A Triple Co-Delivery Liposomal Carrier That Enhances Apoptosis via an Intrinsic Pathway in Melanoma Cells

 , , , ,
, , , ,
Abstract
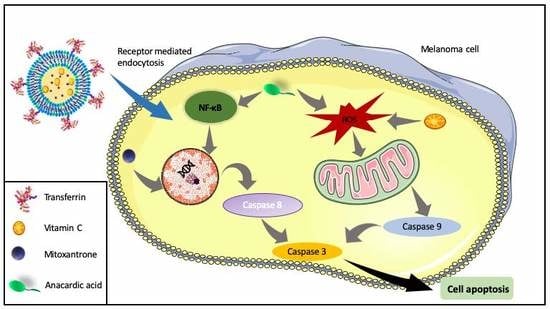
:
1. Introduction
2. Results
2.1. Liposomes Preparation and Characterization
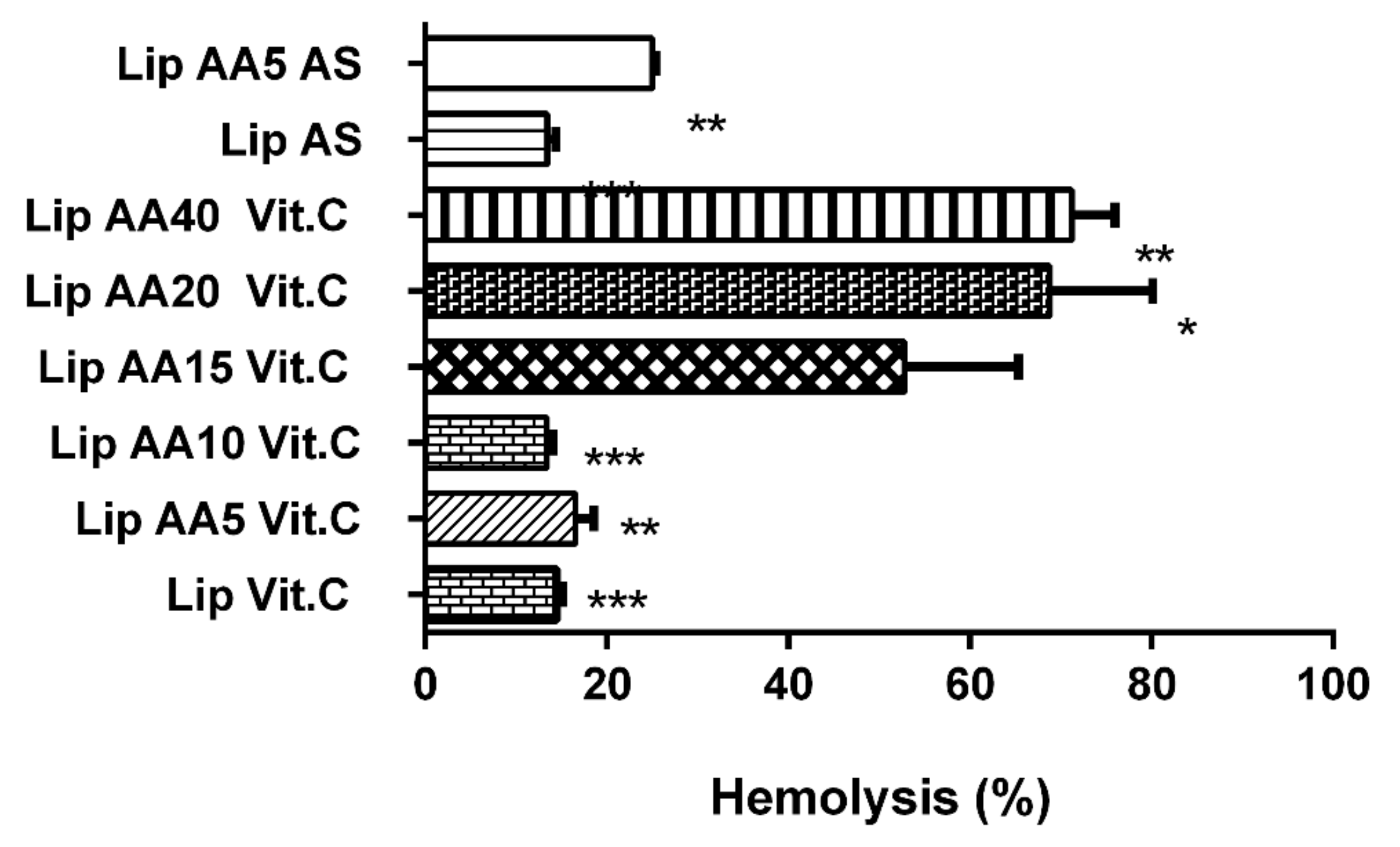
2.2. Liposomes Demonstrate Selectivity toward Melanoma Cells and Low Cardio- and Hepatotoxicity and a Lower Hemolysis Ratio
2.3. ROS Formation Induced by Liposome Formulations
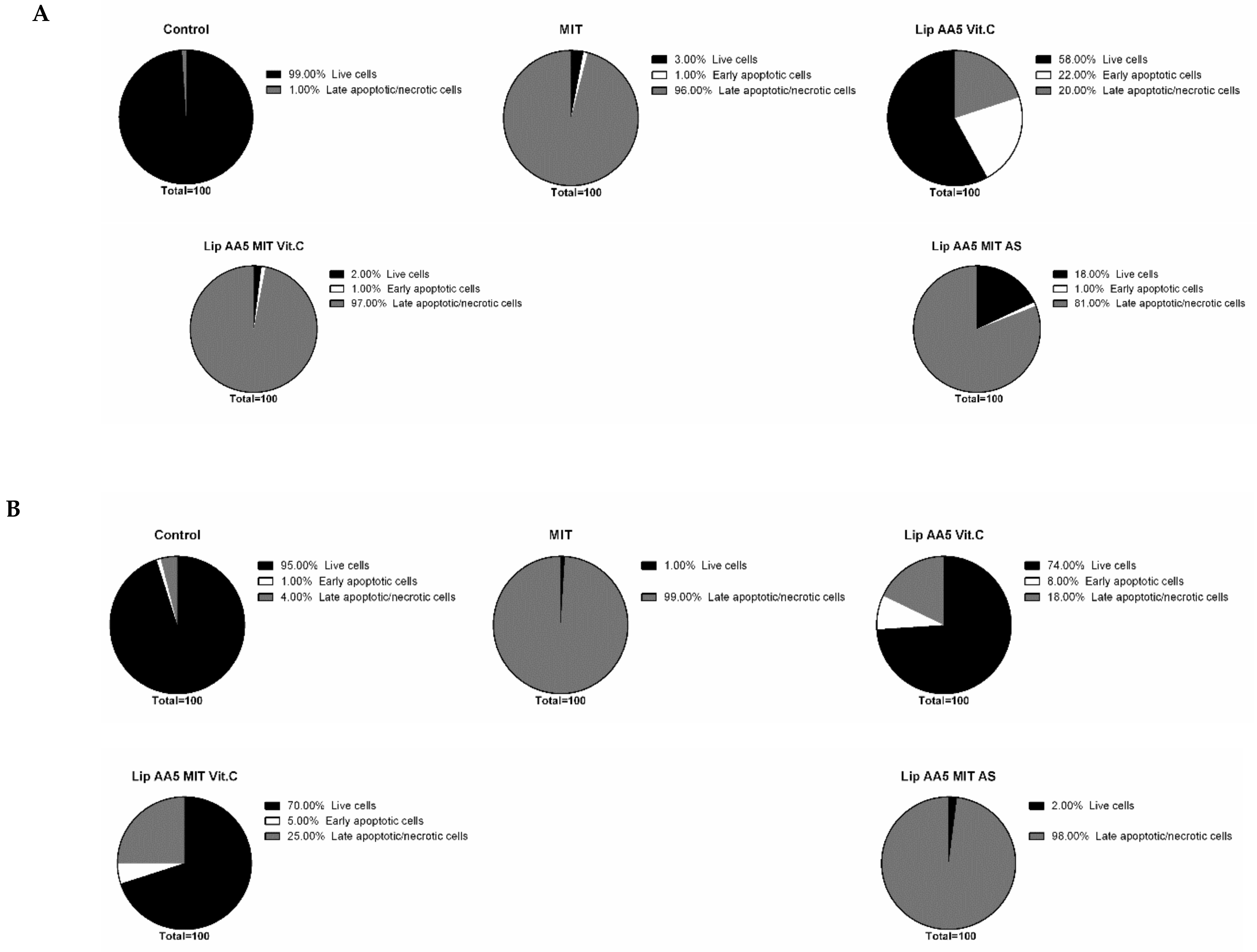
2.4. Liposome Treatment Enhances the Apoptosis
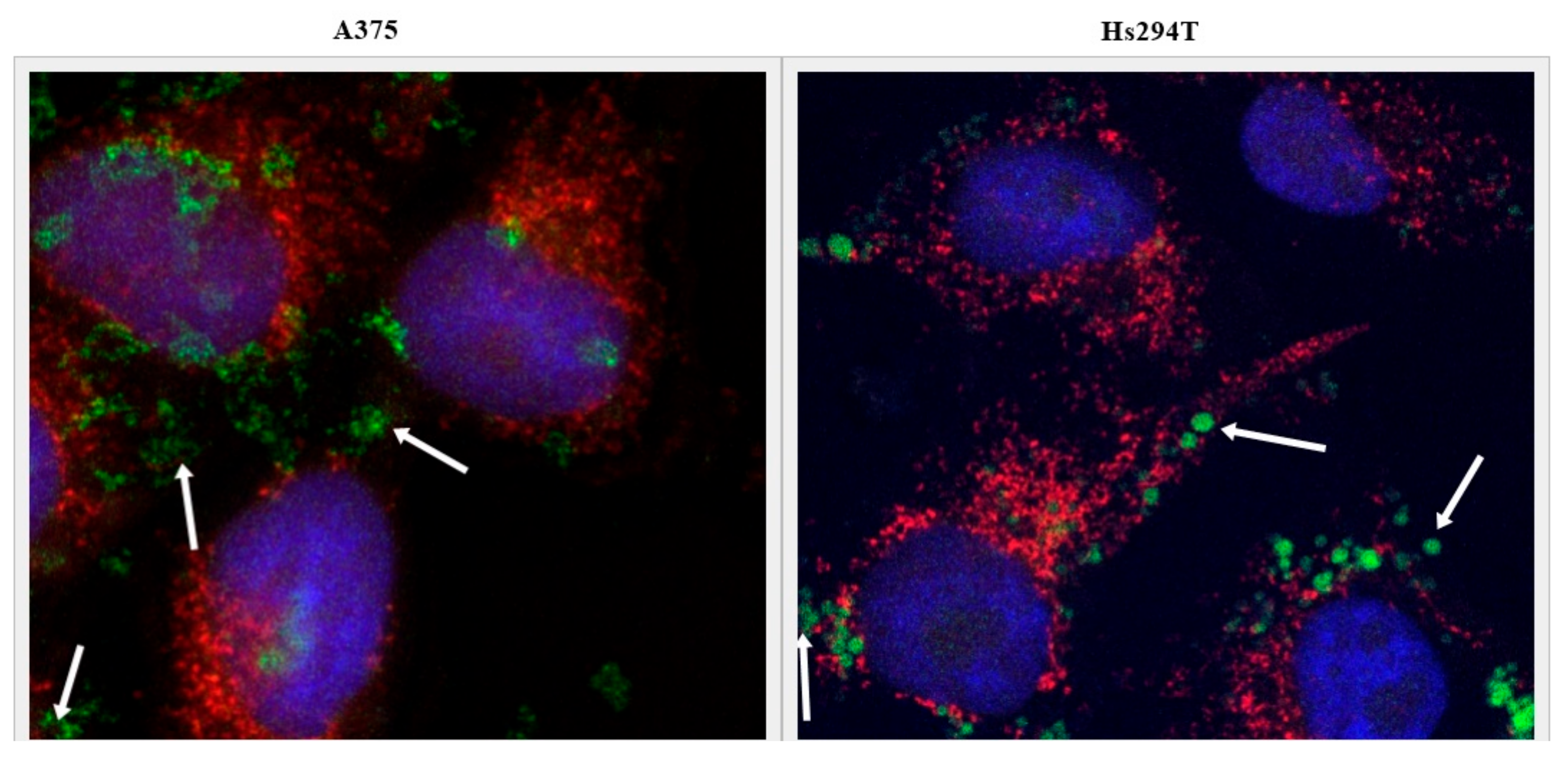
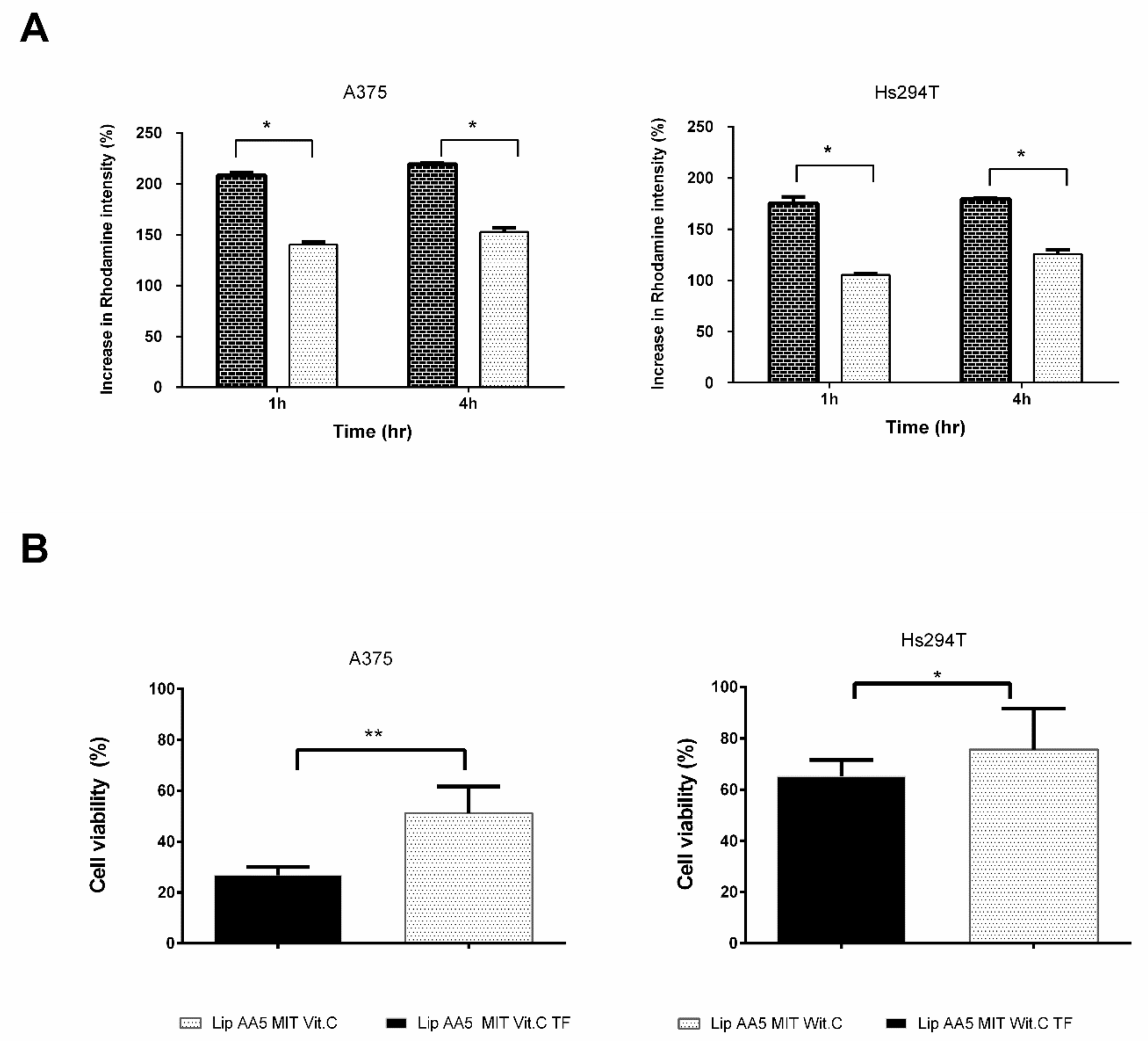
2.5. Tf-Targeted Liposomes Demonstrate Efficient Melanoma Cell Killing
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation and Characterization OF Liposomes
4.3. Targeting of the Liposomes
4.3.1. Synthesis of pNP-PEG-PE
4.3.2. Attachment of Protein
4.4. Ex Vivo Red Blood Cell Hemolysis Assay
4.5. Cell Culture and Treatment
4.6. Cell Viability
4.6.1. MTT Assay
4.6.2. ATP Measurement
4.7. Determination of the Association of Targeted Liposomes with Cells
4.8. Analysis of Apoptosis
4.9. Lactate Dehydrogenase Leakage Assay
4.10. Measurement of Intracellular Reactive Oxygen Species
4.11. Caspase 9 and 3/7 Assays
4.12. Cytochrome C Staining
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haass, N.K.; Smalley, K.S.M.; Herlyn, M. The role of altered cell-cell communication in melanoma progression. J. Mol. Histol. 2004, 35, 309–318. [Google Scholar] [CrossRef]
- Pleshkan, V.V.; Zinov’eva, M.V.; Sverdlov, E.D. Melanoma: Surface markers as the first point of targeted delivery of therapeutic genes in multilevel gene therapy. Mol. Biol. 2011, 45, 416–433. [Google Scholar] [CrossRef]
- Vultur, A.; Herlyn, M. SnapShot: Melanoma. Cancer Cell 2013, 23, 706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggermont, A.M.M.; Kirkwood, J.M. Re-evaluating the role of dacarbazine in metastatic melanoma: What have we learned in 30 years? Eur. J. Cancer 2004, 40, 1825–1836. [Google Scholar] [CrossRef] [PubMed]
- Frinton, E.; Tong, D.; Tan, J.; Read, G.; Kumar, V.; Kennedy, S.; Lim, C.; Board, R.E. Metastatic melanoma: Prognostic factors and survival in patients with brain metastases. J. Neurooncol. 2017, 135, 507–512. [Google Scholar] [CrossRef] [Green Version]
- Dickson, P.V.; Gershenwald, J.E. Staging and prognosis of cutaneous melanoma. Surg. Oncol. Clin. 2011, 20, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Grosso, J.F.; Jure-Kunkel, M.N. CTLA-4 blockade in tumor models: An overview of preclinical and translational research. Cancer Immun. Arch. 2013, 13, 5. [Google Scholar]
- Mishra, H.; Mishra, P.K.; Ekielski, A.; Jaggi, M.; Iqbal, Z.; Talegaonkar, S. Melanoma treatment: From conventional to nanotechnology. J. Cancer Res. Clin. Oncol. 2018, 144, 2283–2302. [Google Scholar] [CrossRef]
- Seremet, T.; Jansen, Y.; Planken, S.; Njimi, H.; Delaunoy, M.; El Housni, H.; Awada, G.; Schwarze, J.K.; Keyaerts, M.; Everaert, H.; et al. Undetectable circulating tumor DNA (ctDNA) levels correlate with favorable outcome in metastatic melanoma patients treated with anti-PD1 therapy. J. Transl. Med. 2019, 17, 303. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Thompson, J.A. PD-1 Blockade in Melanoma: A Promising Start, but a Long Way to Go. JAMA 2016, 315, 1573–1575. [Google Scholar] [CrossRef]
