Replacement of miR-155 Elicits Tumor Suppressive Activity and Antagonizes Bortezomib Resistance in Multiple Myeloma


 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
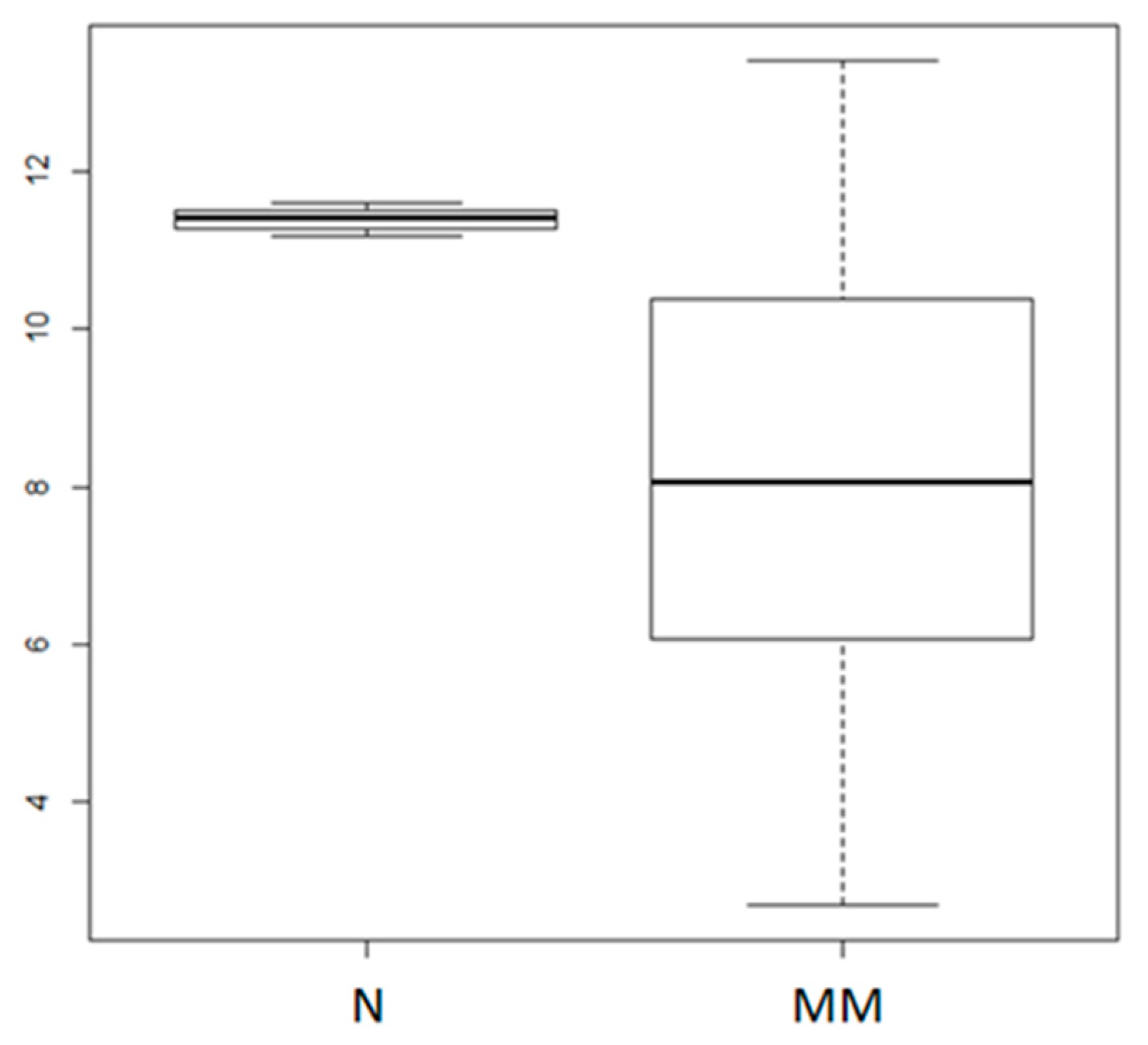
2.1. MiR-155 Expression is Dysregulated in MM
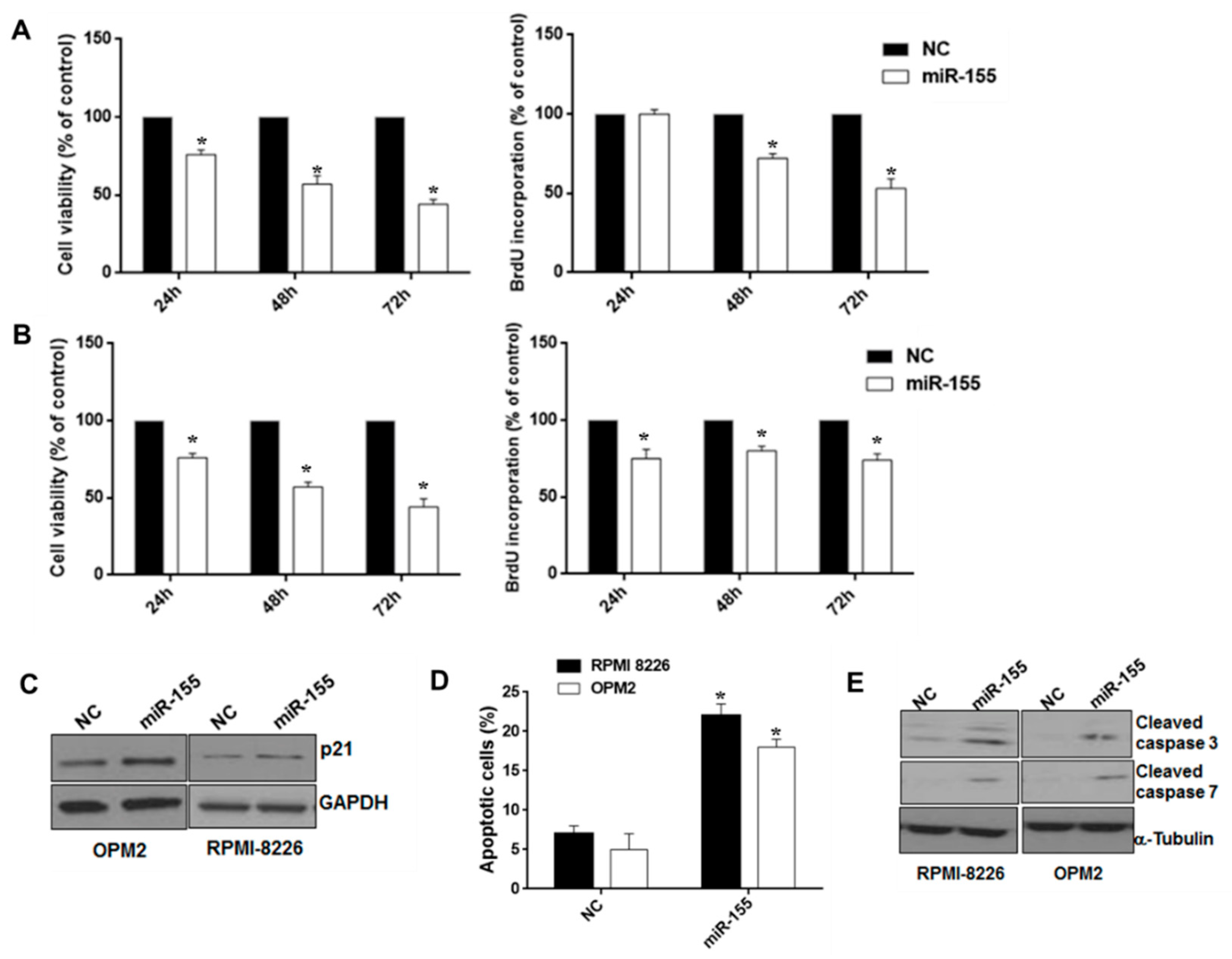
2.2. Synthetic miR-155 Mimics Trigger Anti-Proliferative and Pro-Apoptotic Effects in MM Cells
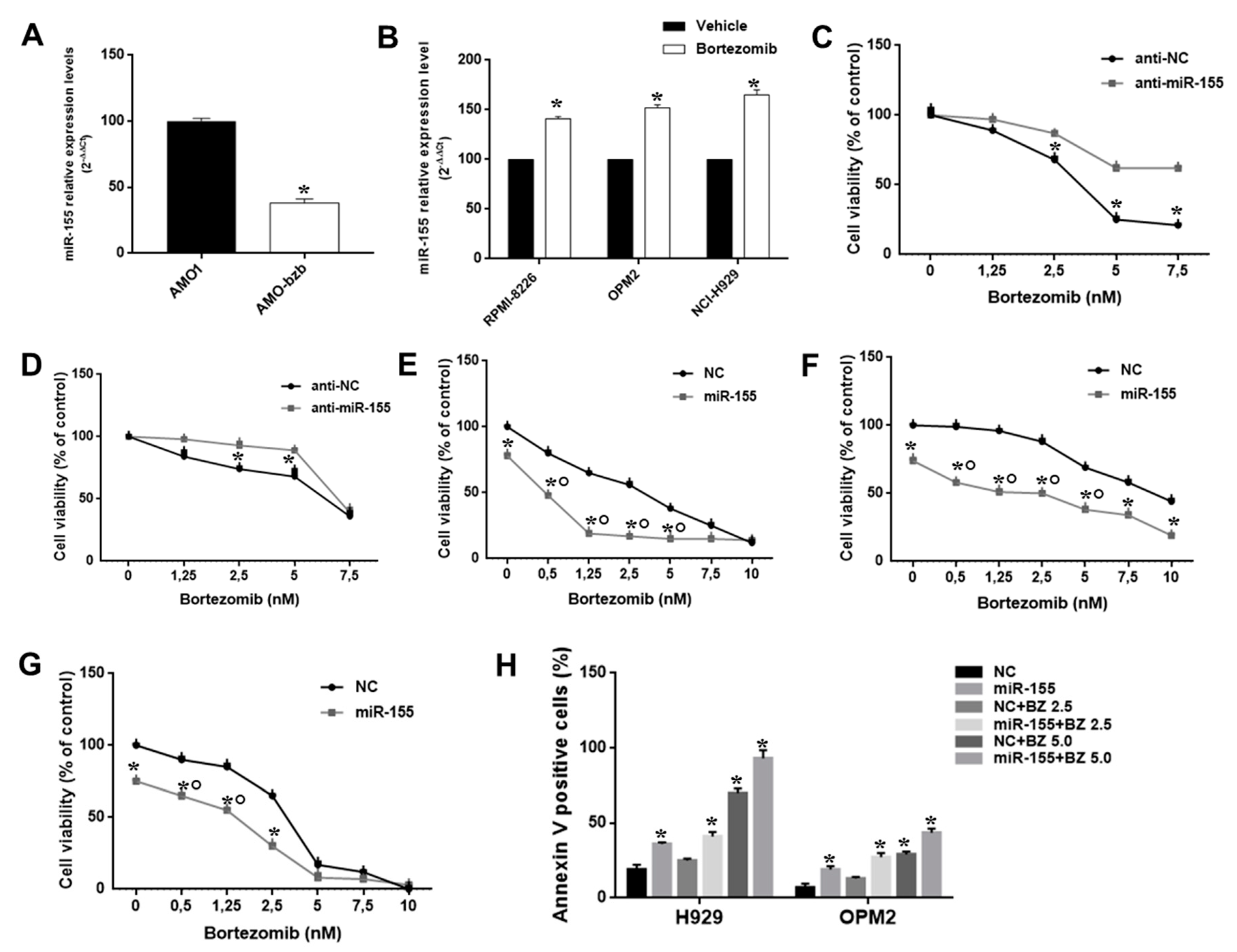
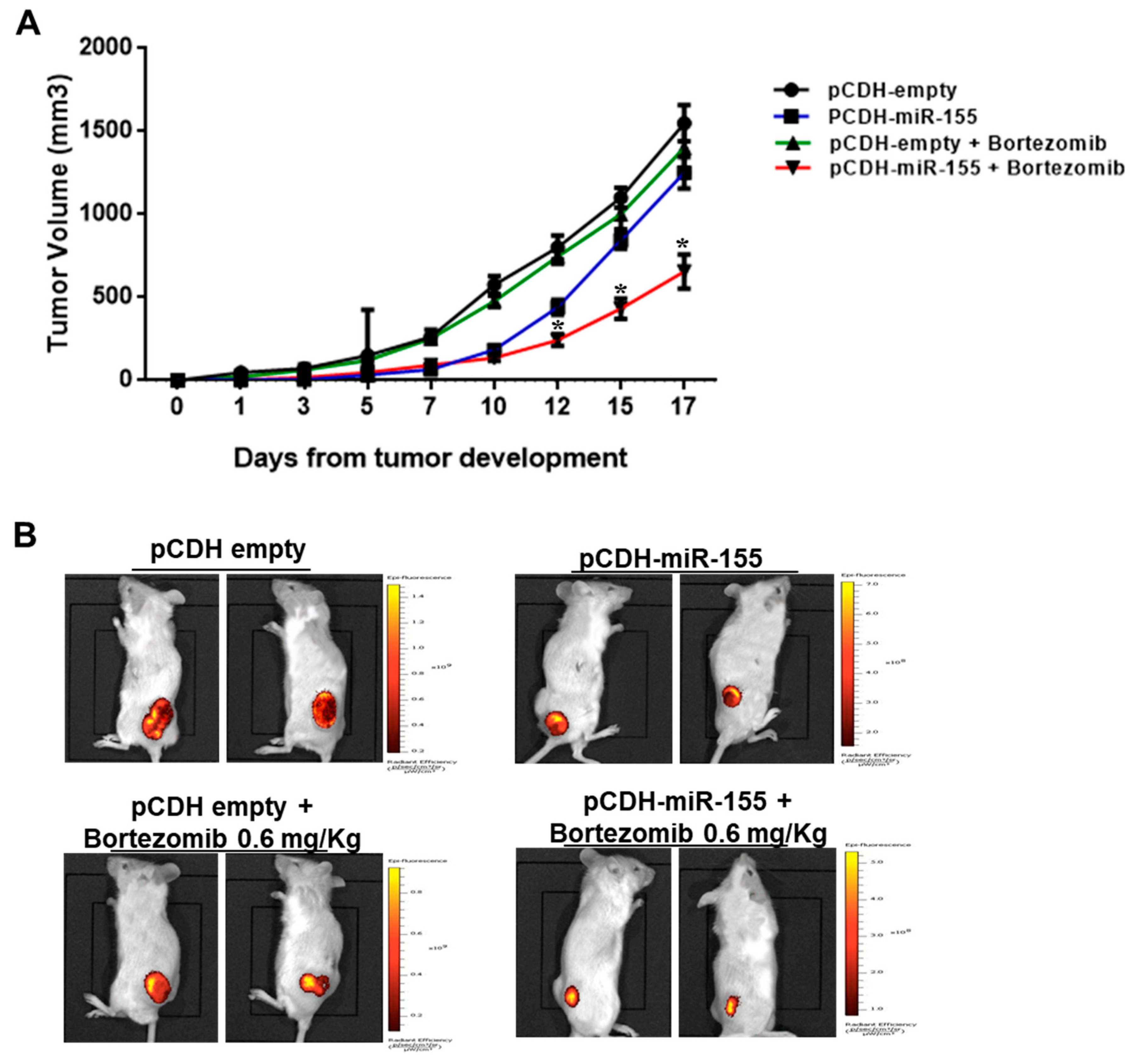
2.3. The miR-155 Modulates the Anti-MM Activity of Bortezomib Both In Vitro and In Vivo
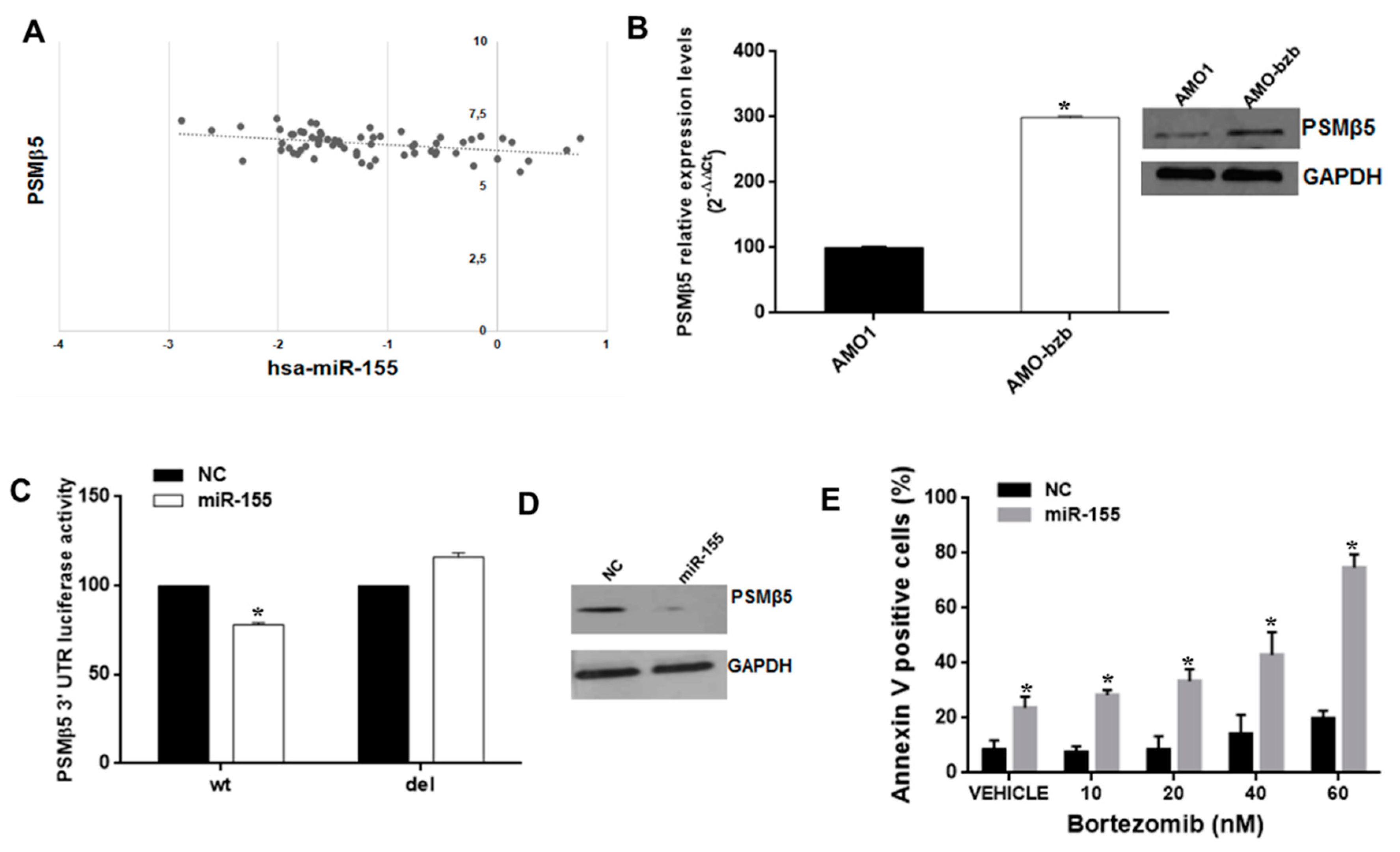
2.4. miR-155 Targets the Proteasome Subunit β5 (PSMβ5) and Sensitizes AMO-bzb Cell Line to Bortezomib
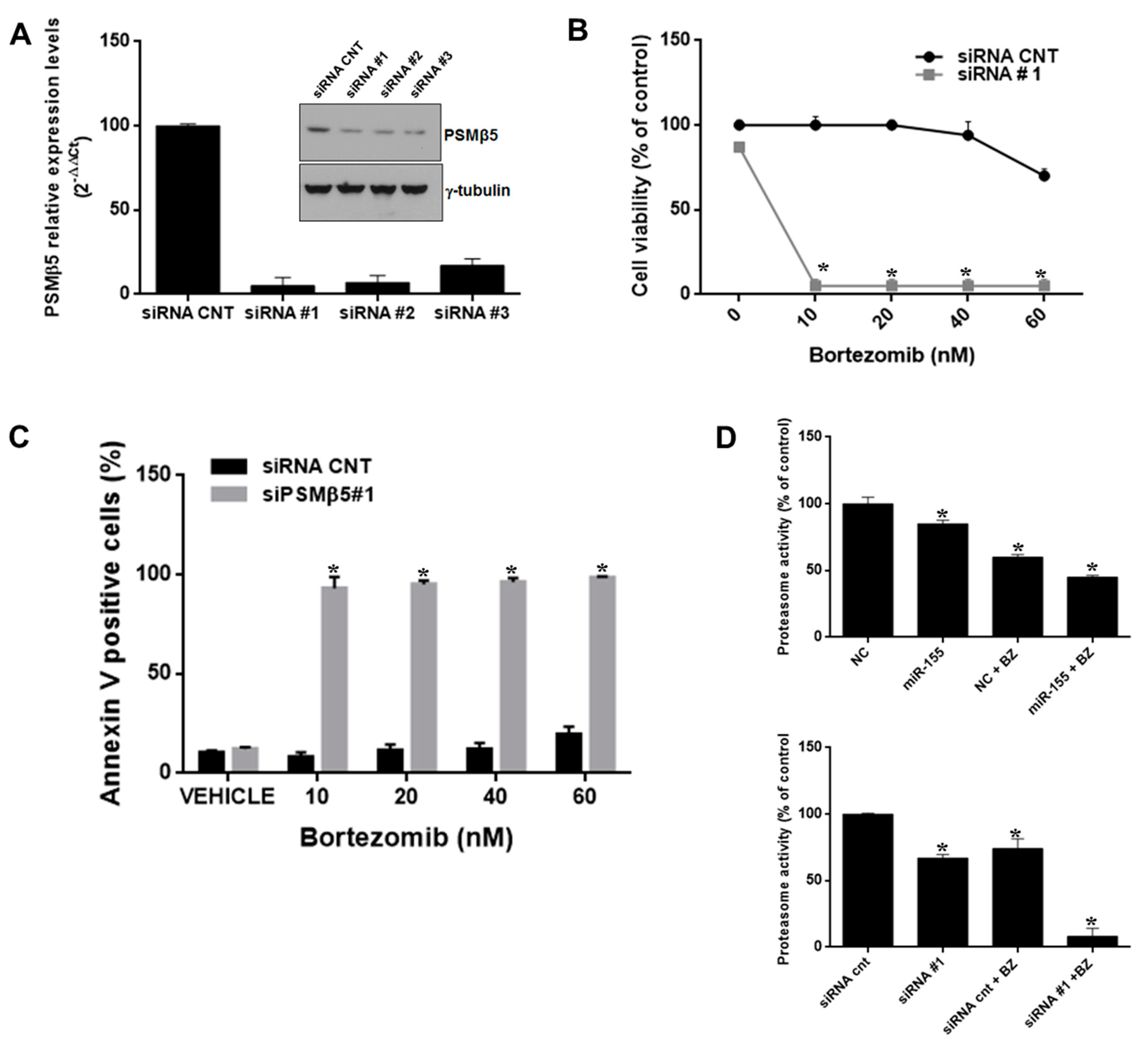
2.5. PSMβ5-Silencing Phenocopies miR-155 Effects on Bortezomib Sensitivity
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Drugs
4.2. Quantitative Real-Time Amplification of miRNAs and mRNAs
4.3. In Vitro Transfection of MM cells
4.4. CCK8, BrdU, and Cell Cycle Assays
4.5. Luciferase Reporter Experiments
4.6. Western Blotting (WB) Analysis
4.7. Animals and In Vivo Model of Human MM
4.8. Proteasome Activity Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anderson, K.C.; Carrasco, R.D. Pathogenesis of myeloma. Ann. Rev. Pathol. 2011, 6, 249–274. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; De Luca, A.; Morelli, E.; Giavaresi, G.; Tagliaferri, P.; Tassone, P.; Amodio, N. MicroRNAs: Novel Crossroads between Myeloma Cells and the Bone Marrow Microenvironment. BioMed Res. Int. 2016, 2016, 6504593. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Rossi, M.; Amodio, N.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P.; Cho, W.C. MicroRNA and multiple myeloma: From laboratory findings to translational therapeutic approaches. Curr. Pharm. Biotechnol. 2014, 15, 459–467. [Google Scholar] [CrossRef]
