Tumor-Associated Macrophages Induce Endocrine Therapy Resistance in ER+ Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
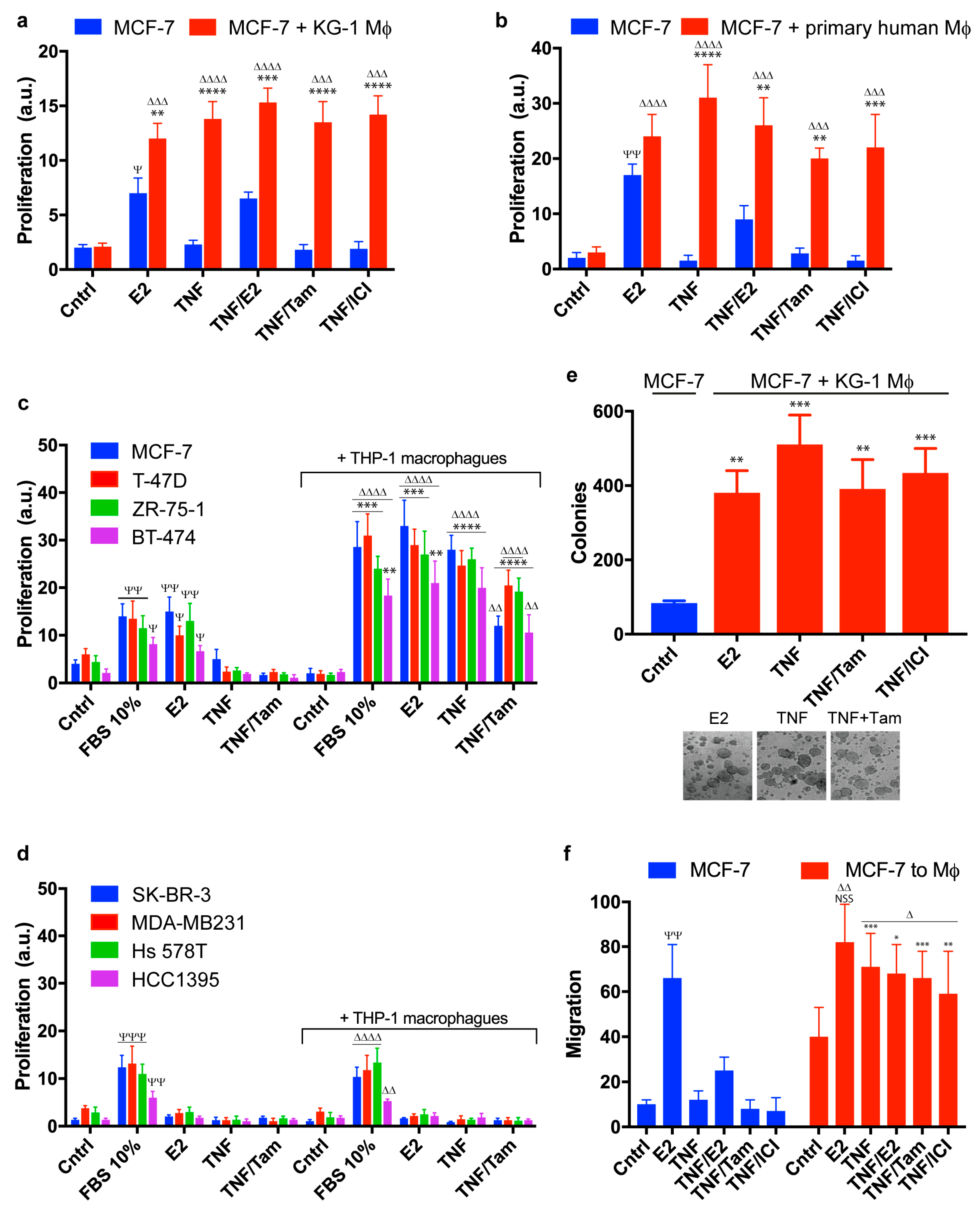
2.1. Macrophages Promote Proliferation, Invasion, and Migration of ER+ Breast Cancer Cells in an Estradiol-Independent Manner
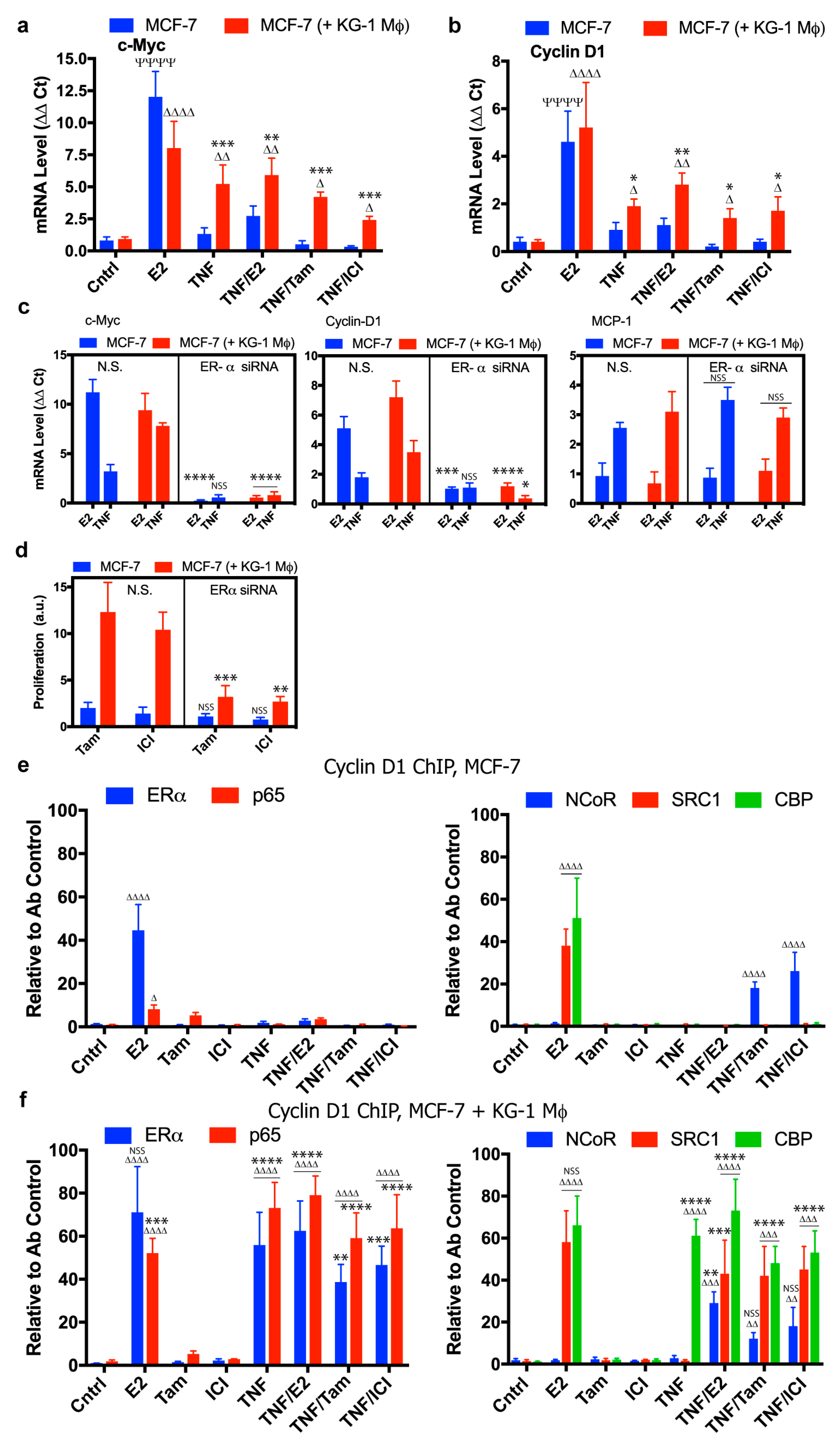
2.2. Modulation of Breast Cancer Proliferative Genes by Co-Culture with Macrophages
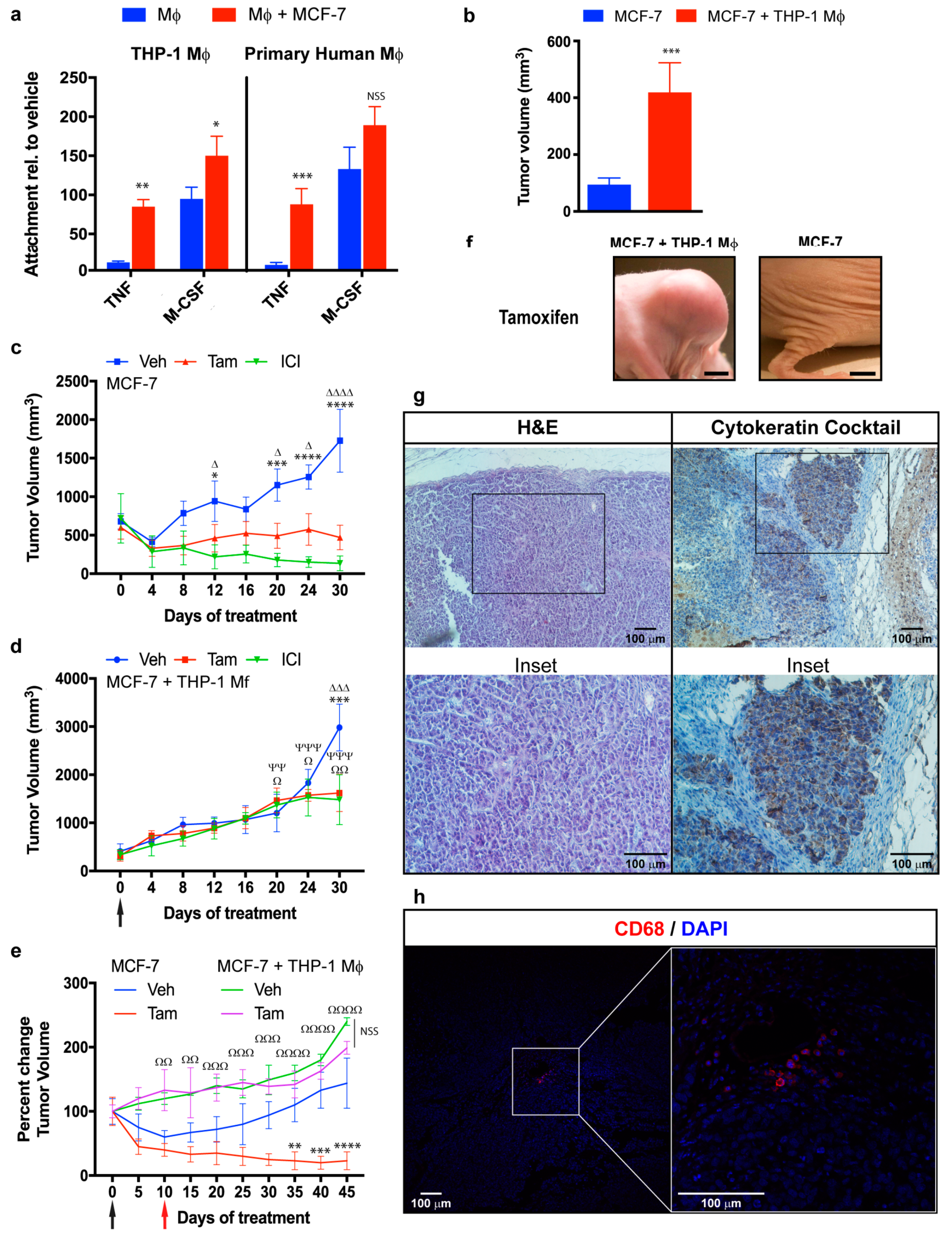
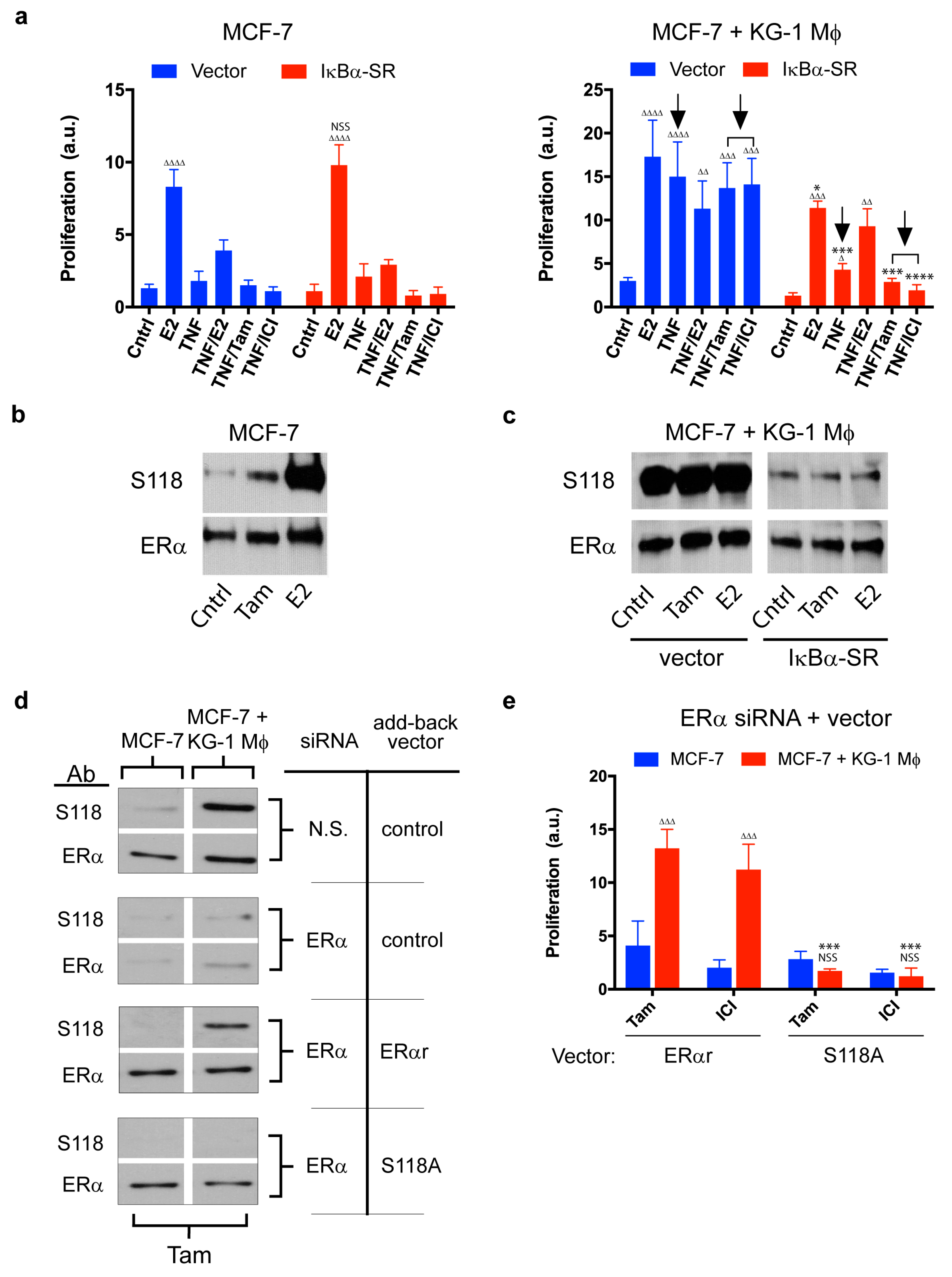
2.3. Role of NF-κB in Macrophage-Mediated MCF-7 Proliferation
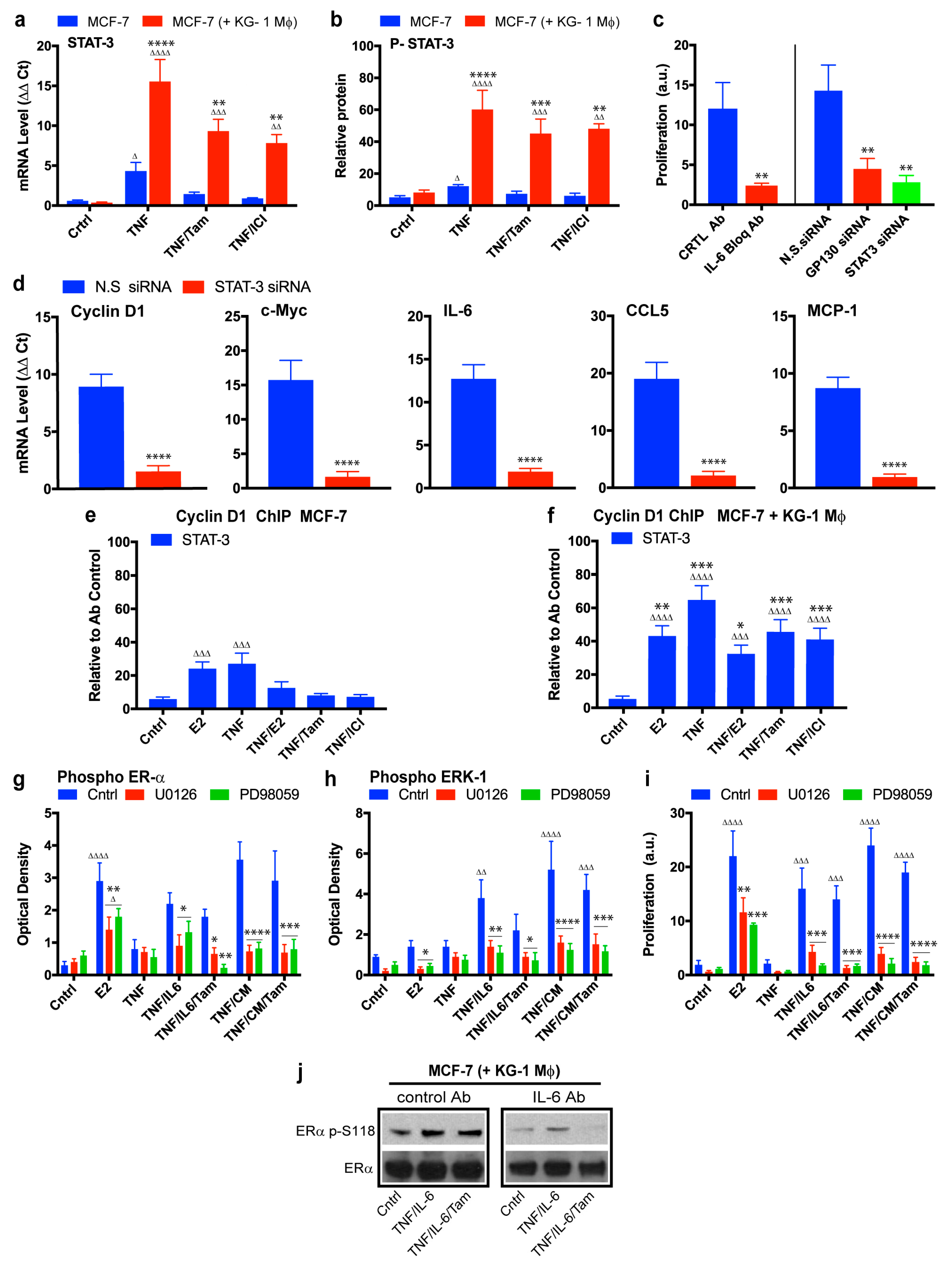
2.4. Role of the IL-6/STAT3 Pathway in Macrophage-Mediated MCF-7 Endocrine Resistance
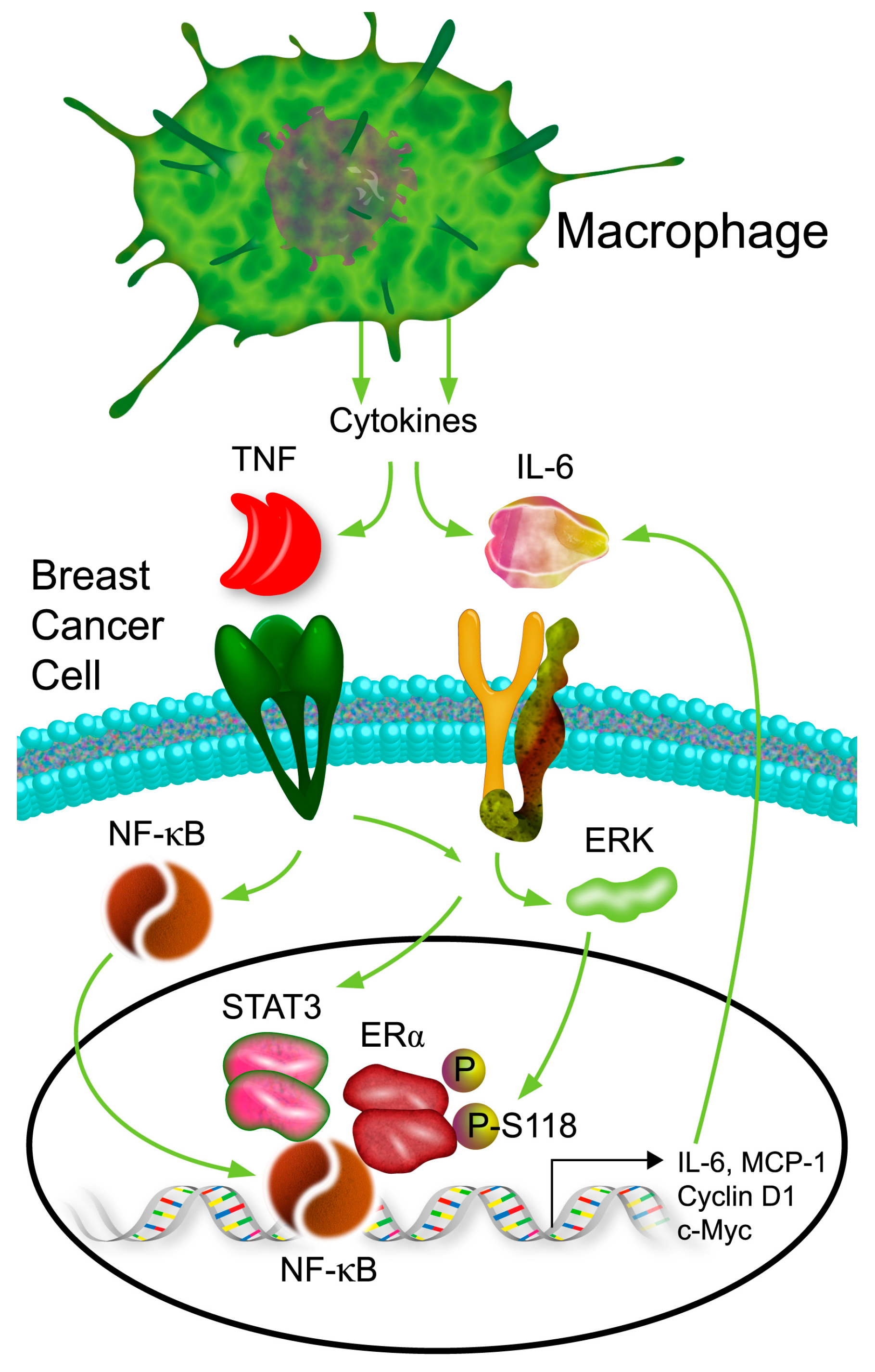
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Primary Human Macrophage Culture and Differentiation
4.3. Differentiation and Conditioning of Macrophages
4.4. Macrophage-Conditioned Medium and Cell Coculture
4.5. Proliferation Assay
4.6. Colony Formation Assay
4.7. Migration Assay
4.8. Transfection of MCF-7 Cells
4.9. siRNA
4.10. Quantitative Real-Time RT-PCR (qPCR)
4.11. Chromatin Immunoprecipitation (ChIP) Assays
4.12. Antibodies Used for IP
4.13. Cytokine, Growth Factor, and Chemokine Levels
4.14. Animal Studies
4.15. Western Blotting Analysis
4.16. Tumor Tissues, Immunohistochemistry, and Immunofluorescence
4.17. Flow Cytometry
4.18. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early Breast Cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Jameera Begam, A.; Jubie, S.; Nanjan, M.J. Estrogen receptor agonists/antagonists in Breast Cancer therapy: A critical review. Bioorgan. Chem. 2017, 71, 257–274. [Google Scholar] [CrossRef] [PubMed]
- McEwan, I.J. Nuclear receptors: One big family. Methods Mol. Biol. 2009, 505, 3–18. [Google Scholar] [PubMed]
- Lubahn, D.B.; Moyer, J.S.; Golding, T.S.; Couse, J.F.; Korach, K.S.; Smithies, O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc. Natl. Acad. Sci. USA 1993, 90, 11162–11166. [Google Scholar] [CrossRef] [PubMed]
- Brisken, C.; O’Malley, B. Hormone action in the mammary gland. Cold Spring Harb. Perspect. Biol. 2010, 2, a003178. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.M.; Albanese, C.; Anderson, C.M.; Hilty, K.; Webb, P.; Uht, R.M.; Price, R.H., Jr.; Pestell, R.G.; Kushner, P.J. Opposing action of estrogen receptors alpha and beta on cyclin d1 gene expression. J. Biol. Chem. 2002, 277, 24353–24360. [Google Scholar] [CrossRef] [PubMed]
- Helguero, L.A.; Faulds, M.H.; Gustafsson, J.A.; Haldosen, L.A. Estrogen receptors alfa (eralpha) and beta (erbeta) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line hc11. Oncogene 2005, 24, 6605–6616. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C.; O’Malley, B.W. Selective estrogen-receptor modulators and antihormonal resistance in Breast Cancer. J. Cin. Oncol. 2007, 25, 5815–5824. [Google Scholar] [CrossRef] [PubMed]
- Howell, A. Fulvestrant (‘faslodex’): Current and future role in Breast Cancer management. Crit. Re. Oncol. 2006, 57, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Bramlett, K.S.; Burris, T.P. Effects of selective estrogen receptor modulators (serms) on coactivator nuclear receptor (nr) box binding to estrogen receptors. Mol. Genet. Metab. 2002, 76, 225–233. [Google Scholar] [CrossRef]