- Brück, T.B.; Brück, D.W. Oxidative metabolism of the anti-cancer agent mitoxantrone by horseradish, lacto-and lignin peroxidase. Biochimie 2011, 93, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Seiter, K. Toxicity of the topoisomerase II inhibitors. Expert Opin. Drug Saf. 2005, 4, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, O.; Kieseier, B.C.; Hartung, H.-P. Therapeutic role of mitoxantrone in multiple sclerosis. Pharmacol. Ther. 2006, 109, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Seifrtová, M.; Havelek, R.; Chmelařová, M.; Cmielová, J.; Muthná, D.; Stoklasová, A.; Zemánková, S.; Rezáčová, M. The effect of ATM and ERK1/2 inhibition on mitoxantrone-induced cell death of leukaemic cells. Folia Biol. (Praha) 2011, 57, 74–81. [Google Scholar]
- Fan, J.-T.; Kuang, B.; Zeng, G.-Z.; Zhao, S.-M.; Ji, C.-J.; Zhang, Y.-M.; Tan, N.-H. Biologically active arborinane-type triterpenoids and anthraquinones from Rubia yunnanensis. J. Nat. Prod. 2011, 74, 2069–2080. [Google Scholar] [CrossRef]
- Wang, J.; Gao, R.; Li, Q.; Xie, S.; Zhao, J.; Wang, C. Synthesis, cytotoxicity, and cell death profile of polyaminoanthraquinones as antitumor agents. Chem. Biol. Drug Des. 2012, 80, 909–917. [Google Scholar] [CrossRef]
- Khan, S.N.; Lal, S.K.; Kumar, P.; Khan, A.U. Effect of mitoxantrone on proliferation dynamics and cell-cycle progression. Biosci. Rep. 2010, 30, 375–381. [Google Scholar] [CrossRef]
- Avasarala, J.R.; Cross, A.H.; Clifford, D.B.; Singer, B.A.; Siegel, B.A.; Abbey, E.E. Rapid onset mitoxantrone-induced cardiotoxicity in secondary progressive multiple sclerosis. Mult. Scler. 2003, 9, 59–62. [Google Scholar] [CrossRef]
- Rossato, L.G.; Costa, V.M.; de Pinho, P.G.; Arbo, M.D.; de Freitas, V.; Vilain, L.; de Lourdes Bastos, M.; Palmeira, C.; Remião, F. The metabolic profile of mitoxantrone and its relation with mitoxantrone-induced cardiotoxicity. Arch. Toxicol. 2013, 87, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Krishnaja, A.P.; Sharma, N.K. Ascorbic acid potentiates mitomycin C-induced micronuclei and sister chromatid exchanges in human peripheral blood lymphocytes in vitro. Teratog. Carcinog. Mutagen. 2003, 23, 99–112. [Google Scholar] [CrossRef]
- Kurbacher, C.M.; Wagner, U.; Kolster, B.; Andreotti, P.E.; Krebs, D.; Bruckner, H.W. Ascorbic acid (vitamin C) improves the antineoplastic activity of doxorubicin, cisplatin, and paclitaxel in human breast carcinoma cells in vitro. Cancer Lett. 1996, 103, 183–189. [Google Scholar] [CrossRef]
- Sorice, A.; Guerriero, E.; Capone, F.; Colonna, G.; Castello, G.; Costantini, S. Ascorbic acid: Its role in immune system and chronic inflammation diseases. Mini Rev. Med. Chem. 2014, 14, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Heaney, M.L.; Gardner, J.R.; Karasavvas, N.; Golde, D.W.; Scheinberg, D.A.; Smith, E.A.; O’Connor, O.A. Vitamin C antagonizes the cytotoxic effects of antineoplastic drugs. Cancer Res. 2008, 68, 8031–8038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrax, J.; Calderon, P.B. The controversial place of vitamin C in cancer treatment. Biochem. Pharmacol. 2008, 76, 1644–1652. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Mamede, A.C.; Tavares, S.D.; Abrantes, A.M.; Trindade, J.; Maia, J.M.; Botelho, M.F. The role of vitamins in cancer: A review. Nutr. Cancer 2011, 63, 479–494. [Google Scholar] [CrossRef]
