Inhibition of BET Proteins and Histone Deacetylase (HDACs): Crossing Roads in Cancer Therapy


Abstract
:1. Introduction
2. HDAC Proteins
3. BET Proteins
4. Molecular Interplay Between HDAC and BET Proteins and Dual Targeting Inhibition Strategies
5. Combining HDAC Inhibitors and BET Protein Inhibitors with Other Anti-Cancer Drugs
5.1. Combination of HDACi with DNA Damaging Agents and PARP Inhibitors
5.2. Combination of HDACi with RTK Pathway Inhibitors
5.3. Combination of HDACi with Proteasome Inhibitors
5.4. Combination of HDACi with TRAIL
5.5. Combination of HDACi with Hormone Therapy
5.6. Combination of HDACi with Immune Checkpoint Inhibitors
5.7. Combination of BETi with DNA-Damaging Agents and PARPi
5.8. Combination of BETi with RTK Pathway Inhibitors
5.9. Combination of BETi with Hormone Therapy
5.10. Combination of BETi with CDK Inhibitors
5.11. Combination of BETi with BCL-2 Inhibitors
5.12. Combination of BETi with Immune Checkpoint Inhibitors
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.W.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer development, progression, and therapy: An epigenetic overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Glass, C.K.; Rosenfeld, M.G. Coactivator and corepressor complexes in nuclear receptor function. Curr. Opin. Genet. Dev. 1999, 9, 140–147. [Google Scholar] [CrossRef]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Gene Dev. 2000, 14, 121–141. [Google Scholar] [PubMed]
- Chou, C.W.; Wu, M.S.; Huang, W.C.; Chen, C.C. HDAC Inhibition Decreases the Expression of EGFR in Colorectal Cancer Cells. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Greer, C.B.; Cecchini, K.R.; Harris, L.N.; Tuck, D.P.; Kim, T.H. HDAC inhibitors induce transcriptional repression of high copy number genes in breast cancer through elongation blockade. Oncogene 2013, 32, 2828–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancisi, V.; Gandolfi, G.; Ambrosetti, D.C.; Ciarrocchi, A. Histone Deacetylase Inhibitors Repress Tumoral Expression of the Proinvasive Factor RUNX2. Cancer Res. 2015, 75, 1868–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.B.; Zang, C.Z.; Cui, K.R.; Schones, D.E.; Barski, A.; Peng, W.Q.; Zhao, K.J. Genome-wide Mapping of HATs and HDACs Reveals Distinct Functions in Active and Inactive Genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.M.; Yao, Y.L.; Sun, J.M.; Davie, J.R.; Seto, E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem. 1997, 272, 28001–28007. [Google Scholar] [CrossRef] [PubMed]
- LaBonte, M.J.; Wilson, P.M.; Fazzone, W.; Groshen, S.; Lenz, H.J.; Ladner, R.D. DNA microarray profiling of genes differentially regulated by the histone deacetylase inhibitors vorinostat and LBH589 in colon cancer cell lines. BMC Med. Genom. 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Doetzlhofer, A.; Rotheneder, H.; Lagger, G.; Koranda, M.; Kurtev, V.; Brosch, G.; Wintersberger, E.; Seiser, C. Histone deacetylase 1 can repress transcription by binding to Sp1. Mol. Cell Biol. 1999, 19, 5504–5511. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Greer, C.B.; Tanaka, Y.; Kim, Y.J.; Xie, P.; Zhang, M.Q.; Park, I.H.; Kim, T.H. Histone Deacetylases Positively Regulate Transcription through the Elongation Machinery. Cell Rep. 2015, 13, 1444–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.N.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadoul, K.; Boyault, C.; Pabion, M.; Khochbin, S. Regulation of protein turnover by acetyltransferases and deacetylases. Biochimie 2008, 90, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Balbas, M.A.; Bauer, U.M.; Nielsen, S.J.; Brehm, A.; Kouzarides, T. Regulation of E2F1 activity by acetylation. EMBO J. 2000, 19, 662–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Menzies, K.J.; Zhang, H.; Katsyuba, E.; Auwerx, J. Protein acetylation in metabolism–– metabolites and cofactors. Nat. Rev. Endocrinol. 2016, 12, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Kwon, H.J.; Yoon, B.I.; Kim, J.H.; Han, S.U.; Joo, H.J.; Kim, D.Y. Expression profile of histone deacetylase 1 in gastric cancer tissues. JPN J. Cancer. Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class IHDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.H.; Laban, M.; Leung, C.H.W.; Lee, L.; Lee, C.K.; Salto-Tellez, M.; Raju, G.C.; Hooi, S.C. Inhibition of histone deacetylase 2 increases apoptosis and p21(Cip1/WAF1) expression, independent of histone deacetylase 1. Cell Death Differ. 2005, 12, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.P.; Gottlicher, M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004, 5, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.H.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.; Iwase, H. HDAC6 expression is correlated with better survival in breast cancer. Clin. Cancer Res. 2004, 10, 6962–6968. [Google Scholar] [CrossRef] [PubMed]
- Niegisch, G.; Knievel, J.; Koch, A.; Hader, C.; Fischer, U.; Albers, P.; Schulz, W.A. Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol. Oncol. 2013, 31, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Hoshino, T.; Redner, R.L.; Kajigaya, S.; Liu, J.M. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc. Natl. Acad. Sci. USA 1998, 95, 10860–10865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.J.; Sternsdorf, T.; Tini, M.; Evans, R.M. Transcriptional regulation in acute promyelocytic leukemia. Oncogene 2001, 20, 7204–7215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, C.Y.; Ngo, L.; Xu, W.S.; Richon, V.M.; Marks, P.A. Histone deacetylase (HDAC) inhibitor activation of p21(WAF1) involves changes in promoter-associated proteins, including HDAC1. Proc. Natl. Acad. Sci. USA 2004, 101, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Kalafut, J.; Okon, E.; Czapinski, J.; Halasa, M.; Przybyszewska, A.; Miziak, P.; Okla, K.; Rivero-Muller, A.; Stepulak, A. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers (Basel) 2019, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Kaletsch, A.; Pinkerneil, M.; Hoffmann, M.J.; Jaguva Vasudevan, A.A.; Wang, C.; Hansen, F.K.; Wiek, C.; Hanenberg, H.; Gertzen, C.; Gohlke, H.; et al. Effects of novel HDAC inhibitors on urothelial carcinoma cells. Clin. Epigenetics 2018, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.A.; Chabner, B.A. Histone Deacetylase Inhibitors in Cancer Therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [PubMed]