- Rossi, M.; Amodio, N.; Di Martino, M.T.; Caracciolo, D.; Tagliaferri, P.; Tassone, P. From target therapy to miRNA therapeutics of human multiple myeloma: Theoretical and technological issues in the evolving scenario. Curr. Drug Targets 2013, 14, 1144–1149. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Di Martino, M.T.; Neri, A.; Tagliaferri, P.; Tassone, P. Non-coding RNA: A novel opportunity for the personalized treatment of multiple myeloma. Expert Opin. Biol. Ther. 2013, 13 (Suppl. S1), S125–S137. [Google Scholar] [CrossRef] [PubMed]
- Orellana, E.A.; Kasinski, A.L. MicroRNAs in Cancer: A Historical Perspective on the Path from Discovery to Therapy. Cancers (Basel) 2015, 7, 1388–1405. [Google Scholar] [CrossRef] [Green Version]
- Di Martino, M.T.; Gulla, A.; Cantafio, M.E.; Lionetti, M.; Leone, E.; Amodio, N.; Guzzi, P.H.; Foresta, U.; Conforti, F.; Cannataro, M.; et al. In vitro and in vivo anti-tumor activity of miR-221/222 inhibitors in multiple myeloma. Oncotarget 2013, 4, 242–255. [Google Scholar] [CrossRef]
- Di Martino, M.T.; Rossi, M.; Caracciolo, D.; Gulla, A.; Tagliaferri, P.; Tassone, P. Mir-221/222 are promising targets for innovative anticancer therapy. Expert Opin. Biol. Ther. 2016, 20, 1099–1108. [Google Scholar] [CrossRef]
- Gulla, A.; Di Martino, M.T.; Gallo Cantafio, M.E.; Morelli, E.; Amodio, N.; Botta, C.; Pitari, M.R.; Lio, S.G.; Britti, D.; Stamato, M.A.; et al. A 13 mer LNA-i-miR-221 Inhibitor Restores Drug Sensitivity in Melphalan-Refractory Multiple Myeloma Cells. Clin. Cancer Res. 2016, 22, 1222–1233. [Google Scholar] [CrossRef] [PubMed]
- Leotta, M.; Biamonte, L.; Raimondi, L.; Ronchetti, D.; Di Martino, M.T.; Botta, C.; Leone, E.; Pitari, M.R.; Neri, A.; Giordano, A.; et al. A p53-dependent tumor suppressor network is induced by selective miR-125a-5p inhibition in multiple myeloma cells. J. Cell. Physiol. 2014, 229, 2106–2116. [Google Scholar] [CrossRef] [PubMed]
