An Effective Multi-Stage Liposomal DNA Origami Nanosystem for In Vivo Cancer Therapy

 ,
,  , and
, and 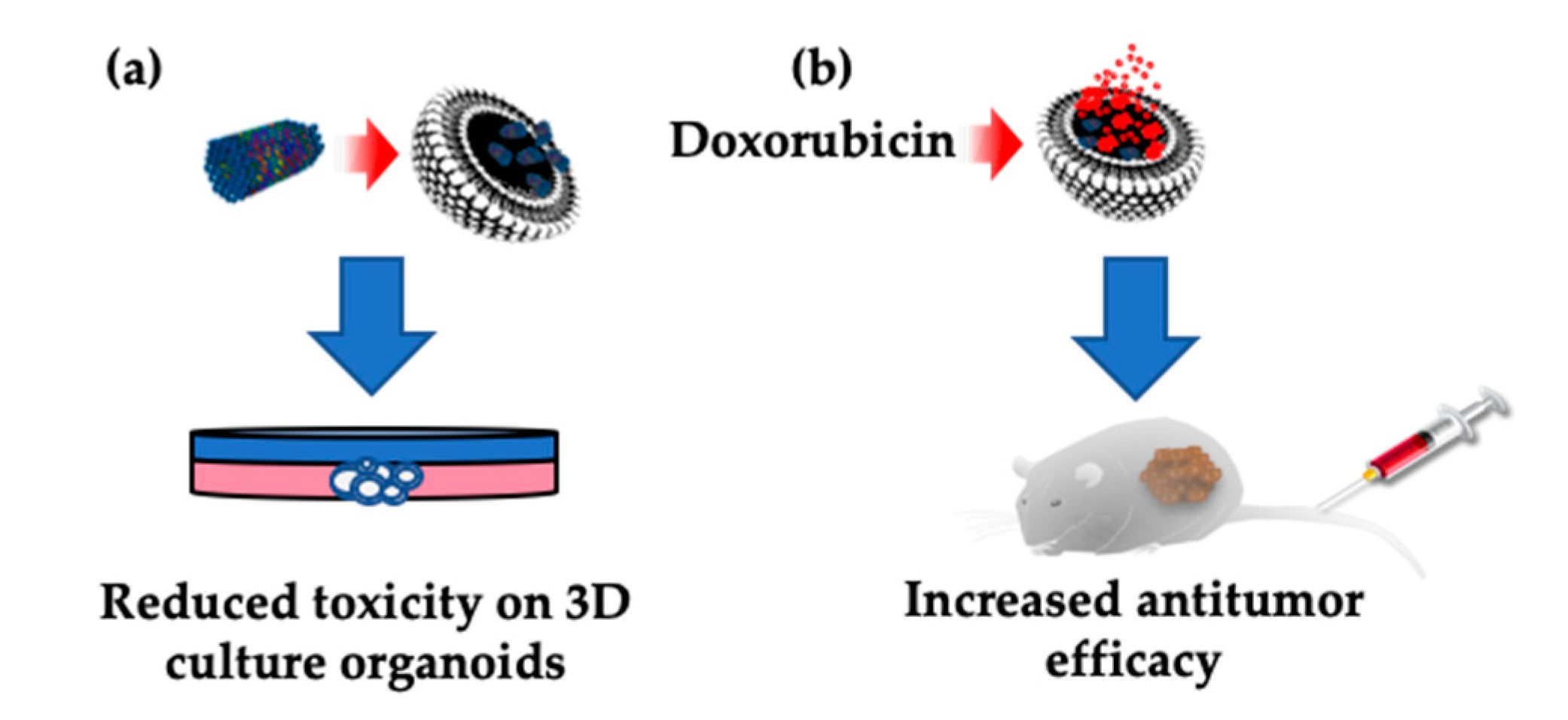{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Synthesis and Characterization
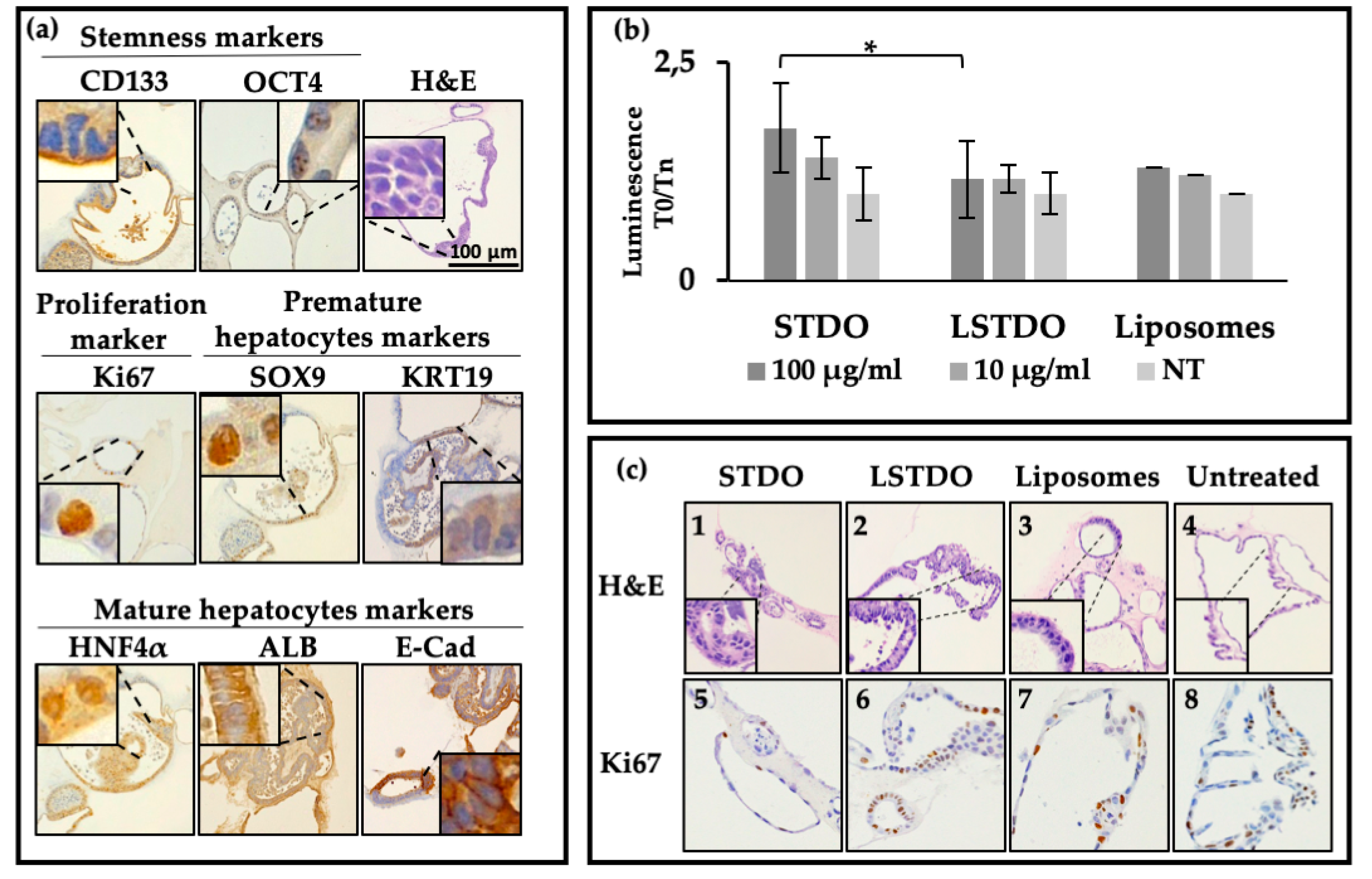
2.2. Liver Organoid Toxicity
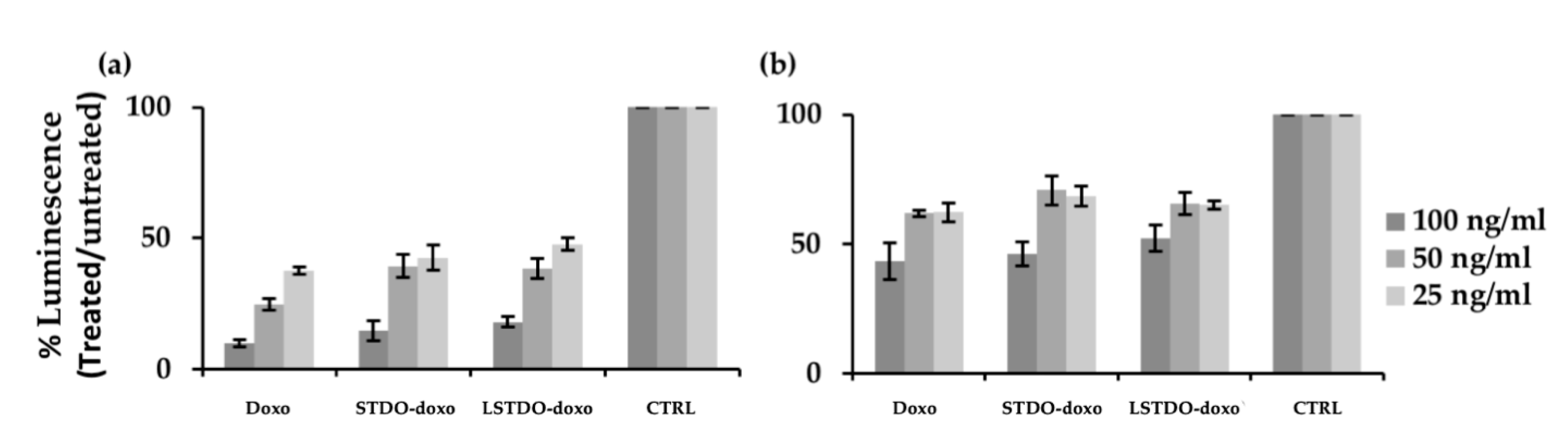
2.3. In Vitro Efficacy
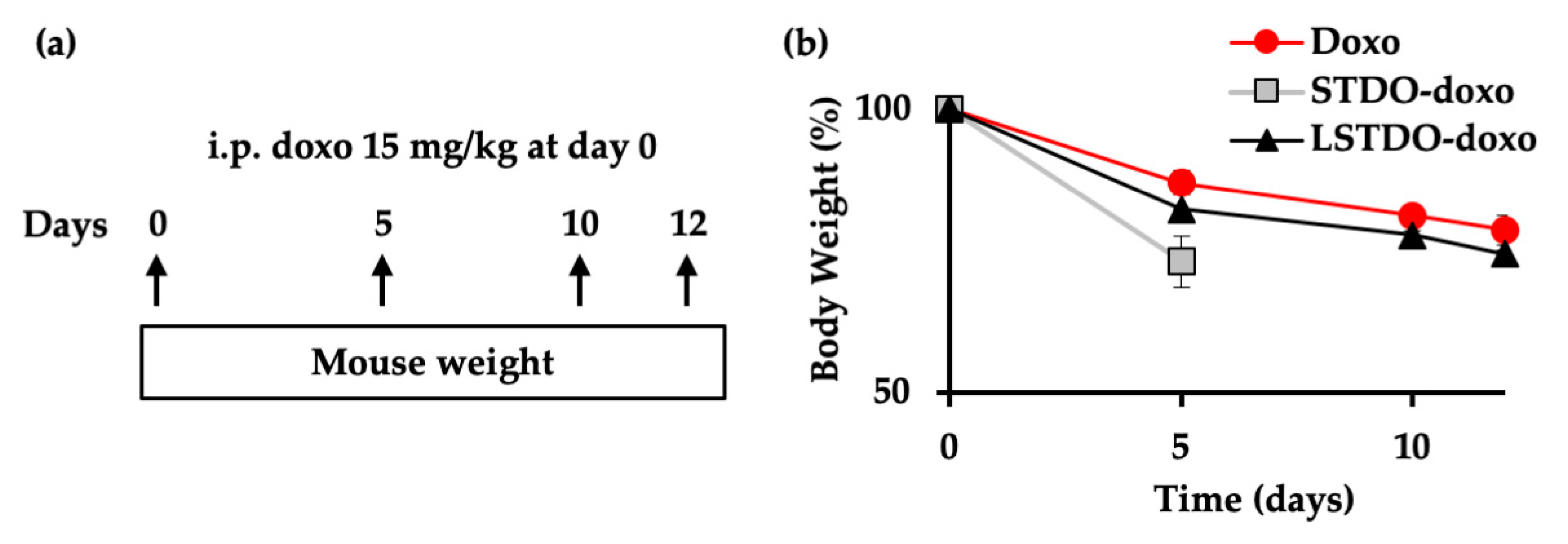
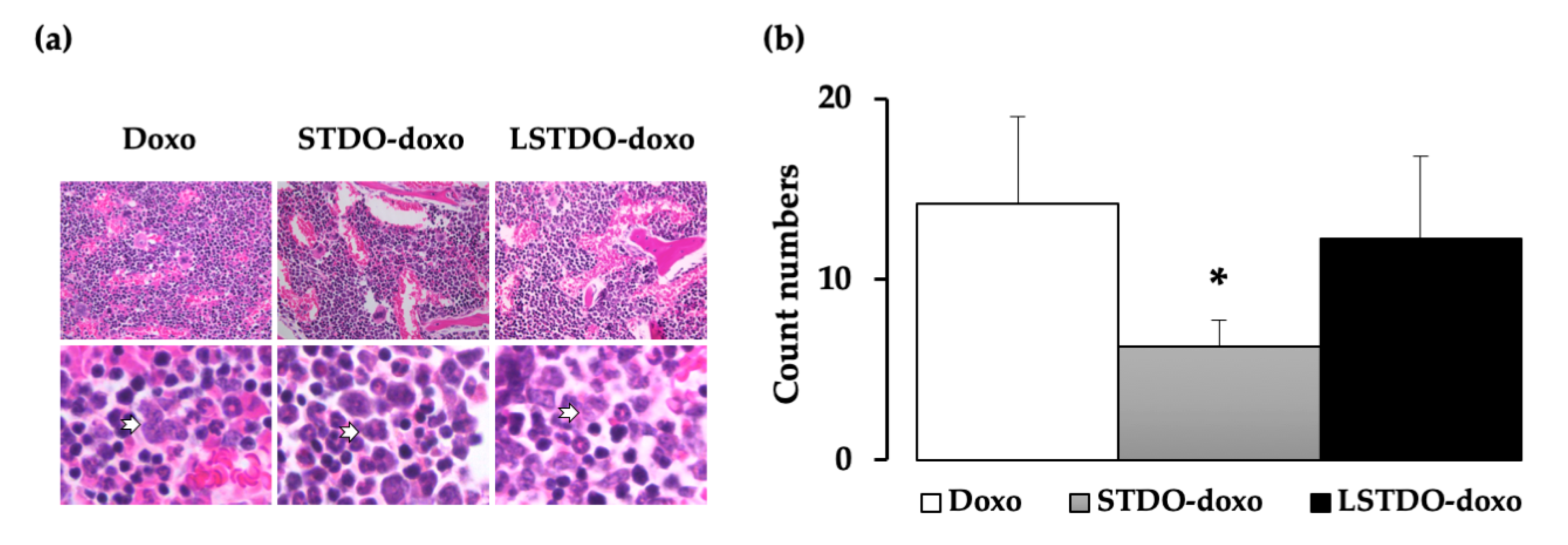
2.4. In Vivo Toxicity
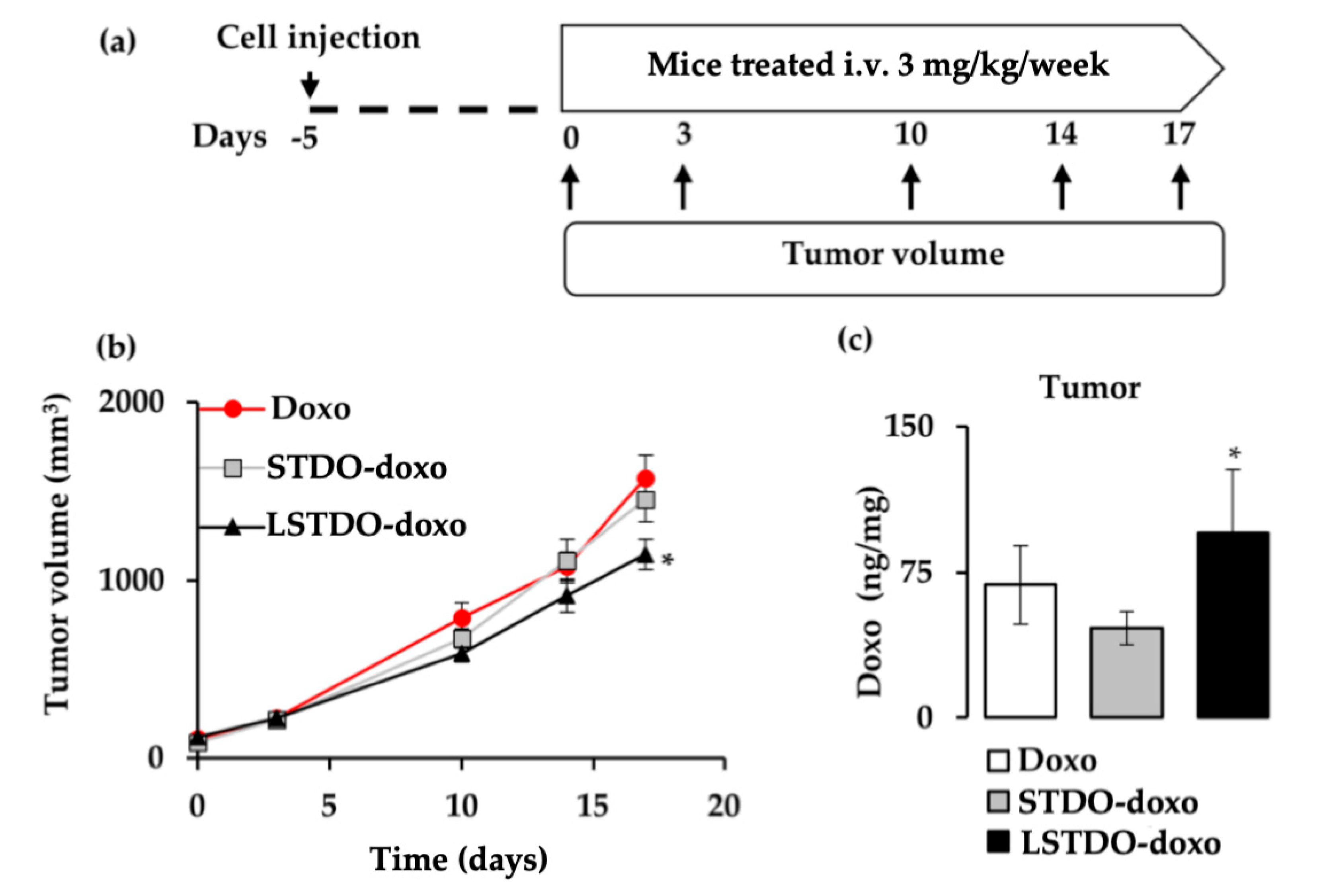
2.5. LSTDO-Doxo is More Effective than Free Doxo In Vivo
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Self-Assembly of DNA Origami
4.3. DNA Origami Purification
4.4. LSTDO Preparation and Purification
4.5. Doxorubicin Loading and Release
4.6. Cell Viability Assay
4.7. Organoid Isolation
4.8. Toxicity Tests on Mouse Liver Organoids
4.9. Histopathology
4.10. Mouse Xenograft
4.11. Biodistribution
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, J.; Fan, C.; Pei, H.; Shi, J.; Huang, Q. Smart drug delivery nanocarriers with self-assembled DNA nanostructures. Adv. Mater. 2013, 25, 4386–4396. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, P.W.K.; Ekani-Nkodo, A.; Papadakis, N.; Kumar, A.; Fygenson, D.K.; Winfree, E. Design and Characterization of Programmable DNA Nanotubes. J. Am. Chem. Soc. 2004, 126, 16344–16352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasciotti, E. Smart cancer therapy with DNA origami. Nat. Biotechnol. 