenic Therapies. Clin. Cancer Res. 2018, 24, 3550–3559. [Google Scholar] [CrossRef] [PubMed] [Green Version]


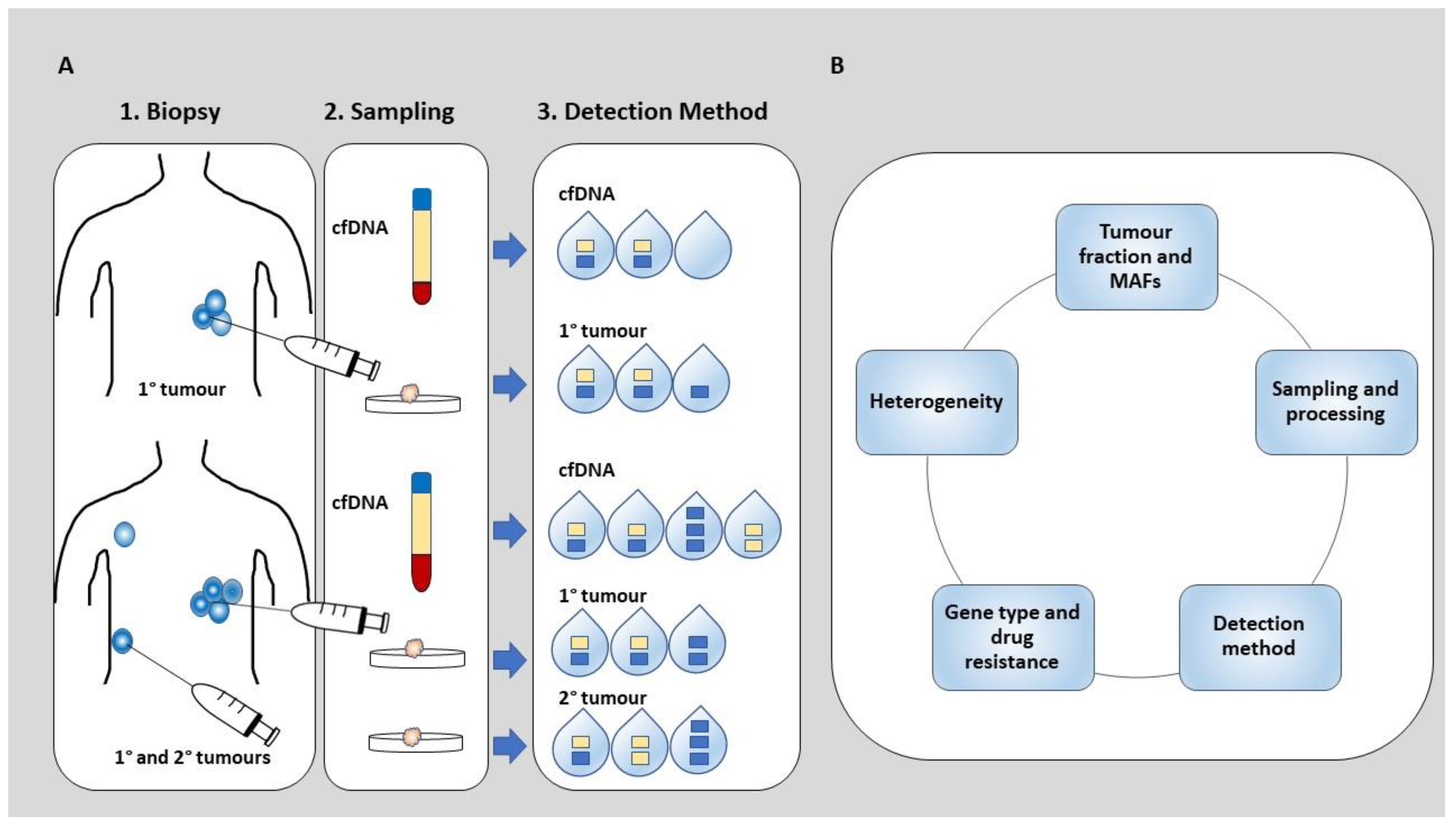{kind=link}
| Author/Cohort Size | Cancer Type | Concordance with Primary or Metastatic Tumour | Driver and Actionable Driver Alterations | Method for Tumour/cfDNA |
|---|---|---|---|---|
| Thompson/102 [15] | NSCLC | cfDNA and primary tumour (51%) compared to cfDNA and metastatic tumour (79%) for all alterations | 50 drivers and 12 resistance alterations | Targeted sequencing |
| Liu/72 [81] | NSCLC | cfDNA and primary (50%) compared to cfDNA and metastatic (65%) in 19 patients | lung cancer panel including EGFR L858R, L861Q, e19 del, e20INS, KRAS G12X, EML4-ALK, RET-KIF5B, BRAF V600E | ARMS-PCR and targeted sequencing/Sequencing and ddPCR |
| Xie/35 [83] | NSCLC | cfDNA and metastatic tumour (73.2 %), cfDNA and primary tumour (68.4%) | 56 lung cancer genes | Targeted sequencing |
| Guo/56 [72] | NSCLC | 54.6% of patients in early stage and 80% in late stage | lung & colon cancer panel (LV103) and lung cancer panel (L82) | Targeted sequencing for both, ddPCR for some cfDNA samples |
| Garcia- Saenz/49 [47] | 6 metastatic and 43 localised breast cancer | 59.1% (overall) 79.8% (for metastatic patients) | PIK3CA mutations | COBAS PIK3CA Mutation Test/ddPCR using (rare PIK3CA Mutation Assays) |
| Tzanikou/56 [84] | Early and metastatic breast cancer | 48.2% (27/56) in early breast cancer, 66.6% (18/27) in metastatic breast cancer | PIK3CA mutations | Custom method and ddPCR |
| Chae/12 [70] | mCRC | For sequencing approaches, 39% for primary and 55% for metastasis in all panel | 21 gene panel including TP53, PIK3CA and KRAS | Targeted sequencing/targeted sequencing, OnTarget assay and ddPCR |
| Kato/55 [85] | Esophageal, gastroesophageal junction, and gastric adenocarcinoma | concordance between ctDNA and primary site vs. cfDNA and metastatic site for TP53: 52.2% vs. 87.5% and for ERBB2: 78.3% vs. 100% | 54-73 gene panel including KRAS, TP53 and PTEN | Sequencing |
| Author/Cohort Size | Cancer Type | Concordance Information | Positive Concordance (MUT/MUT)|Negative Concordance (WT/WT)|Discordance | Driver and Actionable Driver Alterations | Method for Tumour/cfDNA |
|---|---|---|---|---|---|
| Wyatt/45 [10] | MPC | 88.9% in clinically actionable genes | 72 genes including AR, BRCA2, PTEN, PIK3CA and TP53 | WES/targeted sequencing | |
| Vandekerkhove/53 [95] | MPC | 80% in matched samples | Panel of genes including TP53 and DNA repair genes | Targeted sequencing | |