- Sirivech, S.; Frieden, E.; Osaki, S. The release of iron from horse spleen ferritin by reduced flavins. Biochem. J. 1974, 143, 311–315. [Google Scholar] [CrossRef] [Green Version]
- Eliot, H.; Gianni, L.; Myers, C. Oxidative destruction of DNA by the adriamycin-iron complex. Biochemistry 1984, 23, 928–936. [Google Scholar] [CrossRef]
- Muindi, J.R.; Sinha, B.K.; Gianni, L.; Myers, C.E. Hydroxyl radical production and DNA damage induced by anthracycline-iron complex. FEBS Lett. 1984, 172, 226–230. [Google Scholar] [CrossRef] [Green Version]
- Zweier, J.L. Reduction of O2 by iron-adriamycin. J. Biol. Chem. 1984, 259, 6056–6058. [Google Scholar]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanyam, K.; Swaminathan, V.; Ranganathan, A.; Kundu, T.K. Small molecule modulators of histone acetyltransferase p300. J. Biol. Chem. 2003, 278, 19134–19140. [Google Scholar] [CrossRef] [Green Version]
- Sung, B.; Pandey, M.K.; Ahn, K.S.; Yi, T.; Chaturvedi, M.M.; Liu, M.; Aggarwal, B.B. Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-kappaB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-kappaBalpha kinase, leading to potentiation of apoptosis. Blood 2008, 111, 4880–4891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; He, L.; Zhang, L.; Chen, J.; Yi, Z.; Zhang, J.; Liu, M.; Pang, X. Anacardic acid (6-pentadecylsalicylic acid) inhibits tumor angiogenesis by targeting Src/FAK/Rho GTPases signaling pathway. J. Pharmacol. Exp. Ther. 2011, 339, 403–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, S.; Nitoda, T.; Kubo, I. Effects of salicylic acid on mushroom tyrosinase and B16 melanoma cells. Z. Naturforsch. C J. Biosci. 2007, 62, 227–233. [Google Scholar] [CrossRef]
- Park, W.J.; Ma, E. Inhibition of PCAF histone acetyltransferase, cytotoxicity and cell permeability of 2-acylamino-1-(3-or 4-carboxy-phenyl)benzamides. Molecules 2012, 17, 13116–13131. [Google Scholar] [CrossRef]
- Schultz, D.J.; Wickramasinghe, N.S.; Ivanova, M.M.; Isaacs, S.M.; Dougherty, S.M.; Imbert-Fernandez, Y.; Cunningham, A.R.; Chen, C.; Klinge, C.M. Anacardic acid inhibits estrogen receptor alpha-DNA binding and reduces target gene transcription and breast cancer cell proliferation. Mol. Cancer Ther. 2010, 9, 594–605. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Chen, B.; He, L.; Tang, Y.; Jiang, Z.; Yin, G.; Wang, J.; Jiang, X. Anacardic acid (6-pentadecylsalicylic acid) induces apoptosis of prostate cancer cells through inhibition of androgen receptor and activation of p53 signaling. Chin. J. Cancer Res. 2012, 24, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-C.; Hernández-Ledesma, B.; de Lumen, B.O. Cell proliferation inhibitory and apoptosis-inducing properties of anacardic acid and lunasin in human breast cancer MDA-MB-231 cells. Food Chem. 2011, 125, 630–636. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K. ABC proteins protect the human body and maintain optimal health. Biosci. Biotechnol. Biochem. 2011, 75, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabekura, T.; Hiroi, T.; Kawasaki, T.; Uwai, Y. Effects of natural nuclear factor-kappa B inhibitors on anticancer drug efflux transporter human P-glycoprotein. Biomed. Pharmacother. 