- Jaber, B.M.; Gao, T.; Huang, L.; Karmakar, S.; Smith, C.L. The pure estrogen receptor antagonist ici 182,780 promotes a novel interaction of estrogen receptor-alpha with the 3’,5’-cyclic adenosine monophosphate response element-binding protein-binding protein/p300 coactivators. Mol. Endocrinol. 2006, 20, 2695–2710. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C.; Brodie, A.M. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of Breast Cancer. Steroids 2007, 72, 7–25. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S. The Breast Cancer continuum in hormone-receptor–positive Breast Cancer in postmenopausal women: Evolving management options focusing on aromatase inhibitors. Ann. Oncol. 2008, 19, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Augusto, T.V.; Correia-da-Silva, G.; Rodrigues, C.M.P.; Teixeira, N.; Amaral, C. Acquired resistance to aromatase inhibitors: Where we stand! Endocr. Related Cancer 2018, 25, R283–R301. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in Breast Cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Macedo, L.F.; Sabnis, G.; Brodie, A. Preclinical modeling of endocrine response and resistance: Focus on aromatase inhibitors. Cancer 2008, 112, 679–688. [Google Scholar] [CrossRef] [PubMed]
- MacGregor Schafer, J.; Liu, H.; Bentrem, D.J.; Zapf, J.W.; Jordan, V.C. Allosteric silencing of activating function 1 in the 4-hydroxytamoxifen estrogen receptor complex is induced by substituting glycine for aspartate at amino acid 351. Cancer Res. 2000, 60, 5097–5105. [Google Scholar]
- Osborne, C.K.; Shou, J.; Massarweh, S.; Schiff, R. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in Breast Cancer. Clin. Cancer Res. 2005, 11, 865s–870s. [Google Scholar]
- Dihge, L.; Bendahl, P.O.; Grabau, D.; Isola, J.; Lovgren, K.; Ryden, L.; Ferno, M. Epidermal growth factor receptor (egfr) and the estrogen receptor modulator amplified in Breast Cancer (aib1) for predicting clinical outcome after adjuvant tamoxifen in Breast Cancer. Breast Cancer Res. Treat. 2008, 109, 255–262. [Google Scholar] [CrossRef]
- Kurokawa, H.; Lenferink, A.E.G.; Simpson, J.F.; Pisacane, P.I.; Sliwkowski, M.X.; Forbes, J.T.; Arteaga, C.L. Inhibition of her2/neu (erbb-2) and mitogen-activated protein kinases enhances tamoxifen action against her2-overexpressing, tamoxifen-resistant Breast Cancer cells. Cancer Res. 2000, 60, 5887–5894. [Google Scholar]
- Gu, Y.; Chen, T.; Li, G.; Xu, C.; Xu, Z.; Zhang, J.; He, K.; Zheng, L.; Guan, Z.; Su, X.; et al. Lower beclin 1 downregulates her2 expression to enhance tamoxifen sensitivity and predicts a favorable outcome for er positive Breast Cancer. Oncotarget 2017, 8, 52156–52177. [Google Scholar] [CrossRef] [PubMed]
- Fagan, D.H.; Uselman, R.R.; Sachdev, D.; Yee, D. Acquired resistance to tamoxifen is associated with loss of the type i insulin-like growth factor receptor: Implications for Breast Cancer treatment. Cancer Res. 2012, 72, 3372–3380. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, D.C.; Knowles, M.A.; Speirs, V. Mechanisms of fgfr3 actions in endocrine resistant Breast Cancer. Int. J. Cancer 2012, 130, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.L. Endocrine resistance in Breast Cancer: New roles for erbb3 and erbb4. Breast Cancer Res 2011, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, N.; Kim, J.S.; Jiang, J.; Peston, D.; Sinnett, H.D.; Madden, P.; Gee, J.M.; Nicholson, R.I.; Lykkesfeldt, A.E.; Shousha, S.; et al. Phosphorylation of eralpha at serine 118 in primary Breast Cancer and in tamoxifen-resistant tumours is indicative of a complex role for eralpha phosphorylation in Breast Cancer progression. Endocr. Related Cancer 2006, 13, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Skliris, G.P.; Nugent, Z.J.; Rowan, B.G.; Penner, C.R.; Watson, P.H.; Murphy, L.C. A phosphorylation code for oestrogen receptor-alpha predicts clinical outcome to endocrine therapy in Breast Cancer. Endocr. Related Cancer 2010, 17, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Anbalagan, M.; Rowan, B.G. Estrogen receptor alpha phosphorylation and its functional impact in human Breast Cancer. Mol. Cell. Endocrinol. 2015, 418, 264–272. [Google Scholar] [CrossRef]
- Thomas, R.S.; Sarwar, N.; Phoenix, F.; Coombes, R.C.; Ali, S. Phosphorylation at serines 104 and 106 by erk1/2 mapk is important for estrogen receptor-alpha activity. J. Mol. Endocrinol. 2008, 40, 173–184. [Google Scholar] [CrossRef]
- Raj, G.V.; Sareddy, G.R.; Ma, S.; Lee, T.K.; Viswanadhapalli, S.; Li, R.; Liu, X.; Murakami, S.; Chen, C.C.; Lee, W.R.; et al. Estrogen receptor coregulator binding modulators (erxs) effectively target estrogen receptor positive human Breast Cancers. eLife 2017, 6, e26857. [Google Scholar] [CrossRef]
- Groner, A.C.; Brown, M. Role of steroid receptor and coregulator mutations in hormone-dependent cancers. J. Clin. Investig. 2017, 127, 1126–1135. [Google Scholar] [CrossRef]
- Oida, K.; Matsuda, A.; Jung, K.; Xia, Y.; Jang, H.; Amagai, Y.; Ahn, G.; Nishikawa, S.; Ishizaka, S.; Jensen-Jarolim, E.; et al. Nuclear factor-kb plays a critical role in both intrinsic and acquired resistance against endocrine therapy in human Breast Cancer cells. Sci. Rep. 2014, 4, 4057. [Google Scholar] [CrossRef] [PubMed]
- Nakshatri, H.; Bhat-Nakshatri, P.; Martin, D.A.; Goulet, R.J., Jr.; Sledge, G.W., Jr. Constitutive activation of nf-kappab during progression of Breast Cancer to hormone-independent growth. Mol. Cell. Biol. 1997, 17, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Liu, Y.J.; Cui, J.W.; Yang, M.; Liu, X.L.; Li, P.; Wang, Z.; Zhu, L.Z.; Lu, S.Y.; Zou, L.; et al. Aspirin regulation of c-myc and cyclind1 proteins to overcome tamoxifen resistance in estrogen receptor-positive Breast Cancer cells. Oncotarget 2017, 8, 30252–30264. [Google Scholar] [CrossRef] [PubMed]
- Green, A.R.; Aleskandarany, M.A.; Agarwal, D.; Elsheikh, S.; Nolan, C.C.; Diez-Rodriguez, M.; Macmillan, R.D.; Ball, G.R.; Caldas, C.; Madhusudan, S.; et al. Myc functions are specific in biological subtypes of Breast Cancer and confers resistance to endocrine therapy in luminal tumours. Br. J. Cancer 2016, 114, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Waxman, S.; Germain, D. Tamoxifen stimulates the growth of cyclin d1-overexpressing Breast Cancer cells by promoting the activation of signal transducer and activator of transcription 3. Cancer Res. 2008, 68, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.J.; McNeil, C.M.; Musgrove, E.A.; Sutherland, R.L. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: The potential roles of c-myc, cyclin d1 and cycline. Endocr. Related Cancer 2005, 12, S47–S59. [Google Scholar] [CrossRef] [PubMed]