| Grasselli/146 [31] | mCRC | 89.7% | 10.3% (15 cases) concordance | RAS mutations | SoC PCR/ddPCR (BEAMing) |
| Bando/280 [32] | mCRC | 86.4% (242/280) | 82.1% (110/134)|90.4% (132/146)|11% (38/280) | RAS mutations | ddPCR (BEAMing) |
| Garcia-Foncillas/236 [65] | mCRC | 89% (210/236) improved to 92% by re-analysis | 86.30%|92.40%|In lung metastasis cases (tissue only) | RAS mutations | SoC PCR/OncoBEAM |
| Schmiegel/98 [33] | mCRC | 91.8% (90/98) | 90.4% (47/52)|93.5% (43/46)|- | RAS mutations | Sequencing, SOC, ddPCR (BEAMing)/ddPCR (BEAMing) |
| Demuth/28 [75] | mCRC | 79% for Ion Torrent seq.- 89% for ddPCR | KRAS mutations | Genotyping/Sequencing and ddPCR | |
| Spindler/229 [96] | mCRC | 85% | KRAS | Standard methods/ARMS-qPCR | |
| Bachet/425 [97] | mCRC | 71%- 89% | RAS | Standard methods/sequencing | |
| Vidal/115 [98] | mCRC | 93% | RAS | Standard methods/OncoBEAM | |
| Buim/26 [99] | mCRC | 71% | KRAS | Standard methods/pyrosequencing | |
| Thierry/140 [66] | mCRC | 72%, 74% and 87% for KRAS exon 2, KRAS exon 3–4 and BRAF V600E, respectively | 28 mutations including KRAS, BRAF, NRAS | Standard methods/Q-PCR-based-method (IntPlex V) | |
| Wang/184 [100] | mCRC | 93.33% in pre-treatment cohort | KRAS, NRAS, BRAF, PIK3CA | ARMS-based PCR /Firefly | |
| Osumi/101 [101] | mCRC | 77.2% (78/101) for RAS | 23 cases for RAS (discordance) | 14 CRC- related genes including, APC, TP53 and RAS | Standard methods/Sequencing |
| Germano/20 [102] | mCRC | 84.6% (11/13 cases) | RAS, BRAF, ERBB2 | Standard methods/ddPCR | |
| Beije/12 [103] | mCRC | KRAS, PIK3CA and TP53 for OnTarget assay (80%), digital PCR (93%) | 21 CRC gene panel including TP53, PIK3CA and KRAS | Sequencing/Sequencing, OnTarget assay and ddPCR | |
| Kato/94 [104] | CRC | ranging from 63.2% APC to 85.5% BRAF | panel including KRAS, TP53 and APC | Sequencing | |
| Mohamed Suhaimi/44 [105] | CRC | 84.1% for KRAS and 90.9% BRAF | KRAS and BRAF | Genotyping/sanger sequencing, HRM and ASPCR and pyroseqeuncing | |
| Takeshita/35 [44] | MBC | 74.3% (26/35) | 1/35|25/35|9/35 | ESR1 mutations | ddPCR |
| Beaver/29 [48] | Early BC | 14/15 mutations | PIK3CA mutations | Sanger sequencing, ddPCR/ddPCR | |
| Higgins/49 and 60 [45] | MBC (49 retrospective and 60 prospective) | 100% in 41 matched retrospectives, 72.5% in 51 prospectives | 27.5% in 51 prospective samples (discordance) | PIK3CA mutations | Sequencing or BEAMing/ddPCR (BEAMing) |
