Brain Metastases from Lung Cancer: Is MET an Actionable Target?

 ,
,  and
and 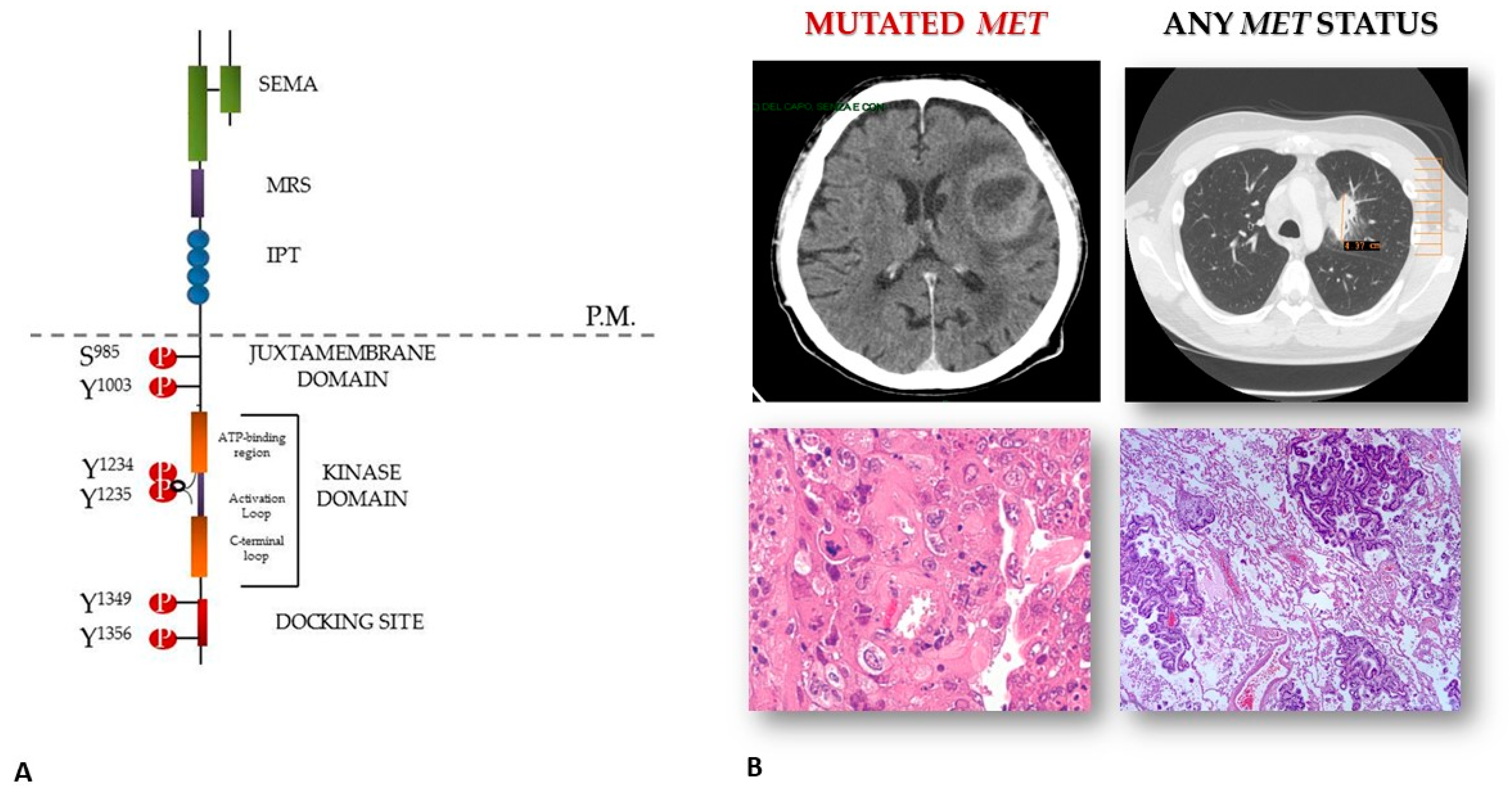{kind=link}
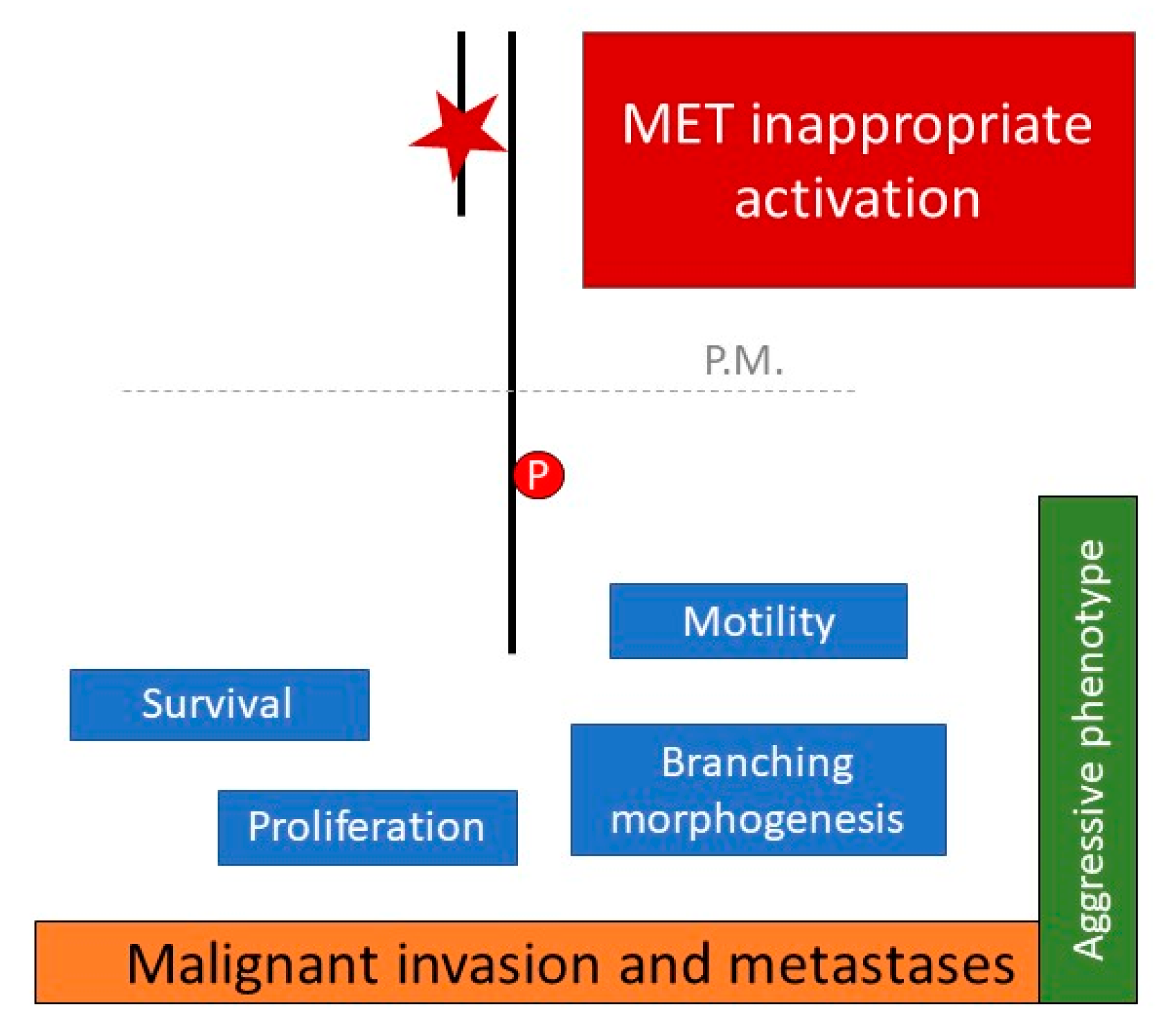{kind=link}
Abstract
:1. Introduction
2. Invasive Growth and Metastatic Spread
3. MET-Driven Invasive Growth in Brain Metastases from NSCLC: A Proposed Hypothesis for a Still Obscure Phenomenon
4. Therapeutic MET Targeting in Brain Metastases
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed]
- Truini, A.; Santos Pereira, P.; Cavazza, A.; Spagnolo, P.; Nosseir, S.; Longo, L.; Jukna, A.; Lococo, F.; Vincenzi, G.; Bogina, G.; et al. Classification of Different Patterns of Pulmonary Adenocarcinomas. Expert Rev. Respir. Med. 2015, 9, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, J.; Govindan, R. Lung Cancer in Never Smokers: A Review. J. Clin. Oncol. 2007, 25, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Chute, C.G.; Greenberg, E.R.; Baron, J.; Korson, R.; Baker, J.; Yates, J. Presenting Conditions of 1539 Population-Based Lung Cancer Patients by Cell Type and Stage in New Hampshire and Vermont. Cancer 1985, 56, 2107–2111. [Google Scholar] [CrossRef]
- Lagerwaard, F.J.; Levendag, P.C.; Nowak, P.J.; Eijkenboom, W.M.; Hanssens, P.E.; Schmitz, P.I. Identification of Prognostic Factors in Patients with Brain Metastases: A Review of 1292 Patients. Int. J. Radiat. Oncol. Biol. Phys. 1999, 43, 795–803. [Google Scholar] [CrossRef]
- Gaspar, L.; Scott, C.; Rotman, M.; Asbell, S.; Phillips, T.; Wasserman, T.; McKenna, W.G.; Byhardt, R. Recursive Partitioning Analysis (RPA) of Prognostic Factors in Three Radiation Therapy Oncology Group (RTOG) Brain Metastases Trials. Int. J. Radiat. Oncol. Biol. Phys. 1997, 37, 745–751. [Google Scholar] [CrossRef]
- Specht, H.M.; Combs, S.E. Stereotactic Radiosurgery of Brain Metastases. J. Neurosurg. Sci. 2016, 60, 357–366. [Google Scholar] [PubMed]
- Gao, H.X.; Huang, S.G.; Du, J.F.; Zhang, X.C.; Jiang, N.; Kang, W.X.; Mao, J.; Zhao, Q. Comparison of Prognostic Indices in NSCLC Patients with Brain Metastases after Radiosurgery. Int. J. Biol. Sci. 2018, 14, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Ryken, T.C.; McDermott, M.; Robinson, P.D.; Ammirati, M.; Andrews, D.W.; Asher, A.L.; Burri, S.H.; Cobbs, C.S.; Gaspar, L.E.; Kondziolka, D.; et al. The role of steroids in the management of brain metastases: A systematic review and evidence-based clinical practice guideline. J. Neuro-Oncol. 2010, 96, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Winkler, F.; Valiente, M.; Manegold, C.; Moyal, E.; Widhalm, G.; Tonn, J.-C.; Zielinski, C. Recent advances in the biology and treatment of brain metastases of non-small cell lung cancer: Summary of a multidisciplinary roundtable discussion. ESMO Open 2018, 3, e000262. [Google Scholar] [CrossRef] [PubMed]