- Lonard, D.M.; O’Malley, B.W. Nuclear receptor coregulators: Judges, juries, and executioners of cellular regulation. Mol. Cell 2007, 27, 691–700. [Google Scholar] [CrossRef]
- McKenna, N.J.; O’Malley, B.W. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002, 108, 465–474. [Google Scholar] [CrossRef]
- Lonard, D.M.; O’Malley, B.W. Expanding functional diversity of the coactivators. Trends Biochem. Sci. 2005, 30, 126–132. [Google Scholar] [CrossRef]
- Rosenfeld, M.G.; Lunyak, V.V.; Glass, C.K. Sensors and signals: A coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006, 20, 1405–1428. [Google Scholar] [CrossRef]
- Lu, R.; Hu, X.; Zhou, J.; Sun, J.; Zhu, A.Z.; Xu, X.; Zheng, H.; Gao, X.; Wang, X.; Jin, H.; et al. Cops5 amplification and overexpression confers tamoxifen-resistance in eralpha-positive Breast Cancer by degradation of ncor. Nat. Commun. 2016, 7, 12044. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.A.; Lee, G.Y.; Bissell, M.J. Targeting the tumor microenvironment. Front. Biosci. 2007, 12, 3468–3474. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, T.; Wood, M. Tumor-stromal cell interactions and opportunities for therapeutic intervention. Curr. Opin. Pharmacol. 2010, 10, 369–374. [Google Scholar] [CrossRef]
- Tsutsui, S.; Yasuda, K.; Suzuki, K.; Tahara, K.; Higashi, H.; Era, S. Macrophage infiltration and its prognostic implications in Breast Cancer: The relationship with vegf expression and microvessel density. Oncol. Rep. 2005, 14, 425–431. [Google Scholar] [CrossRef]
- Choi, J.; Gyamfi, J.; Jang, H.; Koo, J.S. The role of tumor-associated macrophage in Breast Cancer biology. Histol. Histopathol. 2018, 33, 133–145. [Google Scholar] [PubMed]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Bolli, E.; Movahedi, K.; Laoui, D.; Van Ginderachter, J.A. Novel insights in the regulation and function of macrophages in the tumor microenvironment. Curr. Opin. Oncol. 2017, 29, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Austenaa, L.; Natoli, G. A shortcut for early macrophage recruitment into tumors by activated oncogenes. Genes Dev. 2017, 31, 223–225. [Google Scholar] [CrossRef]
- Chen, P.; Bonaldo, P. Role of macrophage polarization in tumor angiogenesis and vessel normalization: Implications for new anticancer therapies. Int. Rev. Cell Mol. Biol. 2013, 301, 1–35. [Google Scholar] [PubMed]
- Caux, C.; Ramos, R.N.; Prendergast, G.C.; Bendriss-Vermare, N.; Menetrier-Caux, C. A milestone review on how macrophages affect tumor growth. Cancer Res. 2016, 76, 6439–6442. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; Yeh, E.S.; Soloff, A.C. Tumor-associated macrophages: Unwitting accomplices in Breast Cancer malignancy. NPJ Breast Cancer 2016, 2, 15025. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Dahlen, E.; Dawe, K.; Ohlsson, L.; Hedlund, G. Dendritic cells and macrophages are the first and major producers of tnf-alpha in pancreatic islets in the nonobese diabetic mouse. J. Immunol. 1998, 160, 3585–3593. [Google Scholar] [PubMed]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-alpha signaling in macrophages. Crit. Rev. Eukaryot. Gene Express. 2010, 20, 87–103. [Google Scholar] [CrossRef]