- Libby, E.N.; Becker, P.S.; Burwick, N.; Green, D.J.; Holmberg, L.; Bensinger, W.I. Panobinostat: A review of trial results and future prospects in multiple myeloma. Expert Rev. Hematol. 2015, 8, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, E538–E548. [Google Scholar] [CrossRef]
- Slingerland, M.; Guchelaar, H.J.; Rosing, H.; Scheulen, M.E.; van Warmerdam, L.J.C.; Beijnen, J.H.; Gelderblom, H. Bioequivalence of Liposome-Entrapped Paclitaxel Easy-To-Use (LEP-ETU) Formulation and Paclitaxel in Polyethoxylated Castor Oil: A Randomized, Two-Period Crossover Study in Patients With Advanced Cancer. Clin. Ther. 2013, 35, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Molife, L.R.; Attard, G.; Fong, P.C.; Karavasilis, V.; Reid, A.H.M.; Patterson, S.; Riggs, C.E.; Higano, C.; Stadler, W.M.; McCulloch, W.; et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann. Oncol. 2010, 21, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Haigentz, M.; Kim, M.; Sarta, C.; Lin, J.; Keresztes, R.S.; Culliney, B.; Gaba, A.G.; Smith, R.V.; Shapiro, G.I.; Chirieac, L.R.; et al. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral. Oncol. 2012, 48, 1281–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luu, T.H.; Morgan, R.J.; Leong, L.; Lim, D.; McNamara, M.; Portnow, J.; Frankel, P.; Smith, D.D.; Doroshow, J.H.; Gandara, D.R.; et al. A Phase II Trial of Vorinostat (Suberoylanilide Hydroxamic Acid) in Metastatic Breast Cancer: A California Cancer Consortium Study. Clin. Cancer Res. 2008, 14, 7138–7142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriniere, J.; Rousseaux, S.; Steuerwald, U.; Soler-Lopez, M.; Curtet, S.; Vitte, A.L.; Govin, J.; Gaucher, J.; Sadoul, K.; Hart, D.J.; et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature 2009, 461, 664. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: going beyond transcriptional regulation. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.W.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 903–916. [Google Scholar] [CrossRef] [PubMed]
- McCleland, M.L.; Mesh, K.; Lorenzana, E.; Chopra, V.S.; Segal, E.; Watanabe, C.; Haley, B.; Maybe, O.; Vaylaoglu, M.; Gnad, F.; et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J. Clin. Invest. 2016, 126, 639–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockwood, W.W.; Zejnullahu, K.; Bradner, J.E.; Varmus, H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 19408–19413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyce, A.; Ganji, G.; Smitheman, K.N.; Chung, C.W.; Korenchuk, S.; Bai, Y.C.; Barbash, O.; Le, B.C.; Craggs, P.D.; McCabe, M.T.; et al. BET Inhibition Silences Expression of MYCN and BCL2 and Induces Cytotoxicity in Neuroblastoma Tumor Models. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Sancisi, V.; Manzotti, G.; Gugnoni, M.; Rossi, T.; Gandolfi, G.; Gobbi, G.; Torricelli, F.; Catellani, F.; do Valle, T.F.; Remondini, D.; et al. RUNX2 expression in thyroid and breast cancer requires the cooperation of three non-redundant enhancers under the control of BRD4 and c-JUN. Nucleic Acids Res. 2017, 45, 11249–11267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Liu, Q.; Acharya, P.; Stengel, K.R.; Sheng, Q.H.; Zhou, X.F.; Kwak, H.; Fischer, M.A.; Bradner, J.E.; Strickland, S.A.; et al. High-Resolution Mapping of RNA Polymerases Identifies Mechanisms of Sensitivity and Resistance to BET Inhibitors in t(8;21) AML. Cell Rep. 2016, 16, 2003–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterlin, B.M. Transcription elongation takes central stage: The P-TEFb connection. Cell Cycle 2010, 9, 2933–2934. [Google Scholar] [CrossRef] [PubMed]
- Lenasi, T.; Barboric, M. P-TEFb stimulates transcription elongation and pre-mRNA splicing through multilateral mechanisms. RNA Biol. 2010, 7, 145–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.Y.; He, N.H.; Zhou, Q. Brd4 recruits P-TER to chromosomes at late mitosis to promote G(1) gene expression and cell cycle progression. Mol. Cell Biol. 2008, 28, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Diribarne, G.; Bensaude, O. 7SK RNA, a non-coding RNA regulating P-TEFb, a general transcription factor. RNA Biol. 2009, 6, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.Y.; Yik, J.H.N.; Chen, R.C.; He, N.H.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Baek, G.; Ramanand, S.G.; Sharp, A.; Gao, Y.P.; Yuan, W.; Welti, J.; Rodrigues, D.N.; Dolling, D.; Figueiredo, I.; et al. BRD4 Promotes DNA Repair and Mediates the Formation of TMPRSS2-ERG Gene Rearrangements in Prostate Cancer. Cell Rep. 2018, 22, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Pike, A.M.; Lee, S.S.; Strong, M.A.; Connelly, C.J.; Greider, C.W. BRD4 inhibitors block telomere elongation. Nucleic Acids Res. 2017, 45, 8403–8410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, C.A.; Miyoshi, I.; Kubonishi, I.; Grier, H.E.; Perez-Atayde, A.R.; Fletcher, J.A. BRD4-NUT fusion oncogene: A novel mechanism in aggressive carcinoma. Cancer Res. 2003, 63, 304–307. [Google Scholar] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J. Targeting epigenetic readers in cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, E196–E204. [Google Scholar] [CrossRef]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, E186–E195. [Google Scholar] [CrossRef]