2015, 70, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.-H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an antioxidant: Evaluation of its role in disease prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Re, A.; Nanni, S.; Aiello, A.; Granata, S.; Colussi, C.; Campostrini, G.; Spallotta, F.; Mattiussi, S.; Pantisano, V.; D’Angelo, C.; et al. Anacardic acid and thyroid hormone enhance cardiomyocytes production from undifferentiated mouse ES cells along functionally distinct pathways. Endocrine 2016, 53, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Seong, Y.-A.; Shin, P.-G.; Kim, G.-D. Anacardic acid induces mitochondrial-mediated apoptosis in the A549 human lung adenocarcinoma cells. Int. J. Oncol. 2013, 42, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Kamath, R.; Jin, S.; Balasubramani, M.; Pandita, T.K.; Rajasekaran, B. Tip60-mediated acetylation activates transcription independent apoptotic activity of Abl. Mol. Cancer 2011, 10, 88. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Jiang, X.; Chen, S.; Price, B.D. Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS Lett. 2006, 580, 4353–4356. [Google Scholar] [CrossRef] [Green Version]
- Ashino, H.; Shimamura, M.; Nakajima, H.; Dombou, M.; Kawanaka, S.; Oikawa, T.; Iwaguchi, T.; Kawashima, S. Novel function of ascorbic acid as an angiostatic factor. Angiogenesis 2003, 6, 259–269. [Google Scholar] [CrossRef]
- Lee, S.K.; Kang, J.S.; Jung, D.J.; Hur, D.Y.; Kim, J.E.; Hahm, E.; Bae, S.; Kim, H.W.; Kim, D.; Cho, B.J.; et al. Vitamin C suppresses proliferation of the human melanoma cell SK-MEL-2 through the inhibition of cyclooxygenase-2 (COX-2) expression and the modulation of insulin-like growth factor II (IGF-II) production. J. Cell Physiol. 2008, 216, 180–188. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Lai, W.-W.; Chou, C.-C.; Kuo, H.-M.; Li, T.-M.; Chung, J.-G.; Yang, J.-H. Sodium ascorbate inhibits growth via the induction of cell cycle arrest and apoptosis in human malignant melanoma A375.S2 cells. Melanoma Res. 2006, 16, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Türkez, H.; Aydin, E. The protective role of ascorbic acid on imazalil-induced genetic damage assessed by the cytogenetic tests. Toxicol. Ind. Health 2012, 28, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, M.; Miyazaki, T.; Masatsuji-Kato, E.; Tsuzuki, T.; Oribe, T.; Miwa, N. Cytoprotection against ischemia-induced DNA cleavages and cell injuries in the rat liver by pro-vitamin C via hydrolytic conversion into ascorbate. Mol. Cell. Biochem. 2003, 252, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Duarte, T.L.; Cooke, M.S.; Jones, G.D.D. Gene expression profiling reveals new protective roles for vitamin C in human skin cells. Free Radic. Biol. Med. 2009, 46, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Urushibara, A.; Yamashita, S.; Shikazono, N.; Yokoya, A.; Katsumura, Y. Chemical repair of base lesions, AP-sites, and strand breaks on plasmid DNA in dilute aqueous solution by ascorbic acid. Biochem. Biophys. Res. Commun. 2013, 434, 341–345. [Google Scholar] [CrossRef]