- Pitari, M.R.; Rossi, M.; Amodio, N.; Botta, C.; Morelli, E.; Federico, C.; Gulla, A.; Caracciolo, D.; Di Martino, M.T.; Arbitrio, M.; et al. Inhibition of miR-21 restores RANKL/OPG ratio in multiple myeloma-derived bone marrow stromal cells and impairs the resorbing activity of mature osteoclasts. Oncotarget 2015, 6, 27343–27358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, E.; Biamonte, L.; Federico, C.; Amodio, N.; Di Martino, M.T.; Gallo Cantafio, M.E.; Manzoni, M.; Scionti, F.; Samur, M.K.; Gulla, A.; et al. Therapeutic vulnerability of multiple myeloma to MIR17PTi, a first-in-class inhibitor of pri-miR-17-92. Blood 2018, 132, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Stamato, M.A.; Gulla, A.M.; Morelli, E.; Romeo, E.; Raimondi, L.; Pitari, M.R.; Ferrandino, I.; Misso, G.; Caraglia, M.; et al. Therapeutic Targeting of miR-29b/HDAC4 Epigenetic Loop in Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 1364–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulciniti, M.; Amodio, N.; Bandi, R.L.; Cagnetta, A.; Samur, M.K.; Acharya, C.; Prabhala, R.; D’Aquila, P.; Bellizzi, D.; Passarino, G.; et al. miR-23b/SP1/c-myc forms a feed-forward loop supporting multiple myeloma cell growth. Blood Cancer J. 2016, 6, e380. [Google Scholar] [CrossRef]
- Stamato, M.A.; Juli, G.; Romeo, E.; Ronchetti, D.; Arbitrio, M.; Caracciolo, D.; Neri, A.; Tagliaferri, P.; Tassone, P.; Amodio, N. Inhibition of EZH2 triggers the tumor suppressive miR-29b network in multiple myeloma. Oncotarget 2017, 8, 106527–106537. [Google Scholar] [CrossRef]
- Caracciolo, D.; Di Martino, M.T.; Amodio, N.; Morelli, E.; Montesano, M.; Botta, C.; Scionti, F.; Talarico, D.; Altomare, E.; Gallo Cantafio, M.E.; et al. miR-22 suppresses DNA ligase III addiction in multiple myeloma. Leukemia 2018. [Google Scholar] [CrossRef]
- Gutierrez, N.C.; Sarasquete, M.E.; Misiewicz-Krzeminska, I.; Delgado, M.; De Las Rivas, J.; Ticona, F.V.; Ferminan, E.; Martin-Jimenez, P.; Chillon, C.; Risueno, A.; et al. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia 2010, 24, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Calura, E.; Bisognin, A.; Manzoni, M.; Todoerti, K.; Taiana, E.; Sales, G.; Morgan, G.J.; Tonon, G.; Amodio, N.; Tassone, P.; et al. Disentangling the microRNA regulatory milieu in multiple myeloma: Integrative genomics analysis