- Kanno, T.; Kanno, Y.; LeRoy, G.; Campos, E.; Sun, H.W.; Brooks, S.R.; Vahedi, G.; Heightman, T.D.; Garcia, B.A.; Reinberg, D.; et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat. Struct. Mol. Biol. 2014, 21, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.M.; Lu, X.D.; Liu, R.Z.; Ai, N.P.; Cao, Z.H.; Li, Y.N.; Liu, J.F.; Yu, B.; Liu, K.; Wang, H.P.; et al. Histone Cross-talk Connects Protein Phosphatase 1 alpha (PP1 alpha) and Histone Deacetylase (HDAC) Pathways to Regulate the Functional Transition of Bromodomain-containing 4 (BRD4) for Inducible Gene Expression. J. Biol. Chem. 2014, 289, 23154–23167. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.H.J.; Yoon, H.G.; Qin, J.; Wong, J.M. Regulation of P-TEFb elongation complex activity by CDK9 acetylation. Mol. Cell Biol. 2007, 27, 4641–4651. [Google Scholar] [CrossRef] [PubMed]
- Pinz, S.; Unser, S.; Buob, D.; Fischer, P.; Jobst, B.; Rascle, A. Deacetylase inhibitors repress STAT5-mediated transcription by interfering with bromodomain and extra-terminal (BET) protein function. Nucleic Acids Res. 2015, 43, 3524–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhadury, J.; Nilsson, L.M.; Muralidharan, S.V.; Green, L.C.; Li, Z.L.; Gesner, E.M.; Hansen, H.C.; Keller, U.B.; McLure, K.G.; Nilsson, J.A. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E2721–E2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahbazi, J.; Liu, P.Y.; Atmadibrata, B.; Bradner, J.E.; Marshall, G.M.; Lock, R.B.; Liu, T. The Bromodomain Inhibitor JQ1 and the Histone Deacetylase Inhibitor Panobinostat Synergistically Reduce N-Myc Expression and Induce Anticancer Effects. Clin. Cancer Res. 2016, 22, 2534–2544. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Sharma, S.; Qi, J.; Valenta, J.A.; Schaub, L.J.; Shah, B.; Peth, K.; Portier, B.P.; Rodriguez, M.; Devaraj, S.G.T.; et al. Highly Active Combination of BRD4 Antagonist and Histone Deacetylase Inhibitor against Human Acute Myelogenous Leukemia Cells. Mol. Cancer Ther. 2014, 13, 1142–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21. [Google Scholar] [CrossRef] [PubMed]
- Holscher, A.S.; Schulz, W.A.; Pinkerneil, M.; Niegisch, G.; Hoffmann, M.J. Combined inhibition of BET proteins and class I HDACs synergistically induces apoptosis in urothelial carcinoma cell lines. Clin. Epigenetics 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, A.; Cullinane, C.; De Paoli-Iseppi, R.; Wilmott, J.S.; Gunatilake, D.; Madore, J.; Strbenac, D.; Yang, J.Y.; Gowrishankar, K.; Tiffen, J.C.; et al. Combining BET and HDAC inhibitors synergistically induces apoptosis of melanoma and suppresses AKT and YAP signaling. Oncotarget 2015, 6, 21507–21521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.R.; Ishida, C.T.; Ishida, W.; Lo, S.F.L.; Zhao, J.F.; Shu, C.; Bianchetti, E.; Kleiner, G.; Sanchez-Quintero, M.J.; Quinzii, C.M.; et al. Combined HDAC and Bromodomain Protein Inhibition Reprograms Tumor Cell Metabolism and Elicits Synthetic Lethality in Glioblastoma. Clin. Cancer Res. 2018, 24, 3941–3954. [Google Scholar] [CrossRef] [PubMed]
- Rascle, A.; Johnston, J.A.; Amati, B. Deacetylase activity is required for recruitment of the basal transcription machinery and transactivation by STAT5. Mol. Cell Biol. 2003, 23, 4162–4173. [Google Scholar] [CrossRef] [PubMed]
- Rascle, A.; Lees, E. Chromatin acetylation and remodeling at the Cis promoter during STAT5-induced transcription. Nucleic Acids Res. 2003, 31, 6882–6890. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.H.; Walker, S.R.; Nelson, E.A.; Cerulli, R.; Xiang, M.; Toniolo, P.A.; Qi, J.; Stone, R.M.; Wadleigh, M.; Bradner, J.E.; et al. Targeting STAT5 in Hematologic Malignancies through Inhibition of the Bromodomain and Extra-Terminal (BET) Bromodomain Protein BRD2. Mol. Cancer Ther. 2014, 13, 1194–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.M.; Hou, S.H.; Chen, H.L.; Ran, T.; Jiang, F.; Bian, Y.Y.; Zhang, D.W.; Zhi, Y.L.; Wang, L.; Zhang, L.; et al. Targeting epigenetic reader and eraser: Rational design, synthesis and in vitro evaluation of dimethylisoxazoles derivatives as BRD4/HDAC dual inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 2931–2935. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.F.; He, L.H.; Zheng, L.; Huang, L.X.; Zhou, Y.Y.; Wang, T.J.; Chen, Y.; Shen, M.S.; Wang, F.; Yang, Z.; et al. Structure-based design, synthesis and in vitro antiproliferative effects studies of novel dual BRD4/HDAC inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 4051–4055. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, S.J.; Soden, P.E.; Angell, D.C.; Bantscheff, M.; Chung, C.W.; Giblin, K.A.; Smithers, N.; Furze, R.C.; Gordon, L.; Drewes, G.; et al. The structure based design of dual HDAC/BET inhibitors as novel epigenetic probes. Medchemcomm 2014, 5, 342–351. [Google Scholar] [CrossRef]
- Amemiya, S.; Yamaguchi, T.; Hashimoto, Y.; Noguchi-Yachide, T. Synthesis and evaluation of novel dual BRD4/HDAC inhibitors. Bioorgan Med. Chem. 2017, 25, 3677–3684. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Manic, G.; Obrist, F.; Vitale, I. Trial watch––inhibiting PARP enzymes for anticancer therapy. Mol. Cell Oncol. 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone Deacetylase Regulation of ATM-Mediated DNA Damage Signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groselj, B.; Sharma, N.L.; Hamdy, F.C.; Kerr, M.; Kiltie, A.E. Histone deacetylase inhibitors as radiosensitisers: Effects on DNA damage signalling and repair. Br. J. Cancer 2013, 108, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Harms, K.L.; Chen, X.B. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 2007, 67, 3145–3152. [Google Scholar] [CrossRef] [PubMed]