- Popper, H.H. Progression and metastasis of lung cancer. Cancer Metast. Rev. 2016, 35, 75–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M.; Trusolino, L.; Boccaccio, C. Known and novel roles of the MET oncogene in cancer: A coherent approach to targeted therapy. Nat. Rev. Cancer 2018, 18, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Faham, N.; Welm, A.L. RON Signaling Is a Key Mediator of Tumor Progression in Many Human Cancers. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.-P.; Zhou, Y.-Q.; Zhang, R.; Wang, M.-H. MSP-RON signalling in cancer: Pathogenesis and therapeutic potential. Nat. Rev. Cancer 2013, 13, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, H.; Okamoto, I.; Okamoto, W.; Tanizaki, J.; Nakagawa, K.; Nishio, K. Targeting MET Amplification as a New Oncogenic Driver. Cancers 2014, 6, 1540–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.-Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, L.; Han, R.; Jiao, L.; Zheng, J.; He, Y. Clinical analysis by next-generation sequencing for NSCLC patients with MET amplification resistant to osimertinib. Lung Cancer 2018, 118, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L. Oncogenic MET as an Effective Therapeutic Target in Non-Small Cell Lung Cancer Resistant to EGFR Inhibitors: The Rise of the Phoenix. Cancer Discov. 2016, 6, 1306–1308. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated with Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. 2016, 34, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.B.; Frampton, G.M.; Suh, J.; Chalmers, Z.R.; Rosenzweig, M.; Erlich, R.L.; Halmos, B.; Goldman, J.; Forde, P.; Leuenberger, K.; et al. Characterization of 298 Patients with Lung Cancer Harboring MET Exon 14 Skipping Alterations. J. Thorac. Oncol. 2016, 11, 1493–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.H.; Yeung, S.F.; Chan, A.W.H.; Chung, L.Y.; Chau, S.L.; Lung, R.W.M.; Tong, C.Y.; Chow, C.; Tin, E.K.Y.; Yu, Y.H.; et al. MET Amplification and Exon 14 Splice Site Mutation Define Unique Molecular Subgroups of Non-Small Cell Lung Carcinoma with Poor Prognosis. Clin. Cancer Res. 2016, 22, 3048–3056. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.D.; Lee, S.E.; Oh, D.-Y.; Yu, D.-B.; Jeong, H.M.; Kim, J.; Hong, S.; Jung, H.S.; Oh, E.; Song, J.-Y.; et al. MET Exon 14 Skipping Mutations in Lung Adenocarcinoma: Clinicopathologic Implications and Prognostic Values. J. Thorac. Oncol. 2017, 12, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Drilon, A.; Fan, P.