- Zhu, P.; Baek, S.H.; Bourk, E.M.; Ohgi, K.A.; Garcia-Bassets, I.; Sanjo, H.; Akira, S.; Kotol, P.F.; Glass, C.K.; Rosenfeld, M.G.; et al. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell 2006, 124, 615–629. [Google Scholar] [CrossRef]
- Balkwill, F. Tumor necrosis factor or tumor promoting factor? Cytokine Growth Factor Rev. 2002, 13, 135–141. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-kappab in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Naugler, W.E.; Karin, M. The wolf in sheep’s clothing: The role of interleukin-6 in immunity, inflammation and cancer. Trends Mol. Med. 2008, 14, 109–119. [Google Scholar] [CrossRef]
- Nettles, K.W.; Gil, G.; Nowak, J.; Metivier, R.; Sharma, V.B.; Greene, G.L. Cbp is a dosage-dependent regulator of nuclear factor-kappab suppression by the estrogen receptor. Mol. Endocrinol. 2008, 22, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Gionet, N.; Jansson, D.; Mader, S.; Pratt, M.A. Nf-kappab and estrogen receptor alpha interactions: Differential function in estrogen receptor-negative and -positive hormone-independent Breast Cancer cells. J. Cell. Biochem. 2009, 107, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.K.; Shi, Q.; Baily, S.; Strickland, I.; Ghosh, S.; Pardee, A.B.; Iglehart, J.D. Nf-kappa b activation in human Breast Cancer specimens and its role in cell proliferation and apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10137–10142. [Google Scholar] [CrossRef] [PubMed]
- Chavey, C.; Bibeau, F.; Gourgou-Bourgade, S.; Burlinchon, S.; Boissiere, F.; Laune, D.; Roques, S.; Lazennec, G. Oestrogen receptor negative Breast Cancers exhibit high cytokine content. Breast Cancer Res. 2007, 9, R15. [Google Scholar] [CrossRef] [PubMed]
- Yaal-Hahoshen, N.; Shina, S.; Leider-Trejo, L.; Barnea, I.; Shabtai, E.L.; Azenshtein, E.; Greenberg, I.; Keydar, I.; Ben-Baruch, A. The chemokine ccl5 as a potential prognostic factor predicting disease progression in stage ii Breast Cancer patients. Clin. Cancer Res. 2006, 12, 4474–4480. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.; Wang, T.C. Inflammation and cancer: Il-6 and stat3 complete the link. Cancer Cell 2009, 15, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Hollmen, M.; Roudnicky, F.; Karaman, S.; Detmar, M. Characterization of macrophage Cancer cell crosstalk in estrogen receptor positive and triple-negative Breast Cancer. Sci. Rep. 2015, 5, 9188. [Google Scholar] [CrossRef]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human Breast Cancer cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef]
- Masuda, M.; Wakasaki, T.; Suzui, M.; Toh, S.; Joe, A.K.; Weinstein, I.B. Stat3 orchestrates tumor development and progression: The achilles’ heel of head and neck cancers? Curr. Cancer Drug Targets 2010, 10, 117–126. [Google Scholar] [CrossRef]
- Jhan, J.R.; Andrechek, E.R. Stat3 accelerates myc induced tumor formation while reducing growth rate in a mouse model of Breast Cancer. Oncotarget 2016, 7, 65797–65807. [Google Scholar] [CrossRef]