- Maggio, S.C.; Rosato, R.R.; Kramer, L.B.; Dai, Y.; Rahmani, M.; Paik, D.S.; Czarnik, A.C.; Payne, S.G.; Spiegel, S.; Grant, S. The histone deacetylase inhibitor MS-275 interacts synergistically with fludarabine to induce apoptosis in human leukemia cells. Cancer Res. 2004, 64, 2590–2600. [Google Scholar] [CrossRef] [PubMed]
- Hajji, N.; Wallenborg, K.; Vlachos, P.; Nyman, U.; Hermanson, O.; Joseph, B. Combinatorial action of the HDAC inhibitor trichostatin A and etoposide induces caspase-mediated AIF-dependent apoptotic cell death in non-small cell lung carcinoma cells. Oncogene 2008, 27, 3134–3144. [Google Scholar] [CrossRef] [PubMed]
- Maiso, P.; Colado, E.; Ocio, E.M.; Garayoa, M.; Martin, J.; Atadja, P.; Pandiella, A.; San-Miguel, J.F. The synergy of panobinostat plus doxorubicin in acute myeloid leukemia suggests a role for HDAC inhibitors in the control of DNA repair. Leukemia 2009, 23, 2265–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, G.; Liu, J.; Ren, W.; Wei, W.; Wang, S.; Lahat, G.; Zhu, Q.S.; Bornmann, W.G.; McConkey, D.J.; Pollock, R.E.; et al. Combining PCI-24781, a Novel Histone Deacetylase Inhibitor, with Chemotherapy for the Treatment of Soft Tissue Sarcoma. Clin. Cancer Res. 2009, 15, 3472–3483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.; Fang, W.Y.; Chang, L.; Gao, W.H.; Shen, Y.; Jia, M.Y.; Zhang, Y.X.; Wang, Y.; Dou, H.B.; Zhang, W.J.; et al. Targeting HDAC3, a new partner protein of AKT in the reversal of chemoresistance in acute myeloid leukemia via DNA damage response. Leukemia 2017, 31, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Chao, O.S.; Goodman, O.B. Synergistic Loss of Prostate Cancer Cell Viability by Coinhibition of HDAC and PARP. Mol. Cancer Res. 2014, 12, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, A.; Im, S.A.; Kim, D.K.; Song, S.H.; Kim, H.J.; Lee, K.H.; Kim, T.Y.; Han, S.W.; Oh, D.Y.; Kim, T.Y.; et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2015, 17. [Google Scholar] [CrossRef] [PubMed]
- Shabason, J.E.; Tofilon, P.J.; Camphausen, K. Grand rounds at the National Institutes of Health: HDAC inhibitors as radiation modifiers, from bench to clinic. J. Cell Mol. Med. 2011, 15, 2735–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, L.; Cuneo, K.C.; Fu, A.; Tu, T.X.; Atadja, P.W.; Hallahan, D.E. Histone deacetylase (HDAC) inhibitor LBH589 increases duration of gamma-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Res. 2006, 66, 11298–11304. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.F.; Wong, P.; Radany, E.; Wong, J.Y.C. HDAC Inhibitor, Valproic Acid, Induces p53-Dependent Radiosensitization of Colon Cancer Cells. Cancer Biother. Radio. 2009, 24, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munshi, A.; Kurland, J.F.; Nishikawa, T.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Ismail, S.; Stevens, C.; Meyn, R.E. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin Cancer Res. 2005, 11, 4912–4922. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Parise, R.A.; Ramananthan, R.K.; Lagattuta, T.F.; Musguire, L.A.; Stoller, R.G.; Potter, D.M.; Argiris, A.E.; Zwiebel, J.A.; Egorin, M.J.; et al. Phase I and pharmacokinetic study of vorinostat, A histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin Cancer Res. 2007, 13, 3605–3610. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; et al. Carboplatin and Paclitaxel in Combination With Either Vorinostat or Placebo for First-Line Therapy of Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.; Marchion, D.; Bicaku, E.; Schmitt, M.; Lee, J.H.; DeConti, R.; Simon, G.; Fishman, M.; Minton, S.; Garrett, C.; et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: A clinical and translational study. J. Clin. Oncol. 2007, 25, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Marchion, D.; Thomas, S.; Egorin, M.; Minton, S.; Springett, G.; Lee, J.H.; Simon, G.; Chiappori, A.; Sullivan, D.; et al. Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br. J. Cancer 2009, 101, 1044–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ree, A.H.; Dueland, S.; Folkvord, S.; Hole, K.H.; Seierstad, T.; Johansen, M.; Abrahamsen, T.W.; Flatmark, K. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol. 2010, 11, 459–464. [Google Scholar] [CrossRef]
- Shi, W.Y.; Palmer, J.D.; Werner-Wasik, M.; Andrews, D.W.; Evans, J.J.; Glass, J.; Kim, L.; Bar-Ad, V.; Judy, K.; Farrell, C.; et al. Phase I trial of panobinostat and fractionated stereotactic re-irradiation therapy for recurrent high grade gliomas. J. Neuro-Oncol. 2016, 127, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Avallone, A.; Piccirillo, M.C.; Delrio, P.; Pecori, B.; Di Gennaro, E.; Aloj, L.; Tatangelo, F.; D’Angelo, V.; Granata, C.; Cavalcanti, E.; et al. Phase 1/2 study of valproic acid and short-course radiotherapy plus capecitabine as preoperative treatment in low-moderate risk rectal cancer-V-shoRT-R3 (Valproic acid - short RadioTherapy––rectum 3rd trial). BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Arlinghaus, L.R.; Cardin, D.B.; Goff, L.; Berlin, J.D.; Parikh, A.; Abramson, R.G.; Yankeelov, T.E.; Hiebert, S.; Merchant, N.; et al. Phase I trial of vorinostat added to chemoradiation with capecitabine in pancreatic cancer. Radiother. Oncol. 2016, 119, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millward, M.; Price, T.; Townsend, A.; Sweeney, C.; Spencer, A.; Sukumaran, S.; Longenecker, A.; Lee, L.; Lay, A.; Sharma, G.; et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest. New Drug 2012, 30, 2303–2317. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Ogura, M.; Tobinai, K.; Ando, K.; Suzuki, T.; Watanabe, T.; Ohmachi, K.; Uchida, T.; Hanson, M.E.; Tanaka, Y.; et al. A phase I study of vorinostat combined with bortezomib in Japanese patients with relapsed or refractory multiple myeloma. Int. J. Hematol. 