- Stasiuk, M.; Kozubek, A. Membrane perturbing properties of natural phenolic and resorcinolic lipids. FEBS Lett. 2008, 582, 3607–3613. [Google Scholar] [CrossRef] [Green Version]
- Jaromin, A.; Zarnowski, R.; Kozubek, A. Emulsions of oil from Adenanthera pavonina L. seeds and their protective effect. Cell Mol. Biol. Lett. 2006, 11, 438–448. [Google Scholar] [CrossRef]
- Jaromin, A.; Kozubek, A.; Suchoszek-Lukaniuk, K.; Malicka-Blaszkiewicz, M.; Peczynska-Czoch, W.; Kaczmarek, L. Liposomal formulation of DIMIQ, potential antitumor indolo[2,3-b]quinoline agent and its cytotoxicity on hepatoma Morris 5123 cells. Drug Deliv. 2008, 15, 49–56. [Google Scholar] [CrossRef]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Borner, C. The Bcl-2 protein family: Sensors and checkpoints for life-or-death decisions. Mol. Immunol. 2003, 39, 615–647. [Google Scholar] [CrossRef]
- Allen, T.M. Long-circulating (sterically stabilized) liposomes for targeted drug delivery. Trends Pharmacol. Sci. 1994, 15, 215–220. [Google Scholar] [CrossRef]
- Regev, R.; Yeheskely-Hayon, D.; Katzir, H.; Eytan, G.D. Transport of anthracyclines and mitoxantrone across membranes by a flip-flop mechanism. Biochem. Pharmacol. 2005, 70, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Mayer, L.D.; Cullis, P.R.; Bally, M.B. The Use of Transmembrane pH Gradient-Driven Drug Encapsulation in the Pharmacodynamic Evaluation of Liposomal Doxorubicin. J. Liposome Res. 1994, 4, 529–553. [Google Scholar] [CrossRef]
- Reszka, R.; Beck, P.; Fichtner, I.; Hentschel, M.; Richter, J.; Kreuter, J. Body distribution of free, liposomal and nanoparticle-associated mitoxantrone in B16-melanoma-bearing mice. J. Pharmacol. Exp. Ther. 1997, 280, 232–237. [Google Scholar] [PubMed]
- Bellosillo, B.; Villamor, N.; López-Guillermo, A.; Marcé, S.; Bosch, F.; Campo, E.; Montserrat, E.; Colomer, D. Spontaneous and drug-induced apoptosis is mediated by conformational changes of Bax and Bak in B-cell chronic lymphocytic leukemia. Blood 2002, 100, 1810–1816. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Targeting cancer cells by exploiting their resistance. Trends Mol. Med. 2003, 9, 307–312. [Google Scholar] [CrossRef]
- Weinberg, E.D. Iron and neoplasia. Biol. Trace Elem. Res. 1981, 3, 55–80. [Google Scholar] [CrossRef]
- Daniels, T.R.; Delgado, T.; Helguera, G.; Penichet, M.L. The transferrin receptor part II: Targeted delivery of therapeutic agents into cancer cells. Clin. Immunol. 2006, 121, 159–176. [Google Scholar] [CrossRef]
- O’Donnell, J.L.; Joyce, M.R.; Shannon, A.M.; Harmey, J.; Geraghty, J.; Bouchier-Hayes, D. Oncological implications of hypoxia inducible factor-1alpha (HIF-1alpha) expression. Cancer Treat. Rev. 2006, 32, 407–416. [Google Scholar] [CrossRef]
- Qing, Y.; Shuo, W.; Zhihua, W.; Huifen, Z.; Ping, L.; Lijiang, L.; Xiaorong, Z.; Liming, C.; Daiwen, X.; Yu, H.; et al. The in vitro antitumor effect and in vivo tumor-specificity distribution of human-mouse chimeric antibody against transferrin receptor. Cancer Immunol. Immunother. 2006, 55, 1111–1121. [Google Scholar] [CrossRef]
- Elliott, R.L.; Stjernholm, R.; Elliott, M.C. Preliminary evaluation of platinum transferrin (MPTC-63) as a potential nontoxic treatment for breast cancer. Cancer Detect. Prev. 1988, 12, 469–480. [Google Scholar] [PubMed]