outlines mixed miRNA-TF circuits and pathway-derived networks modulated in t(4;14) patients. Oncotarget 2016, 7, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferri, P.; Rossi, M.; Di Martino, M.T.; Amodio, N.; Leone, E.; Gulla, A.; Neri, A.; Tassone, P. Promises and challenges of MicroRNA-based treatment of multiple myeloma. Curr. Cancer Drug Targets 2012, 12, 838–846. [Google Scholar] [CrossRef]
- Tassone, P.; Tagliaferri, P. Editorial: New approaches in the treatment of multiple myeloma: From target-based agents to the new era of microRNAs (dedicated to the memory of Prof. Salvatore Venuta). Curr. Cancer Drug Targets 2012, 12, 741–742. [Google Scholar] [CrossRef] [PubMed]
- Misso, G.; Di Martino, M.T.; De Rosa, G.; Farooqi, A.A.; Lombardi, A.; Campani, V.; Zarone, M.R.; Gulla, A.; Tagliaferri, P.; Tassone, P.; et al. Mir-34: A new weapon against cancer? Mol. Ther. Nucleic Acids 2014, 3, e194. [Google Scholar] [CrossRef]
- Lionetti, M.; Agnelli, L.; Lombardi, L.; Tassone, P.; Neri, A. MicroRNAs in the pathobiology of multiple myeloma. Curr. Cancer Drug Targets 2012, 12, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; D’Aquila, P.; Passarino, G.; Tassone, P.; Bellizzi, D. Epigenetic modifications in multiple myeloma: Recent advances on the role of DNA and histone methylation. Expert Opin. Ther. Targets 2017, 21, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Calame, K. MicroRNA-155 function in B Cells. Immunity 2007, 27, 825–827. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.; Dahlberg, J.E. miR-155/BIC as an oncogenic microRNA. Genes Chromosomes Cancer 2006, 45, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Costinean, S.; Zanesi, N.; Pekarsky, Y.; Tili, E.; Volinia, S.; Heerema, N.; Croce, C.M. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7024–7029. [Google Scholar] [CrossRef] [PubMed]