2016, 103, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Friday, B.B.; Anderson, S.K.; Buckner, J.; Yu, C.R.; Giannini, C.; Geoffroy, F.; Schwerkoske, J.; Mazurczak, M.; Gross, H.; Pajon, E.; et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: A north central cancer treatment group study. Neuro-Oncology 2012, 14, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Zibelman, M.; Wong, Y.N.; Devarajan, K.; Malizzia, L.; Corrigan, A.; Olszanski, A.J.; Denlinger, C.S.; Roethke, S.K.; Tetzlaff, C.H.; Plimack, E.R. Phase I study of the mTOR inhibitor ridaforolimus and the HDAC inhibitor vorinostat in advanced renal cell carcinoma and other solid tumors. Invest. New Drug 2015, 33, 1040–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Garrido-Laguna, I.; Naing, A.; Fu, S.Q.; Falchook, G.S.; Piha-Paul, S.A.; Wheler, J.J.; Hong, D.S.; Tsimberidou, A.M.; Subbiah, V.; et al. Phase I dose-escalation study of the mTOR inhibitor sirolimus and the HDAC inhibitor vorinostat in patients with advanced malignancy. Oncotarget 2016, 7, 67521–67531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, J.E.; Haura, E.; Chiappori, A.; Tanvetyanon, T.; Williams, C.C.; Pinder-Schenck, M.; Kish, J.A.; Kreahling, J.; Lush, R.; Neuger, A.; et al. A Phase I, Pharmacokinetic, and Pharmacodynamic Study of Panobinostat, an HDAC Inhibitor, Combined with Erlotinib in Patients with Advanced Aerodigestive Tract Tumors. Clin. Cancer Res. 2014, 20, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.C.; Alumkal, J.J.; Stein, M.N.; Taplin, M.E.; Babb, J.; Barnett, E.S.; Gomez-Pinillos, A.; Liu, X.M.; Moore, D.; DiPaola, R.; et al. Epigenetic Therapy with Panobinostat Combined with Bicalutamide Rechallenge in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Gerber, D.E.; Boothman, D.A.; Fattah, F.J.; Dong, Y.; Zhu, H.; Skelton, R.A.; Priddy, L.L.; Vo, P.; Dowell, J.E.; Sarode, V.; et al. Phase 1 study of romidepsin plus erlotinib in advanced non-small cell lung cancer. Lung Cancer 2015, 90, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngamphaiboon, N.; Dy, G.K.; Ma, W.W.; Zhao, Y.J.; Reungwetwattana, T.; DePaolo, D.; Ding, Y.; Brady, W.; Fetterly, G.; Adjei, A.A. A phase I study of the histone deacetylase (HDAC) inhibitor entinostat, in combination with sorafenib in patients with advanced solid tumors. Invest. New Drug 2015, 33, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Pili, R.; Quinn, D.I.; Hammers, H.J.; Monk, P.; George, S.; Dorff, T.B.; Olencki, T.; Shen, L.; Orillion, A.; Lamonica, D.; et al. Immunomodulation by Entinostat in Renal Cell Carcinoma Patients Receiving High-Dose Interleukin 2: A Multicenter, Single-Arm, Phase I/II Trial (NCI-CTEP#7870). Clin. Cancer Res. 2017, 23, 7199–7208. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [PubMed]
- Rauch, N.; Rukhlenko, O.S.; Kolch, W.; Kholodenko, B.N. MAPK kinase signalling dynamics regulate cell fate decisions and drug resistance. Curr. Opin. Struct. Biol. 2016, 41, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaBonte, M.J.; Wilson, P.M.; Fazzone, W.; Russell, J.; Louie, S.G.; El-Khoueiry, A.; Lenz, H.J.; Ladner, R.D. The Dual EGFR/HER2 Inhibitor Lapatinib Synergistically Enhances the Antitumor Activity of the Histone Deacetylase Inhibitor Panobinostat in Colorectal Cancer Models. Cancer Res. 2011, 71, 3635–3648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.E.; Kim, D.E.; Kim, M.J.; Lee, J.S.; Rho, J.K.; Jeong, S.Y.; Choi, E.K.; Kim, C.S.; Hwang, J.J. Vorinostat enhances gefitinib-induced cell death through reactive oxygen species-dependent cleavage of HSP90 and its clients in non-small cell lung cancer with the EGFR mutation. Oncol. Rep. 2019, 41, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.; Bruzzese, F.; Leone, A.; Gagliardi, A.; Puglia, M.; Di Gennaro, E.; Rocco, M.; Gimigliano, A.; Pucci, B.; Armini, A.; et al. Proteomic analysis identifies differentially expressed proteins after HDAC vorinostat and EGFR inhibitor gefitinib treatments in Hep-2 cancer cells. Proteomics 2011, 11, 3725–3742. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Colburn, N.H. Histone deacetylase inhibition down-regulates cyclin D1 transcription by inhibiting nuclear factor-kappa B/p65 DNA binding. Mol. Cancer Res. 2005, 3, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Chen, C.H.; Wang, J.C.; Tsai, A.C.; Liou, J.P.; Pan, S.L.; Teng, C.M. The HDAC inhibitor, MPT0E028, enhances erlotinib-induced cell death in EGFR-TKI-resistant NSCLC cells. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, S.L.; Cleris, L.; Stirparo, G.G.; Tartari, S.; Saba, E.; Pierdominici, M.; Malorni, W.; Carbone, A.; Anichini, A.; Carlo-Stella, C. BIM upregulation and ROS-dependent necroptosis mediate the antitumor effects of the HDACi Givinostat and Sorafenib in Hodgkin lymphoma cell line xenografts. Leukemia 2014, 28, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Horinaka, M.; Shinnoh, M.; Yoshioka, T.; Miki, T.; Sakai, T. A novel HDAC inhibitor OBP-801 and a PI3K inhibitor LY294002 synergistically induce apoptosis via the suppression of survivin and XIAP in renal cell carcinoma. Int. J. Oncol. 2013, 43, 1080–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wedel, S.; Hudak, L.; Seibel, J.M.; Juengel, E.; Tsaur, I.; Wiesner, C.; Haferkamp, A.; Blaheta, R.A. Inhibitory effects of the HDAC inhibitor valproic acid on prostate cancer growth are enhanced by simultaneous application of the mTOR inhibitor RAD001. Life Sci. 2011, 88, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Medina, E.C.; Esquivel, J.A.; Espitia, C.M.; Smith, S.; Oberheu, K.; Swords, R.; Kelly, K.R.; Mita, M.M.; Mita, A.C.; et al. Vorinostat Enhances the Activity of Temsirolimus in Renal Cell Carcinoma Through Suppression of Survivin Levels. Clin. Cancer Res. 2010, 16, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.W.; Salumbides, B.; Van Erp, K.; Hammers, H.; Qian, D.Z.; Sanni, T.; Atadja, P.; Pili, R. Combination strategy targeting the hypoxia inducible factor-1 alpha with mammalian target of rapamycin and histone deacetylase inhibitors. Clin. Cancer Res. 2008, 14, 3589–3597. [Google Scholar] [CrossRef] [PubMed]