| Chae/45 [70] | BC | 91.0%–94.2% for all genes | 10.8%–15.1% (3.5% for CNAs) positive concordance | Foundation 1/Guardant360 | |
| Board/76 [46] | 46 metastatic, 30 localised BC | 95% in 41 matched samples | 80%|(47%) discordance | PIK3CA mutations | Standard methods/ARMS PCR* |
| Garcia- Saenz/49 [47] | 6 Metastatic and 43 localised BC | 59.1% (overall) 79.8% (for metastatic patients) | PIK3CA mutations | COBAS PIK3CA Mutation Test/ddPCR using (rare PIK3CA Mutation Assays) | |
| Kodahl/66 [49] | PIK3CA- mutated MBC | 83% (20/24 cases) | PIK3CA mutations | ddPCR | |
| Combaret/114 [52] | NB | 100% | 1/1|1/1|0 | ALK; F1174L (e23: 3520, T>C) | ddPCR and targeted sequencing |
| 55 cases | 6 cases|49 cases|4 (cfDNA only), 1 (tumour only) | ALK, F1174L (e23:3522, C>A) | |||
| 58 cases | 12 cases|46 cases|1 (cfDNA only), 1 (tumour only) | ALK; R1275Q (e25:3824, G>A) | |||
| Kurihara/10 [106] | NB | 100% | 2/2|8/8|0 | MYCN | FISH/ddPCR |
| Chen/58 [107] | Stage IA, IB, and IIA NSCLC | 50.4% | Panel of 50 driver alterations including EGFR, KRAS, PIK3CA and TP53 | Targeted sequencing | |
| Sung/126 [36] | NSCLC | 90% (ex19del), and 88.33% (L858R) | EGFR (ex19del and L858R) | Genotyping/Targeted sequencing and ddPCR | |
| Li/164 [108] | NSCLC | 73.6% | EGFR mutations | ARMS | |
| Lee/81 [37] | NSCLC | 86.2% (ex19del) and 87.9% (L858R) | EGFR (ex19del and L858R) | Genotyping/ddPCR | |
| Thompson/102 [15] | NSCLC | 79% (19/24) for actionable EGFR mutations 97.5% across all variants | 60% across all variants | 50 drivers, 12 resistance alterations | Sequencing |
| Jin/69 [109] | NSCLC | 88.2% for EGFR mutations | EGFR Ex19del, L858R, G719S/C, and L861Q, TP53 mutations, amp. of RB1, PIK3CA and MYC | Targeted Sequencing | |
| Yang/73 [68] | NSCLC | 74% (54/73) | 26% (19/73) (discordance) | EGFR mutations | Sequencing/Sequencing and ddPCR |
| Guo/41 [110] | NSCLC | 78.1% | 50 cancer genes including EGFR, KRAS, and TP53 | Targeted sequencing | |
| Villaflor/68 [111] | NSCLC | High concordance for truncal oncogenic drivers, 71% for EGFR | Driver alterations including EGFR | targeted multiplex testing or tissue- based sequencing/Guardant360 | |
| Liu/72 [81] | NSCLC | 54.2% for all clinically actionable alterations, EGFR L858R (93.1%), EGFR e19 del (90.3%), KRAS G12X (96.9%), ALK rearrang. (96.9%) | MET or HER2 CNA in cfDNA but not tumour (discordance) | EGFR L858R,L861Q,e19 del, e20 INS, KRAS G12X, EML4-ALK, RET-KIF5B and BRAF V600E | ARMS-PCR and sequencing/Sequencing (cfDNA also validated by ddPCR) |