- Kluiver, J.; Haralambieva, E.; de Jong, D.; Blokzijl, T.; Jacobs, S.; Kroesen, B.J.; Poppema, S.; van den Berg, A. Lack of BIC and microRNA miR-155 expression in primary cases of Burkitt lymphoma. Genes Chromosomes Cancer 2006, 45, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Nie, H.; Wang, M.; Su, L.P.; Li, J.F.; Yu, Y.Y.; Yan, M.; Qu, Q.L.; Zhu, Z.G.; Liu, B.Y. microRNA-155 is downregulated in gastric cancer cells and involved in cell metastasis. Oncol. Rep. 2012, 27, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Levati, L.; Pagani, E.; Romani, S.; Castiglia, D.; Piccinni, E.; Covaciu, C.; Caporaso, P.; Bondanza, S.; Antonetti, F.R.; Bonmassar, E.; et al. MicroRNA-155 targets the SKI gene in human melanoma cell lines. Pigment Cell Melanoma Res. 2011, 24, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Roldo, C.; Missiaglia, E.; Hagan, J.P.; Falconi, M.; Capelli, P.; Bersani, S.; Calin, G.A.; Volinia, S.; Liu, C.G.; Scarpa, A.; et al. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J. Clin. Oncol. 2006, 24, 4677–4684. [Google Scholar] [CrossRef] [PubMed]
- Cascione, L.; Gasparini, P.; Lovat, F.; Carasi, S.; Pulvirenti, A.; Ferro, A.; Alder, H.; He, G.; Vecchione, A.; Croce, C.M.; et al. Integrated microRNA and mRNA signatures associated with survival in triple negative breast cancer. PLoS ONE 2013, 8, e55910. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Lovat, F.; Fassan, M.; Casadei, L.; Cascione, L.; Jacob, N.K.; Carasi, S.; Palmieri, D.; Costinean, S.; Shapiro, C.L.; et al. Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc. Natl. Acad. Sci. USA 2014, 111, 4536–4541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yu, F.; Jia, X.; Iwanowycz, S.; Wang, Y.; Huang, S.; Ai, W.; Fan, D. MicroRNA-155 deficiency enhances the recruitment and functions of myeloid-derived suppressor cells in tumor microenvironment and promotes solid tumor growth. Int. J. Cancer 2015, 136, E602–E613. [Google Scholar] [CrossRef] [PubMed]