- Subramaniam, K.S.; Omar, I.S.; Kwong, S.C.; Mohamed, Z.; Woo, Y.L.; Mat Adenan, N.A.; Chung, I. Cancer-associated fibroblasts promote endometrial cancer growth via activation of interleukin-6/stat-3/c-myc pathway. Am. J. Cancer Res. 2016, 6, 200–213. [Google Scholar]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated stat3 maintains constitutive nf-kappab activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Bui, Q.T.; Im, J.H.; Jeong, S.B.; Kim, Y.M.; Lim, S.C.; Kim, B.; Kang, K.W. Essential role of notch4/stat3 signaling in epithelial-mesenchymal transition of tamoxifen-resistant human Breast Cancer. Cancer Lett. 2017, 390, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Teymourzadeh, A.; Mansouri, S.; Farahmand, L.; Hosseinzade, A.; Majidzadeh, A.K. Er-alpha36 interactions with cytosolic molecular network in acquired tamoxifen resistance. Clin. Breast Cancer 2017, 17, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Torrens-Mas, M.; Pons, D.G.; Sastre-Serra, J.; Oliver, J.; Roca, P. Sirt3 silencing sensitizes Breast Cancer cells to cytotoxic treatments through an increment in ros production. J. Cell. Biochem. 2017, 118, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Vaziri-Gohar, A.; Zheng, Y.; Houston, K.D. Igf-1 receptor modulates foxo1-mediated tamoxifen response in Breast Cancer cells. Mol. Cancer Res. 2017, 15, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Nakayama, Y.; Yamaguchi, N. Down-regulation of forkhead box protein a1 (foxa1) leads to cancer stem cell-like properties in tamoxifen-resistant Breast Cancer cells through induction of interleukin-6. J. Biol. Chem. 2017, 292, 8136–8148. [Google Scholar] [CrossRef]
- Viedma-Rodriguez, R.; Baiza-Gutman, L.; Salamanca-Gomez, F.; Diaz-Zaragoza, M.; Martinez-Hernandez, G.; Ruiz Esparza-Garrido, R.; Velazquez-Flores, M.A.; Arenas-Aranda, D. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive Breast Cancer. Oncol. Rep. 2014, 32, 3–15. [Google Scholar] [CrossRef]
- Komohara, Y.; Jinushi, M.; Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014, 105, 1–8. [Google Scholar] [CrossRef]
- Almatroodi, S.A.; McDonald, C.F.; Darby, I.A.; Pouniotis, D.S. Characterization of m1/m2 tumour-associated macrophages (tams) and th1/th2 cytokine profiles in patients with nsclc. Cancer Microenviron. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Sawe, R.T.; Kerper, M.; Badve, S.; Li, J.; Sandoval-Cooper, M.; Xie, J.; Shi, Z.; Patel, K.; Chumba, D.; Ofulla, A.; et al. Aggressive Breast Cancer in western kenya has early onset, high proliferation, and immune cell infiltration. BMC Cancer 2016, 16, 204. [Google Scholar] [CrossRef]
- Xuan, Q.J.; Wang, J.X.; Nanding, A.; Wang, Z.P.; Liu, H.; Lian, X.; Zhang, Q.Y. Tumor-associated macrophages are correlated with tamoxifen resistance in the postmenopausal Breast Cancer patients. Pathol. Oncol. Res. 2014, 20, 619–624. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Casella, G.; Garzetti, L.; Gatta, A.T.; Finardi, A.; Maiorino, C.; Ruffini, F.; Martino, G.; Muzio, L.; Furlan, R. Il4 induces il6-producing m2 macrophages associated to inhibition of neuroinflammation in vitro and in vivo. J. Neuroinflamm. 2016, 13, 139. [Google Scholar] [CrossRef]
- Pello, O.M.; De Pizzol, M.; Mirolo, M.; Soucek, L.; Zammataro, L.; Amabile, A.; Doni, A.; Nebuloni, M.; Swigart, L.B.; Evan, G.I.; et al. Role of c-myc in alternative activation of human macrophages and tumor-associated macrophage biology. Blood 2012, 119, 411–421. [Google Scholar] [CrossRef]