-D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET Inhibitors in Patients with Stage IV Lung Adenocarcinomas Harboring MET Mutations Causing Exon 14 Skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, G.; Giaccone, G. C-MET inhibitors for advanced non-small cell lung cancer. Expert Opin. Investig. Drugs 2018, 27, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Reungwetwattana, T.; Liang, Y.; Zhu, V.; Ou, S.-H.I. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The Why, the How, the Who, the Unknown, and the Inevitable. Lung Cancer 2017, 103, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yin, J.; Peng, F. Acquired resistance to crizotinib in advanced lung adenocarcinoma with MET exon 14 skipping. Lung Cancer 2017, 113, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Suzawa, K.; Offin, M.; Lu, D.; Kurzatkowski, C.; Vojnic, M.; Smith, R.S.; Sabari, J.K.; Tai, H.; Mattar, M.; Khodos, I.; et al. Activation of KRAS mediates resistance to targeted therapy in MET exon 14 mutant non-small cell lung cancer. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Yang, N.; Zhang, Y. Novel MET Exon 14 Skipping Treatment-Naïve Lung Adenocarcinoma Presented Primary Resistance to Crizotinib. J. Thorac. Oncol. 2018, 13, e124–e126. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Parsons, D.W.; Silliman, N.; Ptak, J.; Szabo, S.; Saha, S.; Markowitz, S.; Willson, J.K.V.; Parmigiani, G.; Kinzler, K.W.; et al. Mutational analysis of the tyrosine kinome in colorectal cancers. Science 2003, 300, 949. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Stella, G.M.; Senetta, R.; Inghilleri, S.; Verdun di Cantogno, L.; Mantovani, C.; Piloni, D.; Scudeller, L.; Meloni, F.; Papotti, M.; Ricardi, U.; et al. MET mutations are associated with aggressive and radioresistant brain metastatic non-small-cell lung cancer. Neuro-Oncol. 2016, 18, 598–599. [Google Scholar] [CrossRef] [PubMed]
- Milan, M.; Benvenuti, S.; Balderacchi, A.M.; Virzì, A.R.; Gentile, A.; Senetta, R.; Cassoni, P.; Comoglio, P.M.; Stella, G.M. RON tyrosine kinase mutations in brain metastases from lung cancer. ERJ Open Res. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Love, C.A.; Esnouf, R.M.; Jones, E.Y. The sema domain. Curr. Opin. Struct. Biol. 2004, 14, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Kong-Beltran, M.; Stamos, J.; Wickramasinghe, D. The Sema domain of Met is necessary for receptor dimerization and activation. Cancer Cell 2004, 6, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, G.M.; Benvenuti, S.; Gramaglia, D.; Scarpa, A.; Tomezzoli, A.; Cassoni, P.; Senetta, R.; Venesio, T.; Pozzi, E.; Bardelli, A.; et al. MET mutations in cancers of unknown primary origin (CUPs). Hum. Mutat. 