| Schwaederle/88 [112] | NSCLC | 76.5- 80.8 % for EGFR mutations depending on sampling time | 7/26 (EGFR mutations) 53% for all alterations|14/26 (EGFR mutations)|5/26 (EGFR mutations) 2 cfDNA only, 3 tumour only | Mutations in TP53, EGFR, MET, KRAS and ALK | Sequencing or genotyping or no test/Guardant360 |
| Yang/107 [113] | NSCLC | 74.8% (80/107) EGFR 88.8% (95/107) BRAF | EGFR and BRAF mutations | Standard methods/competitive Allele-Specific TaqMan PCR (CastPCR) | |
| Soria- Comes/102 [114] | NSCLC | 87.4% | EGFR mutations | Cobas EGFR assay | |
| Yu/22 [115] | Advanced NSCLC | For 19DEL and L858R (90% and 95%, respectively) | EGFR mutations (19DEL and L858R) | ARMS/ddPCR | |
| Mok/241 [116] | Advanced NSCLC | 88% (209/238) | EGFR mutations | Cobas 4800 FFPET test/Cobas 4800 blood test | |
| Zhu/51 [117] | Advanced NSCLC | 86.73% | EGFR mutations | Standard methods/ddPCR | |
| Yao/39 [118] | Advanced NSCLC | 78.21% (30.5/39) for all genes | 47.43%|30.77%|21.8% | Panel of 40 genes including EGFR, KRAS, PIK3CA, ALK and RET | Targeted sequencing |
| Cui/180 [119] | Advanced NSCLC | 87.8% | 97.3%|85.3% | EGFR mutations | Standard methods/SuperARMS |
| Leighl/282 [120] | Advanced NSCLC | 98.2% for EGFR, ALK, ROS1, BRAF | SoC PCR/Guardant360 | ||
| Wu/50 [121] | Advanced NSCLC | 86% (43/50 cases) | Driver alterations including EGFR, TP53, RB1 | Sequencing | |
| Sim/50 [122] | Advanced NSCLC | 81% for EGFR | BRAF, EGFR, ERBB2, KRAS, NRAS, PIK3CA | Sequencing | |
| Xu/42 [123] | Advanced NSCLC | Overall 76% | EGFR, KRAS, PIK3CA, and TP53 | Targeted sequencing | |
| Reck/1311 [124] | Advanced NSCLC | 89% (in 1162 matched samples) | EGFR mutations | Standard methods of local centres | |
| Jia/150 [125] | Advanced NSCLC | 94.7% for EGFR and RAS | EGFR and KRAS mutations | Standard methods/ddPCR | |
| Veldore/132 [126] | Advanced NSCLC | 96.96% | EGFR mutations | Standard methods/sequencing | |
| Ma/219 [127] | Advanced NSCLC | 82% | EGFR mutations | ARMS | |
| Denis/1311 [128] | Advanced NSCLC | 96% in 126 matched samples | EGFR mutations | Standard methods | |
| Guibert/46 [129] | Advanced NSCLC | ROS1/ALK (8/9), EGFR (9/9), BRAF/MET/HER2 (4/6) | EGFR mutations, ROS1, ALK, BRAF/MET/HER2 | Standard methods/Sequencing and ddPCR | |
| Hahn/19 [90] | mRCC | 8.6% concordance | DNA repair genes (discordance) | Foundation 1/Guardant360 | |
| Howell/51 [130] | HCC | moderate | ARID1A AXIN1, ATM, CTNNB1, HNF1A and TP53 | Targeted sequencing | |