- Krzeminski, P.; Sarasquete, M.E.; Misiewicz-Krzeminska, I.; Corral, R.; Corchete, L.A.; Martin, A.A.; Garcia-Sanz, R.; San Miguel, J.F.; Gutierrez, N.C. Insights into epigenetic regulation of microRNA-155 expression in multiple myeloma. Biochim. Biophys. Acta 2015, 1849, 353–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, C.; Chung, T.H.; Huang, G.; Zhou, J.; Yan, J.; Ahmann, G.J.; Fonseca, R.; Chng, W.J. Genome-wide pharmacologic unmasking identifies tumor suppressive microRNAs in multiple myeloma. Oncotarget 2015, 6, 26508–26518. [Google Scholar] [CrossRef] [Green Version]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Volinia, S.; Costinean, S.; Galasso, M.; Neinast, R.; Santhanam, R.; Parthun, M.R.; Perrotti, D.; Marcucci, G.; Garzon, R.; et al. miR-155 targets histone deacetylase 4 (HDAC4) and impairs transcriptional activity of B-cell lymphoma 6 (BCL6) in the Emu-miR-155 transgenic mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 20047–20052. [Google Scholar] [CrossRef]
- Jelinek, T.; Bezdekova, R.; Zatopkova, M.; Burgos, L.; Simicek, M.; Sevcikova, T.; Paiva, B.; Hajek, R. Current applications of multiparameter flow cytometry in plasma cell disorders. Blood Cancer J. 2018, 8, e621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.C. Oncogenomics to target myeloma in the bone marrow microenvironment. Clin. Cancer Res. 2011, 17, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; San-Miguel, J.F.; Anderson, K.C. Emerging therapies for the treatment of relapsed or refractory multiple myeloma. Eur. J. Haematol. 2011, 86, 1–15. [Google Scholar] [CrossRef]
- Chauhan, D.; Li, G.; Podar, K.; Hideshima, T.; Mitsiades, C.; Schlossman, R.; Munshi, N.; Richardson, P.; Cotter, F.E.; Anderson, K.C. Targeting mitochondria to overcome conventional and bortezomib/proteasome inhibitor PS-341 resistance in multiple myeloma (MM) cells. Blood 2004, 104, 2458–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amodio, N.; Di Martino, M.T.; Foresta, U.; Leone, E.; Lionetti, M.; Leotta, M.; Gulla, A.M.; Pitari, M.R.; Conforti, F.; Rossi, M.; et al. miR-29b sensitizes multiple myeloma cells to bortezomib-induced apoptosis through the activation of a feedback loop with the transcription factor Sp1. Cell Death Dis. 2012, 3, e436. [Google Scholar] [CrossRef] [PubMed]