- Beyer, U.; Roth, T.; Schumacher, P.; Maier, G.; Unold, A.; Frahm, A.W.; Fiebig, H.H.; Unger, C.; Kratz, F. Synthesis and in vitro efficacy of transferrin conjugates of the anticancer drug chlorambucil. J. Med. Chem. 1998, 41, 2701–2708. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Shiramoto, S.; Miyashita, M.; Fujishima, Y.; Kaneo, Y. Tumor targeting based on the effect of enhanced permeability and retention (EPR) and the mechanism of receptor-mediated endocytosis (RME). Int. J. Pharm. 2004, 277, 39–61. [Google Scholar] [CrossRef] [PubMed]
- Raso, V.; Basala, M. A highly cytotoxic human transferrin-ricin A chain conjugate used to select receptor-modified cells. J. Biol. Chem. 1984, 259, 1143–1149. [Google Scholar]
- O’Keefe, D.O.; Draper, R.K. Characterization of a transferrin-diphtheria toxin conjugate. J. Biol. Chem. 1985, 260, 932–937. [Google Scholar]
- Nakase, I.; Lai, H.; Singh, N.P.; Sasaki, T. Anticancer properties of artemisinin derivatives and their targeted delivery by transferrin conjugation. Int. J. Pharm. 2008, 354, 28–33. [Google Scholar] [CrossRef]
- Ward, C.M.; Read, M.L.; Seymour, L.W. Systemic circulation of poly(L-lysine)/DNA vectors is influenced by polycation molecular weight and type of DNA: Differential circulation in mice and rats and the implications for human gene therapy. Blood 2001, 97, 2221–2229. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.X.; Szoka, F.C. The influence of polymer structure on the interactions of cationic polymers with DNA and morphology of the resulting complexes. Gene Ther. 1997, 4, 823–832. [Google Scholar] [CrossRef] [Green Version]
- White, S.; Taetle, R.; Seligman, P.A.; Rutherford, M.; Trowbridge, I.S. Combinations of anti-transferrin receptor monoclonal antibodies inhibit human tumor cell growth in vitro and in vivo: Evidence for synergistic antiproliferative effects. Cancer Res. 1990, 50, 6295–6301. [Google Scholar]
- Nagasawa, H.; Uto, Y.; Kirk, K.L.; Hori, H. Design of hypoxia-targeting drugs as new cancer chemotherapeutics. Biol. Pharm. Bull. 2006, 29, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Ang, S.O.; Chen, H.; Hirota, K.; Gordeuk, V.R.; Jelinek, J.; Guan, Y.; Liu, E.; Sergueeva, A.I.; Miasnikova, G.Y.; Mole, D.; et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002, 32, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Liu, Y.; Li, Y.; Peng, J.; Li, L.; Liu, J.; Shen, X.; Shen, G.; Tu, Y. Hypoxia: Dual effect on the expression of transferrin receptor in human melanoma A375 cell line. Exp. Dermatol. 2007, 16, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.S.; Carvalho, S.; Cabral, J.; Reis, C.A.; Gärtner, F. Canine tumors: A spontaneous animal model of human carcinogenesis. Transl. Res. 2012, 159, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, W.; Yu, W.; Xie, Y.; Zhang, X.; Zhang, Y.; Ma, X. Development of an in vitro multicellular tumor spheroid model using microencapsulation and its application in anticancer drug screening and testing. Biotechnol. Prog. 2005, 21, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Becher, O.J.; Holland, E.C. Genetically engineered models have advantages over xenografts for preclinical studies. Cancer Res. 2006, 66, 3355–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, S.J.; Sun, C.-H.; Tromberg, B.J.; Cristini, V.; De Magalhães, N.; Hirschberg, H. Multicell tumor spheroids in photodynamic therapy. Lasers Surg. Med. 2006, 38, 555–564. [Google Scholar] [CrossRef]