| Bernard/194 [131] | PDAC (localised or metastatic) | >95% for KRAS in surgically resected tissue | KRAS | ddPCR | |
| Cohen/221 [93] | PDAC | 100% | KRAS mutations | Sequencing | |
| Pishvaian/34 [94] | Pancreatic cancer | Low concordance | Panels including KRAS and TP53 | Foundation 1/Guardant360 | |
| Kinugasa/75 [132] | Pancreatic cancer | 77.3% (58/75) | KRAS | PCR-PHFA/ddPCR | |
| Gangadhar/25 [133] | Advanced melanoma | 81.8% (9/11) | 61 gene panel including BRAF, NRAS and KIT | Standard methods/Sequencing | |
| Haselmann/634 [134] | Melanoma | BRAFV600 (92.3%–94.5%) | BRAF | SoC PCR/BEAMing | |
| Tang/58 [135] | Melanoma | 70.2% | BRAF | Standard methods/3D ddPCR | |
| Pinzani/55 [136] | Melanoma | 80% | BRAF | Allele-specific RT-PCR | |
| Calapre/24 [137] | Advanced melanoma | 80% (in a subgroup of 7 matching tissue and cfDNA) | 30 melanoma genes including BRAF, NRAS, NF1 and TERT | Targeted sequencing (ddPCR for some cfDNA cases) | |
| Sandulache/23 [138] | Anaplastic thyroid carcinoma | high for BRAF, PIK3CA, NRAS, and PTEN and moderate for TP53 | Highest discordance in post-treatment patients | 50 gene panel for tissue, 70 gene panel for cfDNA, including BRAF, NRAS, TP53 and PIK3CA | Sequencing |
| Author/Cohort Size | Cancer Type | Concordance or Discordance Information | Driver and Actionable Driver Alterations | Method for Tumour/cfDNA |
|---|---|---|---|---|
| Kim/75 [139] | CRC, melanoma gastrointestinal stromal tumour, renal cell carcinoma, gastric cancer, sarcoma and 4 other cancers | 85.9% when all detected mutations considered across all tumour types | Panel of 54 cancer genes | Sequencing |
| Rachiglio/79 [140] | 44 metastatic NSCLC and 35 mCRC | High concordance for EGFR (17/22) and lower concordance for other drivers | ALK, EGFR, ERBB2, ERBB4, FGFR1, FGFR2, FGFR3, MET, DDR2, KRAS, PIK3CA, BRAF, AKT1, PTEN, NRAS, MAP2K1, STK11, NOTCH1, CTNNB1, SMAD4, FBXW7, TP53 | Sequencing/Sequencing and ddPCR |
| Phallen/200 [141] | Breast, colorectal, Lung, Ovarian cancer | High concordance | 58 cancer related genes including drivers | Sequencing (TEC-Seq) |
| Riviere/213 [9] | colorectal adenocarcinoma, appendiceal adenocarcinoma, hepatocellular carcinoma, pancreatic ductal adenocarcinoma | 96% KRAS amplification, 94% MYC amplification, 95% KRAS G12V, 91% EGFR amplification 96% overall concordance on gene level | Panel of 68 genes including KRAS amplification, MYC amplification, KRAS G12V, EGFR amplification | Guardant360 panel |