- Rastgoo, N.; Pourabdollah, M.; Abdi, J.; Reece, D.; Chang, H. Dysregulation of EZH2/miR-138 axis contributes to drug resistance in multiple myeloma by downregulating RBPMS. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Stamato, M.A.; Juli, G.; Morelli, E.; Fulciniti, M.; Manzoni, M.; Taiana, E.; Agnelli, L.; Cantafio, M.E.G.; Romeo, E.; et al. Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia 2018, 32, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Raimondi, L.; Juli, G.; Stamato, M.A.; Caracciolo, D.; Tagliaferri, P.; Tassone, P. MALAT1: A druggable long non-coding RNA for targeted anti-cancer approaches. J. Hematol. Oncol. 2018, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, D.A.; et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef]
- Chang, S.; Sharan, S.K. Epigenetic control of an oncogenic microRNA, miR-155, by BRCA1. Oncotarget 2012, 3, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Mendillo, M.L.; Zhao, J.; Carette, J.E.; Merrill, P.H.; Cikes, D.; Varadarajan, M.; van Diemen, F.R.; Penninger, J.M.; Goldberg, A.L.; et al. Compromising the 19S proteasome complex protects cells from reduced flux through the proteasome. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.X.; Kortum, K.M.; Zhu, Y.X.; Bruins, L.A.; Jedlowski, P.; Votruba, P.G.; Luo, M.; Stewart, R.A.; Ahmann, J.; Braggio, E.; et al. CRISPR Genome-Wide Screening Identifies Dependence on the Proteasome Subunit PSMC6 for Bortezomib Sensitivity in Multiple Myeloma. Mol. Cancer Ther. 2017, 16, 2862–2870. [Google Scholar] [CrossRef] [PubMed]
- Besse, A.; Stolze, S.C.; Rasche, L.; Weinhold, N.; Morgan, G.J.; Kraus, M.; Bader, J.; Overkleeft, H.S.; Besse, L.; Driessen, C. Carfilzomib resistance due to ABCB1/MDR1 overexpression is overcome by nelfinavir and lopinavir in multiple myeloma. Leukemia 2018, 32, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Li, G.; Shringarpure, R.; Podar, K.; Ohtake, Y.; Hideshima, T.; Anderson, K.C. Blockade of Hsp27 overcomes Bortezomib/proteasome inhibitor PS-341 resistance in lymphoma cells. Cancer Res. 2003, 63, 6174–6177. [Google Scholar] [PubMed]