2018, 36, 234–235. [Google Scholar] [CrossRef] [PubMed]
- Palazzolo, S.; Bayda, S.; Hadla, M.; Caligiuri, I.; Corona, G.; Toffoli, G.; Rizzolio, F. The Clinical translation of Organic Nanomaterials for Cancer Therapy: A Focus on Polymeric Nanoparticles, Micelles, Liposomes and Exosomes. Curr. Med. Chem. 2018, 25, 4224–4268. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.X.; Shaw, A.; Zeng, X.; Benson, E.; Nyström, A.M.; Högberg, B. DNA origami delivery system for cancer therapy with tunable release properties. ACS Nano 2012, 6, 8684–8691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Song, C.; Nangreave, J.; Liu, X.; Lin, L.; Qiu, D.; Wang, Z.G.; Zou, G.; Liang, X.; Yan, H.; et al. DNA origami as a carrier for circumvention of drug resistance. J. Am. Chem. Soc. 2012, 134, 13396–13403. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, Q.; Li, N.; Dai, L.; Liu, Q.; Song, L.; Wang, J.; Li, Y.; Tian, J.; Ding, B.; et al. DNA origami as an in vivo drug delivery vehicle for cancer therapy. ACS Nano 2014, 8, 6633–6643. [Google Scholar] [CrossRef]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Ponnuswamy, N.; Bastings, M.M.C.; Nathwani, B.; Ryu, J.H.; Chou, L.Y.T.; Vinther, M.; Li, W.A.; Anastassacos, F.M.; Mooney, D.J.; Shih, W.M. Oligolysine-based coating protects DNA nanostructures from low-salt denaturation and nuclease degradation. Nat. Commun. 2017, 8, 15654. [Google Scholar] [CrossRef]
- Linko, V.; Mikkilä, J.; Kostiainen, M.A. Packaging DNA Origami into Viral Protein Cages. Methods Mol. Biol. 2018, 1776, 267–277. [Google Scholar]
- Kiviaho, J.K.; Linko, V.; Ora, A.; Tiainen, T.; Järvihaavisto, E.; Mikkilä, J.; Tenhu, H.; Nonappa; Kostiainen, M.A. Cationic polymers for DNA origami coating—Examining their binding efficiency and tuning the enzymatic reaction rates. Nanoscale 2016, 8, 11674–11680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrault, S.D.; Shih, W.M. Virus-inspired membrane encapsulation of DNA nanostructures to achieve in vivo stability. ACS Nano 2014, 8, 5132–5140. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.J.W.; Semple, S.C.; Klimuk, S.K.; Ansell, S.; Maurer, N.; Cullis, P.R. Characterization of the drug retention and pharmacokinetic properties of liposomal nanoparticles containing dihydrosphingomyelin. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1121–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofheinz, R.D.; Gnad-Vogt, S.U.; Beyer, U.; Hochhaus, A. Liposomal encapsulated anti-cancer drugs. Anticancer Drugs 2005, 16, 691–707. [Google Scholar] [CrossRef]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Barenholz, Y.C. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Palazzolo, S.; Hadla, M.; Spena, C.R.; Bayda, S.; Kumar, V.; Lo Re, F.; Adeel, M.; Caligiuri, I.; Romano, F.; Corona, G.; et al. Proof-of-Concept Multistage Biomimetic Liposomal DNA Origami Nanosystem for the Remote Loading of Doxorubicin. ACS Med. Chem. Lett. 2019, 10, 517–521. [Google Scholar] [CrossRef]
- Stahl, E.; Martin, T.G.; Praetorius, F.; Dietz, H. Facile and Scalable Preparation of Pure and Dense DNA Origami Solutions. Angew. Chem. Int. Ed. 2014, 53, 12735–12740. [Google Scholar] [CrossRef] [Green Version]
- Bayda, S.; Hadla, M.; Palazzolo, S.; Kumar, V.; Caligiuri, I.; Ambrosi, E.; Pontoglio, E.; Agostini, M.; Tuccinardi, T.; Benedetti, A.; et al. Bottom-up synthesis of carbon nanoparticles with higher doxorubicin efficacy. J. Control. Release 2017, 248, 144–152. [Google Scholar] [CrossRef]
- Garnier, D.; Li, R.; Delbos, F.; Fourrier, A.; Collet, C.; Guguen-Guillouzo, C.; Chesné, C.; Nguyen, T.H. Expansion of human primary hepatocytes in vitro through their amplification as liver progenitors in a 3D organoid system. Sci. Rep. 2018, 8, 8222. [Google Scholar] [CrossRef]