| Jovelet/334 [76] | thoracic, gastrointestinal, breast, head and neck, gynaecologic and urologic cancers | On a gene level only 173/347 mutations corresponded between cfDNA and tumour tissue, 174/347 discordant mutations | Panel of 50 cancer hotspots V2 (CHP2) including TP53, KRAS, PIK3CA, EGFR, APC | Sequencing |
| Leary/91 [56] | Colorectal or breast cancer | Good concordance for cancer driver genes such as ERBB2 and CDK6 | Chromosomal alterations including rearrangements of CDK6 and ERBB2 loci | Sequencing |
| Toor/28 [89] | advanced stage gastrointestinal and lung malignancies | 7% for lung subgroup, 8% for gastrointestinal subgroup (90% positive concordance), high discordance with respect to driver and actionable alteration | Caris or paradigm panels/Guardant360 panel | |
| Baumgartner/80 [142] | appendix cancer, colorectal, peritoneal mesothelioma, small bowel, cholangiocarcinoma, ovarian, and testicular cancer | Overall, positive, and negative concordance was 96.7%, 35.3%, and 96.6% (in 15 cases with matched samples) | Panel of genes including TP53 and KRAS | Sequencing |
| Kato/55 [85] | Esophageal, gastroesophageal junction, and gastric adenocarcinoma | 61.3% (TP53 alterations) to 87.1% (KRAS alterations) | 54-73 gene panel Including KRAS, TP53 and PTEN | Sequencing |
| Perkins/105 [143] | Colorectal, melanoma, breast, prostate, ovarian, NSCLC, mesothelioma, sarcoma, glioblastoma, ACUP, cholangiocarcinoma, and cervical, endometrial, duodenal, esophageal, pancreatic and renal cancers | Overall 60% (25/42) | BRAF, KRAS, NRAS, HRAS, MET, AKT, PIK3CA, KIT | Standard methods/Mass Spectrometry TypePLEX and OncoCarta panel (v1.0) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahangiri, L.; Hurst, T. Assessing the Concordance of Genomic Alterations between Circulating-Free DNA and Tumour Tissue in Cancer Patients. Cancers 2019, 11, 1938. https://doi.org/10.3390/cancers11121938
Jahangiri L, Hurst T. Assessing the Concordance of Genomic Alterations between Circulating-Free DNA and Tumour Tissue in Cancer Patients. Cancers. 2019; 11(12):1938. https://doi.org/10.3390/cancers11121938
Chicago/Turabian StyleJahangiri, Leila, and Tara Hurst. 2019. "Assessing the Concordance of Genomic Alterations between Circulating-Free DNA and Tumour Tissue in Cancer Patients" Cancers 11, no. 12: 1938. https://doi.org/10.3390/cancers11121938
APA StyleJahangiri, L., & Hurst, T. (2019). Assessing the Concordance of Genomic Alterations between Circulating-Free DNA and Tumour Tissue in Cancer Patients. Cancers, 11(12), 1938. https://doi.org/10.3390/cancers11121938