- Ruckrich, T.; Kraus, M.; Gogel, J.; Beck, A.; Ovaa, H.; Verdoes, M.; Overkleeft, H.S.; Kalbacher, H.; Driessen, C. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia 2009, 23, 1098–1105. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Marvaso, G.; Barone, A.; Amodio, N.; Raimondi, L.; Agosti, V.; Altomare, E.; Scotti, V.; Lombardi, A.; Bianco, R.; Bianco, C.; et al. Sphingosine analog fingolimod (FTY720) increases radiation sensitivity of human breast cancer cells in vitro. Cancer Biol. Ther. 2014, 15, 797–805. [Google Scholar] [CrossRef] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amodio, N.; Gallo Cantafio, M.E.; Botta, C.; Agosti, V.; Federico, C.; Caracciolo, D.; Ronchetti, D.; Rossi, M.; Driessen, C.; Neri, A.; et al. Replacement of miR-155 Elicits Tumor Suppressive Activity and Antagonizes Bortezomib Resistance in Multiple Myeloma. Cancers 2019, 11, 236. https://doi.org/10.3390/cancers11020236
Amodio N, Gallo Cantafio ME, Botta C, Agosti V, Federico C, Caracciolo D, Ronchetti D, Rossi M, Driessen C, Neri A, et al. Replacement of miR-155 Elicits Tumor Suppressive Activity and Antagonizes Bortezomib Resistance in Multiple Myeloma. Cancers. 2019; 11(2):236. https://doi.org/10.3390/cancers11020236
Chicago/Turabian StyleAmodio, Nicola, Maria Eugenia Gallo Cantafio, Cirino Botta, Valter Agosti, Cinzia Federico, Daniele Caracciolo, Domenica Ronchetti, Marco Rossi, Christoph Driessen, Antonino Neri, and et al. 2019. "Replacement of miR-155 Elicits Tumor Suppressive Activity and Antagonizes Bortezomib Resistance in Multiple Myeloma" Cancers 11, no. 2: 236. https://doi.org/10.3390/cancers11020236
APA StyleAmodio, N., Gallo Cantafio, M. E., Botta, C., Agosti, V., Federico, C., Caracciolo, D., Ronchetti, D., Rossi, M., Driessen, C., Neri, A., Tagliaferri, P., & Tassone, P. (2019). Replacement of miR-155 Elicits Tumor Suppressive Activity and Antagonizes Bortezomib Resistance in Multiple Myeloma. Cancers, 11(2), 236. https://doi.org/10.3390/cancers11020236