2011, 32, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M.; Boccaccio, C. Scatter factors and invasive growth. Semin. Cancer Biol. 2001, 11, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Stella, G.M.; Benvenuti, S.; Gentile, A.; Comoglio, P.M. MET Activation and Physical Dynamics of the Metastatic Process: The Paradigm of Cancers of Unknown Primary Origin. EBioMedicine 2017, 24, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Serlin, Y.; Shelef, I.; Knyazer, B.; Friedman, A. Anatomy and physiology of the blood-brain barrier. Semin. Cell Dev. Biol. 2015, 38, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Almutairi, M.M.A.; Gong, C.; Xu, Y.G.; Chang, Y.; Shi, H. Factors controlling permeability of the blood-brain barrier. Cell. Mol. Life Sci. 2016, 73, 57–77. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Mikitsh, J.L.; Chacko, A.-M. Pathways for small molecule delivery to the central nervous system across the blood-brain barrier. Perspect. Med. Chem. 2014, 6, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Tiwary, S.; Morales, J.E.; Kwiatkowski, S.C.; Lang, F.F.; Rao, G.; McCarty, J.H. Metastatic Brain Tumors Disrupt the Blood-Brain Barrier and Alter Lipid Metabolism by Inhibiting Expression of the Endothelial Cell Fatty Acid Transporter Mfsd2a. Sci. Rep. 2018, 8, 8267. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Liu, Y.; Sharma, S.; Wu, K.; Chan, M.D.; Lo, H.-W.; Carpenter, R.L.; Metheny-Barlow, L.J.; Zhou, X.; Qasem, S.A.; et al. Activation of the c-Met Pathway Mobilizes an Inflammatory Network in the Brain Microenvironment to Promote Brain Metastasis of Breast Cancer. Cancer Res. 2016, 76, 4970–4980. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.; Finocchiaro, G.; Belda-Iniesta, C.; Recht, L.; Brandes, A.A.; Pineda, E.; Mikkelsen, T.; Chinot, O.L.; Balana, C.; Macdonald, D.R.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab Plus Bevacizumab Versus Placebo Plus Bevacizumab in Patients with Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O6-Methylguanine-DNA Methyltransferase Biomarker Analyses. J. Clin. Oncol. 2017, 35, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, T.T.; Grandal, M.M.; Skartved, N.J.Ø.; Hald, R.; Alifrangis, L.; Koefoed, K.; Lindsted, T.; Fröhlich, C.; Pollmann, S.E.; Eriksen, K.W.; et al. Sym015: A Highly Efficacious Antibody Mixture against MET-Amplified Tumors. Clin. Cancer Res. 2017, 23, 5923–5935. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Goetsch, L.; Tucker, L.; Zhang, Q.; Gonzalez, A.; Vaidya, K.S.; Oleksijew, A.; Boghaert, E.; Song, M.; Sokolova, I.; et al. Anti-c-Met monoclonal antibody ABT-700 breaks oncogene addiction in tumors with MET amplification. BMC Cancer 2016, 16, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Mandiyan, S.; Robinson, B.S.; McMahon, G. Antitumor Properties of an IgG2-Enhanced Next-Generation MET Monoclonal Antibody That Degrades Wild-Type and Mutant MET Receptors. Cancer Res. 2016, 76, 5788–5797. [Google Scholar] [CrossRef] [PubMed]