- Santini, M.T.; Rainaldi, G.; Indovina, P.L. Apoptosis, cell adhesion and the extracellular matrix in the three-dimensional growth of multicellular tumor spheroids. Crit. Rev. Oncol. Hematol. 2000, 36, 75–87. [Google Scholar] [CrossRef]
- Anada, T.; Masuda, T.; Honda, Y.; Fukuda, J.; Arai, F.; Fukuda, T.; Suzuki, O. Three-dimensional cell culture device utilizing thin membrane deformation by decompression. Sens. Actuators B Chem. 2010, 147, 376–379. [Google Scholar] [CrossRef]
- Legut, M.; Lipka, D.; Filipczak, N.; Piwoni, A.; Kozubek, A.; Gubernator, J. Anacardic acid enhances the anticancer activity of liposomal mitoxantrone towards melanoma cell lines-in vitro studies. Int. J. Nanomed. 2014, 9, 653–668. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.C. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 1980, 104, 10–14. [Google Scholar] [CrossRef]
- Biswas, S.; Deshpande, P.P.; Navarro, G.; Dodwadkar, N.S.; Torchilin, V.P. Lipid modified triblock PAMAM-based nanocarriers for siRNA drug co-delivery. Biomaterials 2013, 34, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaromin, A.; Korycińska, M.; Piętka-Ottlik, M.; Musiał, W.; Peczyńska-Czoch, W.; Kaczmarek, Ł.; Kozubek, A. Membrane perturbations induced by new analogs of neocryptolepine. Biol. Pharm. Bull. 2012, 35, 1432–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | ||||
| Amount of Anacardic Acid in Liposome Membrane (mol%) | Diameter (nm) | Zeta Potential (mV) | PDI | Encapsulation Efficiency (%) |
| 0 | 112 ± 1.2 | 0.87 ± 0.39 | 0.036 | 99.5 |
| 5 | 111 ± 1.5 | −4.31 ± 0.49 | 0.042 | 99.6 |
| 10 | 115 ± 2.3 | −5.42 ± 0.63 | 0.051 | 98.9 |
| 15 | 112 ± 0.8 | −5.74 ± 1.1 | 0.046 | 99.2 |
| 20 | 105 ± 2.5 | −4.12 ± 0.9 | 0.032 | 99.2 |
| 40 | 110 ± 2.1 | −2.81 ± 0.5 | 0.06 | 98.6 |
| B | ||||
| Liposomal Formulation | Diameter (nm) | Zeta Potential (mV) | PDI | Encapsulation Efficiency (%) |
| LipAA5 Vit. C | 111 ± 1.5 | −4.31 ± 0.49 | 0.042 | 99.0 |
| LipAA5 Vit. C TF | 119 ± 1.5 | −3.71 ± 0.5 | 0.05 | 89.9 |
| Liposomal Formulation | Cell Line | |||||
|---|---|---|---|---|---|---|
| A375 | Hs294T | NHDF | ||||
| IC50 48 h | IC50 72 h | IC50 48 h | IC50 72 h | IC50 48 h | IC50 72 h | |
| Lip Vit.C | 286.5 ± 31 | 158.37 ± 35 | 74.92 ± 7.5 | 43.95 ± 0.3 | 174.3 ± 7.3 | 122.06 ± 9 |
| LipAA5 Vit.C | 15.4 ± 3 | 12.63 ± 5 | 30.43 ± 6.65 | 0.52 ± 0.03 | 188.5 ± 20.8 | 152.93 ± 11 |
| Lip AA10 Vit.C | 17 ± 0.3 | 58.71 ± 7.4 | 3.54 ± 0.5 | 1 ± 0.03 | 23.4 ± 1.5 | 20.01 ± 0.46 |
| Lip AA15 Vit.C | 14.8 ± 0.08 | 1.78 ± 0.5 | 1.93 ± 0.5 | 5.59 ± 1.34 | 19.57 ± 0.8 | 19.32 ± 0.93 |
| Lip AA20 Vit.C | 17.7 ± 2.1 | 2.71 ± 0.55 | 1.7 ± 0.4 | 0.6 ± 0.04 | 26.87 ± 3 | 12.12 ± 2.8 |
| Lip AA40 Vit.C | 1.35 ± 0.2 | 0.93 ± 0.09 | 1.26 ± 0.07 | 0.29 ± 0.06 | 20 ± 1.3 | 9.1 ± 0.84 |
| Lip AS | 240.5 ± 57 | 99.06 ± 14.2 | 123.15 ± 7 | 86.33 ± 6.8 | 44.5 ± 3.6 | 6 ± 1.1 |
| Lip AA5 AS | 36.1 ± 6 | 92.37 ± 18.9 | 109.43 ± 7.7 | 55.64 ± 9.4 | 58.36 ± 6.1 | 5.4 ± 1.3 |
| Lip MIT Vit.C | 50.44 ± 4.3 | 10.58 ± 3 | 19.52 ± 7.6 | 10.3 ± 2 | 41.25 ± 3.21 | 20.48 ± 4.25 |