- Foulds, C.E. Disrupting a negative feedback loop drives endocrine therapy-resistant Breast Cancer. Proc. Natl. Acad. Sci. USA 2018, 115, 8236–8238. [Google Scholar] [CrossRef]
- Toaldo, C.B.; Alexi, X.; Beelen, K.; Kok, M.; Hauptmann, M.; Jansen, M.; Berns, E.; Neefjes, J.; Linn, S.; Michalides, R.; et al. Protein kinase a-induced tamoxifen resistance is mediated by anchoring protein akap13. BMC Cancer 2015, 15, 588. [Google Scholar]
- Ha, H.; Debnath, B.; Neamati, N. Role of the cxcl8-cxcr1/2 axis in cancer and inflammatory diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef]
- Kim, S.; Jeon, M.; Lee, J.E.; Nam, S.J. Mek activity controls il-8 expression in tamoxifen-resistant mcf-7 Breast Cancer cells. Oncol. Rep. 2016, 35, 2398–2404. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.; Gao, Y.; Shao, J.; Zhang, X.J.; Yao, Z. Reciprocal regulation of 17beta-estradiol, interleukin-6 and interleukin-8 during growth and progression of epithelial ovarian cancer. Cytokine 2009, 46, 382–391. [Google Scholar] [CrossRef]
- Hinz, M.; Krappmann, D.; Eichten, A.; Heder, A.; Scheidereit, C.; Strauss, M. Nf-kappab function in growth control: Regulation of cyclin d1 expression and g0/g1-to-s-phase transition. Mol. Cell. Biol. 1999, 19, 2690–2698. [Google Scholar] [CrossRef]
- Witzel, I.I.; Koh, L.F.; Perkins, N.D. Regulation of cyclin d1 gene expression. Biochem. Soc. Trans. 2010, 38, 217–222. [Google Scholar] [CrossRef]
- Duplessis, T.T.; Williams, C.C.; Hill, S.M.; Rowan, B.G. Phosphorylation of estrogen receptor alpha at serine 118 directs recruitment of promoter complexes and gene-specific transcription. Endocrinology 2011, 152, 2517–2526. [Google Scholar] [CrossRef]
- Liu, X.F.; Bagchi, M.K. Recruitment of distinct chromatin-modifying complexes by tamoxifen-complexed estrogen receptor at natural target gene promoters in vivo. J. Biol. Chem. 2004, 279, 15050–15058. [Google Scholar] [CrossRef]
- McBryan, J.; Theissen, S.M.; Byrne, C.; Hughes, E.; Cocchiglia, S.; Sande, S.; O’Hara, J.; Tibbitts, P.; Hill, A.D.; Young, L.S. Metastatic progression with resistance to aromatase inhibitors is driven by the steroid receptor coactivator src-1. Cancer Res. 2012, 72, 548–559. [Google Scholar] [CrossRef]
- McCartan, D.; Bolger, J.C.; Fagan, A.; Byrne, C.; Hao, Y.; Qin, L.; McIlroy, M.; Xu, J.; Hill, A.D.; Gaora, P.Ó.; et al. Global characterization of the src-1 transcriptome identifies adam22 as an er-independent mediator of endocrine-resistant Breast Cancer. Cancer Res. 2012, 72, 220–229. [Google Scholar] [CrossRef]
- Evans, M.J.; Eckert, A.; Lai, K.; Adelman, S.J.; Harnish, D.C. Reciprocal antagonism between estrogen receptor and nf-kappab activity in vivo. Circul. Res. 2001, 89, 823–830. [Google Scholar] [CrossRef]
- Saji, H.; Koike, M.; Yamori, T.; Saji, S.; Seiki, M.; Matsushima, K.; Toi, M. Significant correlation of monocyte chemoattractant protein-1 expression with neovascularization and progression of breast carcinoma. Cancer 2001, 92, 1085–1091. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Steiner, M.S.; Pound, C.R. Phase iia clinical trial to test the efficacy and safety of toremifene in men with high-grade prostatic intraepithelial neoplasia. Clin. Prostate Cancer 2003, 2, 24–31. [Google Scholar] [CrossRef]