- Négrier, S.; Moriceau, G.; Attignon, V.; Haddad, V.; Pissaloux, D.; Guerin, N.; Carrie, C. Activity of cabozantinib in radioresistant brain metastases from renal cell carcinoma: Two case reports. J. Med. Case Rep. 2018, 12, 351. [Google Scholar] [CrossRef] [PubMed]
- Klempner, S.J.; Borghei, A.; Hakimian, B.; Ali, S.M.; Ou, S.-H.I. Intracranial Activity of Cabozantinib in MET Exon 14-Positive NSCLC with Brain Metastases. J. Thorac. Oncol. 2017, 12, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xiao, P.; Ye, Y.; Liu, P.; Han, L.; Dong, L.; She, C.; Yu, J. Rapid response of brain metastasis to crizotinib in a patient with KLC1-ALK fusion and MET gene amplification positive non-small cell lung cancer: A case report. Cancer Biol. Med. 2017, 14, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, L.D.; Aranda, R.; Lee, M.; Tovar, E.A.; Essenburg, C.J.; Madaj, Z.; Chiang, H.; Briere, D.; Hallin, J.; Lopez-Casas, P.P.; et al. Glesatinib Exhibits Antitumor Activity in Lung Cancer Models and Patients Harboring MET Exon 14 Mutations and Overcomes Mutation-mediated Resistance to Type I MET Inhibitors in Nonclinical Models. Clin. Cancer Res. 2017, 23, 6661–6672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Marti, A.; Felip, E.; Matito, J.; Mereu, E.; Navarro, A.; Cedrés, S.; Pardo, N.; Martinez de Castro, A.; Remon, J.; Miquel, J.M.; et al. Dual MET and ERBB inhibition overcomes intratumor plasticity in osimertinib-resistant-advanced non-small-cell lung cancer (NSCLC). Ann. Oncol. 2017, 28, 2451–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breindel, J.L.; Haskins, J.W.; Cowell, E.P.; Zhao, M.; Nguyen, D.X.; Stern, D.F. EGF receptor activates MET through MAPK to enhance non-small cell lung carcinoma invasion and brain metastasis. Cancer Res. 2013, 73, 5053–5065. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Welsh, E.A.; Fang, B.; Bai, Y.; Kinose, F.; Eschrich, S.A.; Koomen, J.M.; Haura, E.B. Phosphoproteomics Reveals MAPK Inhibitors Enhance MET- and EGFR-Driven AKT Signaling in KRAS-Mutant Lung Cancer. Mol. Cancer Res. 2016, 14, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Kaufman, M.D.; Leary, C.B.; Turner, B.A.; Wise, S.C.; Ahn, Y.M.; Booth, R.J.; Caldwell, T.M.; Ensinger, C.L.; Hood, M.M.; et al. Altiratinib Inhibits Tumor Growth, Invasion, Angiogenesis, and Microenvironment-Mediated Drug Resistance via Balanced Inhibition of MET, TIE2, and VEGFR2. Mol. Cancer Ther. 2015, 14, 2023–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Mu, Q.; Bao, Z.; Chen, Y.; Liu, Y.; Chen, J.; Wang, K.; Wang, Z.; Nam, Y.; Jiang, B.; et al. Mutational Landscape of Secondary Glioblastoma Guides MET-Targeted Trial in Brain Tumor. Cell 2018, 175, 1665–1678. [Google Scholar] [CrossRef] [PubMed]