- Min, Y.; Caster, J.M.; Eblan, M.J.; Wang, A.Z. Clinical Translation of Nanomedicine. Chem. Rev. 2015, 115, 11147–11190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toffoli, G.; Hadla, M.; Corona, G.; Caligiuri, I.; Palazzolo, S.; Semeraro, S.; Gamini, A.; Canzonieri, V.; Rizzolio, F. Exosomal doxorubicin reduces the cardiac toxicity of doxorubicin. Nanomedicine 2015, 10, 2963–2971. [Google Scholar] [CrossRef] [PubMed]
- Hadla, M.; Palazzolo, S.; Corona, G.; Caligiuri, I.; Canzonieri, V.; Toffoli, G.; Rizzolio, F. Exosomes increase the therapeutic index of doxorubicin in breast and ovarian cancer mouse models. Nanomedicine 2016, 11, 2431–2441. [Google Scholar] [CrossRef] [PubMed]
- Perrault, S.D.; Shih, W.M. Lipid Membrane Encapsulation of a 3D DNA Nano Octahedron. Methods Mol. Biol. 2017, 1500, 165–184. [Google Scholar] [PubMed]
- Greene, R.F.; Collins, J.M.; Jenkins, J.F.; Speyer, J.L.; Myers, C.E. Plasma pharmacokinetics of adriamycin and adriamycinol: Implications for the design of in vitro experiments and treatment protocols. Cancer Res. 1983, 43, 3417–3421. [Google Scholar]
- Liu, M.; Fu, J.; Hejesen, C.; Yang, Y.; Woodbury, N.W.; Gothelf, K.; Liu, Y.; Yan, H. A DNA tweezer-actuated enzyme nanoreactor. Nat. Commun. 2013, 4, 2127. [Google Scholar] [CrossRef] [Green Version]