| Lip AA5 MIT Vit.C | 0.4 ± 0.05 | 0.24 ± 0.1 | 1.69 ± 0.3 | 0.66 ± 0.13 | 84.04 ± 12.7 | 44.19 ± 5.7 |
| Lip AA10 MIT Vit.C | 0.47 ± 0.02 | 0.39 ± 0.02 | 1.88 ± 0.33 | 0.94 ± 0.04 | 4.7 ± 0.43 | 1. ± 0.04 |
| Lip AA15 MIT Vit.C | 0.37 ± 0.07 | 0.39 ± 0.01 | 0.77 ± 0.05 | 0.18 ± 0.005 | 1.5 ± 0.28 | 0.9 ± 0.006 |
| Lip AA20 MIT Vit.C | 0.46 ± 0.03 | 0.28 ± 0.002 | 0.18 ± 0.001 | 0.1 ± 0.007 | 0.84 ± 0.03 | 0.23 ± 0.009 |
| Lip AA40 MIT Vit.C | 0.04 ± 0.003 | 0.02 ± 0.002 | 0.12 ± 0.06 | 0.07 ± 0.006 | 0.3 ± 0.02 | 0.19 ± 0.004 |
| Lip MIT AS | 3.87 ± 1.34 | 32.5 ± 2.51 | 17.71 ± 1.22 | 7.11 ± 1.42 | 40.53 ± 6.2 | 1.03 ± 0.06 |
| Lip AA5 MIT AS | 19.88 ± 4.15 | 0.6 ± 0.2 | 16.6 ± 1.7 | 1.19 ± 0.26 | 35.92 ± 3.15 | 1 ± 0.28 |
| MIT | 0.22 ± 0.014 | 0.075 ± 0.001 | 0.165 ± 0.03 | 0.13 ± 0.02 | 0.43 ± 0.02 | 0.15 ± 0.02 |
| Liposomal Formulation | Cell Line | ||
|---|---|---|---|
| A375 | Hs294T | NHDF | |
| Lip AA5 MIT AS | 0.011 | 0.085 | 0.366 |
| Lip AA5 MIT Vit.C | <0.01 | 0.194 | 57.064 |
| Formulation Name/Abbreviation | HSPC | AA | DSPE-PEG2000 | Cholesterol | Drug/Lipid Ratio | Ion Gradient Generator |
|---|---|---|---|---|---|---|
| Lip MIT Vit. C | 55 | 0 | 5 | 40 | 0.2 | Ammonium ascorbate |
| Lip AA5 MIT Vit. C | 55 | 5 | 5 | 35 | 0.2 | Ammonium ascorbate |
| Lip AA10 MIT Vit. C | 55 | 10 | 5 | 30 | 0.2 | Ammonium ascorbate |
| Lip AA15 MIT Vit. C | 55 | 15 | 5 | 25 | 0.2 | Ammonium ascorbate |
| Lip AA20 MIT Vit. C | 55 | 20 | 5 | 20 | 0.2 | Ammonium ascorbate |
| Lip AA40 MIT Vit. C | 55 | 40 | 5 | 0 | 0.2 | Ammonium ascorbate |
| Lip AA5 Vit. C | 55 | 5 | 5 | 35 | 0 | Ammonium ascorbate |
| Lip AA10 Vit. C | 55 | 10 | 5 | 30 | 0 | Ammonium ascorbate |
| Lip AA15 Vit. C | 55 | 15 | 5 | 25 | 0 | Ammonium ascorbate |
| Lip AA20 Vit. C | 55 | 20 | 5 | 20 | 0 | Ammonium ascorbate |
| Lip AA40 Vit. C | 55 | 40 | 5 | 0 | 0 | Ammonium ascorbate |
| Lip AS | 55 | 0 | 5 | 40 | 0 | Ammonium sulfate |
| Lip MIT AS | 55 | 0 | 5 | 40 | 0.2 | Ammonium sulfate |
| Lip AA5 AS | 55 | 5 | 5 | 35 | 0 | Ammonium sulfate |
| Lip AA5 MIT AS | 55 | 5 | 5 | 35 | 0.2 | Ammonium sulfate |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filipczak, N.; Jaromin, A.; Piwoni, A.; Mahmud, M.; Sarisozen, C.; Torchilin, V.; Gubernator, J. A Triple Co-Delivery Liposomal Carrier That Enhances Apoptosis via an Intrinsic Pathway in Melanoma Cells. Cancers 2019, 11, 1982. https://doi.org/10.3390/cancers11121982
Filipczak N, Jaromin A, Piwoni A, Mahmud M, Sarisozen C, Torchilin V, Gubernator J. A Triple Co-Delivery Liposomal Carrier That Enhances Apoptosis via an Intrinsic Pathway in Melanoma Cells. Cancers. 2019; 11(12):1982. https://doi.org/10.3390/cancers11121982
Chicago/Turabian StyleFilipczak, Nina, Anna Jaromin, Adriana Piwoni, Mohamed Mahmud, Can Sarisozen, Vladimir Torchilin, and Jerzy Gubernator. 2019. "A Triple Co-Delivery Liposomal Carrier That Enhances Apoptosis via an Intrinsic Pathway in Melanoma Cells" Cancers 11, no. 12: 1982. https://doi.org/10.3390/cancers11121982
APA StyleFilipczak, N., Jaromin, A., Piwoni, A., Mahmud, M., Sarisozen, C., Torchilin, V., & Gubernator, J. (2019). A Triple Co-Delivery Liposomal Carrier That Enhances Apoptosis via an Intrinsic Pathway in Melanoma Cells. Cancers, 11(12), 1982. https://doi.org/10.3390/cancers11121982