- Tomao, S.; Romiti, A.; Massidda, B.; Ionta, M.T.; Farris, A.; Zullo, A.; Brescia, A.; Santuari, L.; Frati, L. A phase ii study of gemcitabine and tamoxifen in advanced pancreatic cancer. AntiCancer Res. 2002, 22, 2361–2364. [Google Scholar]
- McClay, E.F.; Bogart, J.; Herndon, J.E., 2nd; Watson, D.; Evans, L.; Seagren, S.L.; Green, M.R. A phase iii trial evaluating the combination of cisplatin, etoposide, and radiation therapy with or without tamoxifen in patients with limited-stage small cell lung cancer: Cancer and leukemia group b study (9235). Am. J. Cin. Oncol. 2005, 28, 81–90. [Google Scholar] [CrossRef]
- Wagner, U.; du Bois, A.; Pfisterer, J.; Huober, J.; Loibl, S.; Lück, H.J.; Sehouli, J.; Gropp, M.; Stähle, A.; Schmalfeldt, B.; Meier, W. Gefitinib in combination with tamoxifen in patients with ovarian cancer refractory or resistant to platinum-taxane based therapy A phase ii trial of the ago ovarian cancer study group (ago-ovar 2.6). Gynecol. Oncol. 2007, 105, 132–137. [Google Scholar] [CrossRef]
- Lens, M.B.; Reiman, T.; Husain, A.F. Use of tamoxifen in the treatment of malignant melanoma. Cancer 2003, 98, 1355–1361. [Google Scholar] [CrossRef]
- Battaglia, M.; Pozzi, D.; Grimaldi, S.; Parasassi, T. Hoechst 33258 staining for detecting mycoplasma contamination in cell cultures: A method for reducing fluorescence photobleaching. Biotech. Histochem. 1994, 69, 152–156. [Google Scholar] [CrossRef]
- Peredeltchouk, M.; David, S.A.; Bhattacharya, B.; Volokhov, D.V.; Chizhikov, V. Detection of mycoplasma contamination in cell substrates using reverse transcription-pcr assays. J. Appl. Microbiol. 2011, 110, 54–60. [Google Scholar] [CrossRef]
- Plesner, A. Increasing the yield of human mononuclear cells and low serum conditions for in vitro generation of macrophages with m-csf. J. Immunol. Methods 2003, 279, 287–295. [Google Scholar] [CrossRef]
- Genin, M.; Clement, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and m2 macrophages derived from thp-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015, 15, 577. [Google Scholar] [CrossRef]
- Falasca, M.; Raimondi, C.; Maffucci, T. Boyden chamber. Methods Mol. Biol. 2011, 769, 87–95. [Google Scholar]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castellaro, A.M.; Rodriguez-Baili, M.C.; Di Tada, C.E.; Gil, G.A. Tumor-Associated Macrophages Induce Endocrine Therapy Resistance in ER+ Breast Cancer Cells. Cancers 2019, 11, 189. https://doi.org/10.3390/cancers11020189
Castellaro AM, Rodriguez-Baili MC, Di Tada CE, Gil GA. Tumor-Associated Macrophages Induce Endocrine Therapy Resistance in ER+ Breast Cancer Cells. Cancers. 2019; 11(2):189. https://doi.org/10.3390/cancers11020189
Chicago/Turabian StyleCastellaro, Andrés M., María C. Rodriguez-Baili, Cecilia E. Di Tada, and Germán A. Gil. 2019. "Tumor-Associated Macrophages Induce Endocrine Therapy Resistance in ER+ Breast Cancer Cells" Cancers 11, no. 2: 189. https://doi.org/10.3390/cancers11020189
APA StyleCastellaro, A. M., Rodriguez-Baili, M. C., Di Tada, C. E., & Gil, G. A. (2019). Tumor-Associated Macrophages Induce Endocrine Therapy Resistance in ER+ Breast Cancer Cells. Cancers, 11(2), 189. https://doi.org/10.3390/cancers11020189