- Kocabey, S.; Meinl, H.; MacPherson, I.S.; Cassinelli, V.; Manetto, A.; Rothenfusser, S.; Liedl, T.; Lichtenegger, F.S. Cellular Uptake of Tile-Assembled DNA Nanotubes. Nanomaterials 2014, 5, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Sobczak, J.P.J.; Martin, T.G.; Gerling, T.; Dietz, H. Rapid folding of DNA into nanoscale shapes at constant temperature. Science 2012, 338, 1458–1461. [Google Scholar] [CrossRef]
- Fu, Y.; Zeng, D.; Chao, J.; Jin, Y.; Zhang, Z.; Liu, H.; Li, D.; Ma, H.; Huang, Q.; Gothelf, K.V.; et al. Single-step rapid assembly of DNA origami nanostructures for addressable nanoscale bioreactors. J. Am. Chem. Soc. 2013, 135, 696–702. [Google Scholar] [CrossRef]
- Auvinen, H.; Zhang, H.; Kopilow, A.; Niemelä, E.H.; Nummelin, S.; Correia, A.; Santos, H.A.; Linko, V.; Kostiainen, M.A. Protein Coating of DNA Nanostructures for Enhanced Stability and Immunocompatibility. Adv. Healthc. Mater. 2017, 6, 1700692. [Google Scholar] [CrossRef] [Green Version]
- Bandak, S.; Goren, D.; Horowitz, A.; Tzemach, D.; Gabizon, A. Pharmacological studies of cisplatin encapsulated in long-circulating liposomes in mouse tumor models. Anticancer Drugs 1999, 10, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Stappenbeck, T.S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 2013, 8, 2471–2482. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palazzolo, S.; Hadla, M.; Russo Spena, C.; Caligiuri, I.; Rotondo, R.; Adeel, M.; Kumar, V.; Corona, G.; Canzonieri, V.; Toffoli, G.; et al. An Effective Multi-Stage Liposomal DNA Origami Nanosystem for In Vivo Cancer Therapy. Cancers 2019, 11, 1997. https://doi.org/10.3390/cancers11121997
Palazzolo S, Hadla M, Russo Spena C, Caligiuri I, Rotondo R, Adeel M, Kumar V, Corona G, Canzonieri V, Toffoli G, et al. An Effective Multi-Stage Liposomal DNA Origami Nanosystem for In Vivo Cancer Therapy. Cancers. 2019; 11(12):1997. https://doi.org/10.3390/cancers11121997
Chicago/Turabian StylePalazzolo, Stefano, Mohamad Hadla, Concetta Russo Spena, Isabella Caligiuri, Rossella Rotondo, Muhammad Adeel, Vinit Kumar, Giuseppe Corona, Vincenzo Canzonieri, Giuseppe Toffoli, and et al. 2019. "An Effective Multi-Stage Liposomal DNA Origami Nanosystem for In Vivo Cancer Therapy" Cancers 11, no. 12: 1997. https://doi.org/10.3390/cancers11121997
APA StylePalazzolo, S., Hadla, M., Russo Spena, C., Caligiuri, I., Rotondo, R., Adeel, M., Kumar, V., Corona, G., Canzonieri, V., Toffoli, G., & Rizzolio, F. (2019). An Effective Multi-Stage Liposomal DNA Origami Nanosystem for In Vivo Cancer Therapy. Cancers, 11(12), 1997. https://doi.org/10.3390/cancers11121997