- Glodde, N.; Bald, T.; van den Boorn-Konijnenberg, D.; Nakamura, K.; O’Donnell, J.S.; Szczepanski, S.; Brandes, M.; Eickhoff, S.; Das, I.; Shridhar, N.; et al. Reactive Neutrophil Responses Dependent on the Receptor Tyrosine Kinase c-MET Limit Cancer Immunotherapy. Immunity 2017, 47, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Papaccio, F.; Della Corte, C.M.; Viscardi, G.; Di Liello, R.; Esposito, G.; Sparano, F.; Ciardiello, F.; Morgillo, F. HGF/MET and the Immune System: Relevance for Cancer Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3595. [Google Scholar] [CrossRef] [PubMed]
- Hübel, J.; Hieronymus, T. HGF/Met-Signaling Contributes to Immune Regulation by Modulating Tolerogenic and Motogenic Properties of Dendritic Cells. Biomedicines 2015, 3, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, J.-H.; Birchmeier, C.; Zenke, M.; Hieronymus, T. The HGF Receptor/Met Tyrosine Kinase Is a Key Regulator of Dendritic Cell Migration in Skin Immunity. J. Immunol. 2012, 189, 1699–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Lee, H.W.; Kim, Y.; Lee, Y.; Choi, Y.-S.; Kim, K.H.; Jin, J.; Lee, J.; Joo, K.M.; Nam, D.-H. Radiosensitization of brain metastasis by targeting c-MET. Lab. Investig. 2013, 93, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bacco, F.; Luraghi, P.; Medico, E.; Reato, G.; Girolami, F.; Perera, T.; Gabriele, P.; Comoglio, P.M.; Boccaccio, C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst. 2011, 103, 645–661. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stella, G.M.; Corino, A.; Berzero, G.; Kolling, S.; Filippi, A.R.; Benvenuti, S. Brain Metastases from Lung Cancer: Is MET an Actionable Target? Cancers 2019, 11, 271. https://doi.org/10.3390/cancers11030271
Stella GM, Corino A, Berzero G, Kolling S, Filippi AR, Benvenuti S. Brain Metastases from Lung Cancer: Is MET an Actionable Target? Cancers. 2019; 11(3):271. https://doi.org/10.3390/cancers11030271
Chicago/Turabian StyleStella, Giulia M., Alessandra Corino, Giulia Berzero, Stefan Kolling, Andrea R. Filippi, and Silvia Benvenuti. 2019. "Brain Metastases from Lung Cancer: Is MET an Actionable Target?" Cancers 11, no. 3: 271. https://doi.org/10.3390/cancers11030271
APA StyleStella, G. M., Corino, A., Berzero, G., Kolling, S., Filippi, A. R., & Benvenuti, S. (2019). Brain Metastases from Lung Cancer: Is MET an Actionable Target? Cancers, 11(3), 271. https://doi.org/10.3390/cancers11030271