- Malone, C.F.; Emerson, C.; Ingraham, R.; Barbosa, W.; Guerra, S.; Yoon, H.; Liu, L.L.; Michor, F.; Haigis, M.; Macleod, K.F.; et al. mTOR and HDAC Inhibitors Converge on the TXNIP/Thioredoxin Pathway to Cause Catastrophic Oxidative Stress and Regression of RAS-Driven Tumors. Cancer Discov. 2017, 7, 1450–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, J.K.; Michalowski, A.M.; Gamache, B.J.; DuBois, W.; Patel, J.; Zhang, K.; Gary, J.; Zhang, S.L.; Gaikwad, S.; Connors, D.; et al. Cooperative Targets of Combined mTOR/HDAC Inhibition Promote MYC Degradation. Mol. Cancer Ther. 2017, 16, 2008–2021. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, R.; Catley, L.P.; Hideshima, T.; Lentzsch, S.; Mitsiades, C.S.; Mitsiades, N.; Neuberg, D.; Goloubeva, O.; Pien, C.S.; Adams, J.; et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002, 62, 4996–5000. [Google Scholar] [PubMed]
- Pei, X.Y.; Dai, Y.; Grant, S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, D.R.; Jane, E.P.; Agostino, N.R.; DiDomenico, J.D.; Pollack, I.F. Bortezomib-induced sensitization of malignant human glioma cells to vorinostat-induced apoptosis depends on reactive oxygen species production, mitochondrial dysfunction, Noxa upregulation, Mcl-1 cleavage, and DNA damage. Mol. Carcinog. 2013, 52, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Chiang, A.K.S. Combination of proteasome and class I HDAC inhibitors induces apoptosis of NPC cells through an HDAC6-independent ER stress-induced mechanism. Int. J. Cancer 2014, 135, 2950–2961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C.; et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Carew, J.S.; Pino, M.S.; Highshaw, R.A.; Andtbacka, R.H.I.; Dunner, K.; Pal, A.; Bornmann, W.G.; Chiao, P.J.; Huang, P.; et al. Aggresome disruption: A novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006, 66, 3773–3781. [Google Scholar] [CrossRef] [PubMed]
- Buglio, D.; Mamidipudi, V.; Khaskhely, N.M.; Brady, H.; Heise, C.; Besterman, J.; Martell, R.E.; MacBeth, K.; Younes, A. The class-I HDAC inhibitor MGCD0103 induces apoptosis in Hodgkin lymphoma cell lines and synergizes with proteasome inhibitors by an HDAC6-independent mechanism. Br. J. Haematol. 2010, 151, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, J.L.; Holler, N.; Reynard, S.; Vinciguerra, P.; Schneider, P.; Juo, P.; Blenis, J.; Tschopp, J. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat. Cell Biol. 2000, 2, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Okamoto, K.; Yonehara, S. Sensitization of osteosarcoma cells to death receptor-mediated apoptosis by HDAC inhibitors through downregulation of cellular FLIP. Cell Death Differ. 2005, 12, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway (vol 11, pg 71, 2005). Nat. Med. 2005, 11, 71. [Google Scholar] [CrossRef]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Alvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 2005, 11, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; MacFarlane, M.; Harper, N.; Wheat, L.M.C.; Dyer, M.J.S.; Cohen, G.M. Histone deacetylase inhibitors potentiate TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in lymphoid malignancies. Cell Death Differ. 2004, 11, S193–S206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulm, M.; Ramesh, A.V.; McNamara, K.M.; Ponnusamy, S.; Sasano, H.; Narayanan, R. Therapeutic advances in hormone-dependent cancers: focus on prostate, breast and ovarian cancers. Endocr. Connect 2019, 8, R10–R26. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.W.; Phillips, D.L.; Ferguson, A.T.; Nelson, W.G.; Herman, J.G.; Davidson, N.E. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001, 61, 7025–7029. [Google Scholar] [PubMed]
- Restall, C.; Doherty, J.; Bin Liu, H.; Genovese, R.; Paiman, L.; Byron, K.A.; Anderson, R.L.; Dear, A.E. A novel histone deacetylase inhibitor augments tamoxifen-mediated attenuation of breast carcinoma growth. Int. J. Cancer 2009, 125, 483–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabnis, G.J.; Goloubeva, O.; Chumsri, S.; Nguyen, N.; Sukumar, S.; Brodie, A.M.H. Functional Activation of the Estrogen Receptor-alpha and Aromatase by the HDAC Inhibitor Entinostat Sensitizes ER-Negative Tumors to Letrozole. Cancer Res. 2011, 71, 1893–1903. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, G.J.; Goloubeva, O.G.; Kazi, A.A.; Shah, P.; Brodie, A.H. HDAC Inhibitor Entinostat Restores Responsiveness of Letrozole-Resistant MCF-7Ca Xenografts to Aromatase Inhibitors through Modulation of Her-2. Mol. Cancer Ther. 2013, 12, 2804–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrocco, D.L.; Tilley, W.D.; Bianco-Miotto, T.; Evdokiou, A.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; Butler, L.M. Suberoylanilide hydroxamic acid (vorinostat) represses androgen receptor expression and acts synergistically with an androgen receptor antagonist to inhibit prostate cancer cell proliferation. Mol. Cancer Ther. 2007, 6, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkman, M.; Iljin, K.; Halonen, P.; Sara, H.; Kaivanto, E.; Nees, M.; Kallioniemi, O.P. Defining the molecular action of HDAC inhibitors and synergism with androgen deprivation in ERG-positive prostate cancer. Int. J. Cancer 2008, 123, 2774–2781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Liu, Y.; Cheng, Y.; Wei, Y.; Wei, X. Immune checkpoint blockade and its combination therapy with small-molecule inhibitors for cancer treatment. Biochim. Biophys. Acta Rev. Cancer 2018, 1871, 199–224. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Orillion, A.; Pili, R. Histone deacetylase inhibitors as immunomodulators in cancer therapeutics. Epigenomics-Uk 2016, 8, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, A.J.; West, A.; Banks, K.M.; Haynes, N.M.; Teng, M.W.; Smyth, M.J.; Johnstone, R.W. Eradication of solid tumors using histone deacetylase inhibitors combined with immune-stimulating antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 4141–4146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, K.; Wang, G.; Li, W.; Zhang, L.; Wang, R.; Huang, Y.; Du, L.; Jiang, J.; Wu, C.; He, X.; et al. Histone deacetylase inhibitors prevent activation-induced cell death and promote anti-tumor immunity. Oncogene 2015, 34, 5960–5970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, D.M.; Sodre, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Kirkwood, J.; Dent, P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget 2017, 8, 83155–83170. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Skora, A.D.; Li, Z.B.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.A.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [PubMed]
- Klingbeil, O.; Lesche, R.; Gelato, K.A.; Haendler, B.; Lejeune, P. Inhibition of BET bromodomain-dependent XIAP and FLIP expression sensitizes KRAS-mutated NSCLC to pro-apoptotic agents. Cell Death Dis. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teicher, B.A.; Silvers, T.; Selby, M.; Delosh, R.; Laudeman, J.; Ogle, C.; Reinhart, R.; Parchment, R.; Krushkal, J.; Sonkin, D.; et al. Small cell lung carcinoma cell line screen of etoposide/carboplatin plus a third agent. Cancer Med.-US 2017, 6, 1952–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanellato, I.; Colangelo, D.; Osella, D. JQ1, a BET Inhibitor, Synergizes with Cisplatin and Induces Apoptosis in Highly Chemoresistant Malignant Pleural Mesothelioma Cells. Curr. Cancer Drug Tar. 2018, 18, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Zhu, H.R.; Lee, J.H.; Kossenkov, A.V.; Wu, S.Y.; Wickramasinghe, J.M.; Yin, X.F.; Palozola, K.C.; Gardini, A.; Showe, L.C.; et al. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res. 2016, 76, 6320–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zhang, Y.Y.; Shan, W.W.; Hu, Z.Y.; Yuan, J.; Pi, J.J.; Wang, Y.Y.; Fan, L.L.; Tang, Z.Q.; Li, C.S.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.H.; Vellano, C.P.; Ju, Z.L.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401. [Google Scholar] [CrossRef] [PubMed]
- Mio, C.; Gerratana, L.; Bolis, M.; Caponnetto, F.; Zanello, A.; Barbina, M.; Di Loreto, C.; Garattini, E.; Damante, G.; Puglisi, F. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer 2019, 144, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.; Brand, T.M.; O’Keefe, R.A.; Lee, E.D.; Zeng, Y.; Kemmer, J.D.; Li, H.; Grandis, J.R.; Bhola, N.E. BET Inhibition Overcomes Receptor Tyrosine Kinase-Mediated Cetuximab Resistance in HNSCC. Cancer Res. 2018, 78, 4331–4343. [Google Scholar] [CrossRef] [PubMed]
- Stuhlmiller, T.J.; Miller, S.M.; Zawistowski, J.S.; Nakamura, K.; Beltran, A.S.; Duncan, J.S.; Angus, S.P.; Collins, K.A.L.; Granger, D.A.; Reuther, R.A.; et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Rep. 2015, 11, 390–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.W.; Li, S.; Hai, J.; Wang, X.; Chen, T.; Quinn, M.M.; Gao, P.; Zhang, Y.X.; Ji, H.B.; Cross, D.A.E.; et al. Targeting HER2 Aberrations in Non-Small Cell Lung Cancer with Osimertinib. Clin. Cancer Res. 2018, 24, 2594–2604. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.X.; Buczkowski, K.A.; Zhang, Y.X.; Asahina, H.; Beauchamp, E.M.; Terai, H.; Li, Y.Y.; Meyerson, M.; Wong, K.K.; Hammerman, P.S. NSCLC Driven by DDR2 Mutation Is Sensitive to Dasatinib and JQ1 Combination Therapy. Mol. Cancer Ther. 2015, 14, 2382–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, K.R.; Crawford, L.; Tsui, E.; Manchester, H.E.; Maertens, O.; Liu, X.; Liberti, M.V.; Magpusao, A.N.; Stein, E.M.; Tingley, J.P.; et al. Melanoma Therapeutic Strategies that Select against Resistance by Exploiting MYC-Driven Evolutionary Convergence. Cell Rep. 2017, 21, 2796–2812. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Hattori, N.; Iida, N.; Yamashita, S.; Mori, A.; Kimura, K.; Yoshino, T.; Ushijima, T. Targeting of super-enhancers and mutant BRAF can suppress growth of BRAF-mutant colon cancer cells via repression of MAPK signaling pathway. Cancer Lett. 2017, 402, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Paoluzzi, L.; Hanniford, D.; Sokolova, E.; Osman, I.; Darvishian, F.; Wang, J.; Bradner, J.E.; Hernando, E. BET and BRAF inhibitors act synergistically against BRAF-mutant melanoma. Cancer Med. 2016, 5, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.F.; Wang, L.H.; Neitzel, L.R.; Loganathan, S.N.; Tang, N.; Qin, L.L.; Crispi, E.E.; Guo, Y.; Knapp, S.; Beauchamp, R.D.; et al. The MAPK Pathway Regulates Intrinsic Resistance to BET Inhibitors in Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Zhang, Z.F.; Ma, P.F.; An, S.M.; Shen, Y.; Zhu, L.; Zhuang, G.L. Concomitant BET and MAPK blockade for effective treatment of ovarian cancer. Oncotarget 2016, 7, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Wyce, A.; Matteo, J.J.; Foley, S.W.; Felitsky, D.J.; Rajapurkar, S.R.; Zhang, X.P.; Musso, M.C.; Korenchuk, S.; Karpinich, N.O.; Keenan, K.M.; et al. MEK inhibitors overcome resistance to BET inhibition across a number of solid and hematologic cancers. Oncogenesis 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Echevarria-Vargas, I.M.; Reyes-Uribe, P.I.; Guterres, A.N.; Yin, X.F.; Kossenkov, A.V.; Liu, Q.; Zhang, G.; Krepler, C.; Cheng, C.; Wei, Z.; et al. Co-targeting BET and MEK as salvage therapy for MAPK and checkpoint inhibitor-resistant melanoma. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Laddha, S.V.; Tang, L.; Vosburgh, E.; Levine, A.J.; Normant, E.; Sandy, P.; Harris, C.R.; Chan, C.S.; Xu, E.Y. The bromodomain and extra-terminal inhibitor CPI203 enhances the antiproliferative effects of rapamycin on human neuroendocrine tumors. Cell Death Dis. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Qi, J.; Bradner, J.E.; Said, J.W.; Doan, N.B.; Forscher, C.; Yang, H.; Koeffler, H.P. Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. Int. J. Cancer 2015, 136, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudio, E.; Tarantelli, C.; Ponzoni, M.; Odore, E.; Rezai, K.; Bernasconi, E.; Cascione, L.; Rinaldi, A.; Stathis, A.; Riveiro, E.; et al. Bromodomain inhibitor OTX015 (MK-8628) combined with targeted agents shows strong in vivo antitumor activity in lymphoma. Oncotarget 2016, 7, 58142–58147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, R.; Riveiro, M.E.; Astorgues-Xerri, L.; Odore, E.; Rezai, K.; Erba, E.; Panini, N.; Rinaldi, A.; Kwee, I.; Beltrame, L.; et al. The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget 2017, 8, 7598–7613. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.; Berger, D.; Zielinski, C.C.; Valent, P.; Grunt, T.W. Hitting two oncogenic machineries in cancer cells: cooperative effects of the multi-kinase inhibitor ponatinib and the BET bromodomain blockers JQ1 or dBET1 on human carcinoma cells. Oncotarget 2018, 9, 26491–26506. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.J.; Malik, R.; Cieslik, M.; Yang, R.D.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Zhang, Z.; Shea, M.J.; Creighton, C.J.; Coarfa, C.; Hilsenbeck, S.G.; Lanz, R.; He, B.; Wang, L.; Fu, X.Y.; et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014, 24, 809–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, S.; Biarnes, M.C.; Clarke, R.; Jordan, V.C. Inhibition of BET proteins impairs estrogen-mediated growth and transcription in breast cancers by pausing RNA polymerase advancement. Breast Cancer Res. 2015, 150, 265–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, S.R.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.K.; Taylor, S.; Gupte, A.; Sharp, P.P.; Walia, M.; Walsh, N.C.; Zannettino, A.C.W.; Chalk, A.M.; Burns, C.J.; Walkley, C.R. BET inhibitors induce apoptosis through a MYC independent mechanism and synergise with CDK inhibitors to kill osteosarcoma cells. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Moreno, N.; Holsten, T.; Mertins, J.; Zhogbi, A.; Johann, P.; Kool, M.; Meisterernst, M.; Kerl, K. Combined BRD4 and CDK9 inhibition as a new therapeutic approach in malignant rhabdoid tumors. Oncotarget 2017, 8, 84986–84995. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.; Gollavilli, P.N.; Wang, S.M.; Asangani, I.A. Resistance to BET Inhibitor Leads to Alternative Therapeutic Vulnerabilities in Castration-Resistant Prostate Cancer. Cell Rep. 2018, 22, 2236–2245. [Google Scholar] [CrossRef] [PubMed]
- Bolin, S.; Borgenvik, A.; Persson, C.U.; Sundstrom, A.; Qi, J.; Bradner, J.E.; Weiss, W.A.; Cho, Y.J.; Weishaupt, H.; Swartling, F.J. Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene 2018, 37, 2850–2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durbin, A.D.; Zimmerman, M.W.; Dharia, N.V.; Abraham, B.J.; Iniguez, A.B.; Weichert-Leahey, N.; He, S.N.; Krill-Burger, J.M.; Root, D.E.; Vazquez, F.; et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet. 2018, 50, 1240. [Google Scholar] [CrossRef] [PubMed]
- Tomska, K.; Kurilov, R.; Lee, K.S.; Hullein, J.; Lukas, M.; Sellner, L.; Walther, T.; Wagner, L.; Oles, M.; Brors, B.; et al. Drug-based perturbation screen uncovers synergistic drug combinations in Burkitt lymphoma. Sci. Rep. 2018, 8, 12046. [Google Scholar] [CrossRef] [PubMed]
- Knight, T.; Luedtke, D.; Edwards, H.; Taub, J.W.; Ge, Y. A delicate balance––The BCL-2 family and its role in apoptosis, oncogenesis, and cancer therapeutics. Biochem. Pharmacol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Peirs, S.; Frismantas, V.; Matthijssens, F.; Van Loocke, W.; Pieters, T.; Vandamme, N.; Lintermans, B.; Dobay, M.P.; Berx, G.; Poppe, B.; et al. Targeting BET proteins improves the therapeutic efficacy of BCL-2 inhibition in T-cell acute lymphoblastic leukemia. Leukemia 2017, 31, 2037–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, M.H.; Lin, X.Y.; Albert, D.H.; Li, L.M.; Lam, L.T.; Faivre, E.J.; Warder, S.E.; Huang, X.L.; Wilcox, D.; Donawho, C.K.; et al. Preclinical Characterization of BET Family Bromodomain Inhibitor ABBV-075 Suggests Combination Therapeutic Strategies. Cancer Res. 2017, 77, 2976–2989. [Google Scholar] [CrossRef] [PubMed]
- Ishida, C.T.; Bianchetti, E.; Shu, C.; Halatsch, M.E.; Westhoff, M.A.; Karpel-Massler, G.; Siegelin, M.D. BH3-mimetics and BET-inhibitors elicit enhanced lethality in malignant glioma. Oncotarget 2017, 8, 29558–29573. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.T.; Lin, X.Y.; Faivre, E.J.; Yang, Z.P.; Huang, X.L.; Wilcox, D.M.; Bellin, R.J.; Jin, S.; Tahir, S.K.; Mitten, M.; et al. Vulnerability of Small-Cell Lung Cancer to Apoptosis Induced by the Combination of BET Bromodomain Proteins and BCL2 Inhibitors. Mol. Cancer Ther. 2017, 16, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Adeegbe, D.O.; Liu, S.W.; Hattersley, M.M.; Bowden, M.; Zhou, C.S.W.; Li, S.; Vlahos, R.; Grondine, M.; Dolgalev, I.; Ivanova, E.V.; et al. BET Bromodomain Inhibition Cooperates with PD-1 Blockade to Facilitate Antitumor Response in Kras-Mutant Non-Small Cell Lung Cancer. Cancer Immunol. Res. 2018, 6, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.L.; Stiff, A.; Duggan, M.; Wesolowski, R.; Carson, W.E.; Friedman, A. Modeling combination therapy for breast cancer with BET and immune checkpoint inhibitors. Proc. Natl. Acad. Sci. USA 2018, 115, 5534–5539. [Google Scholar] [CrossRef] [PubMed]



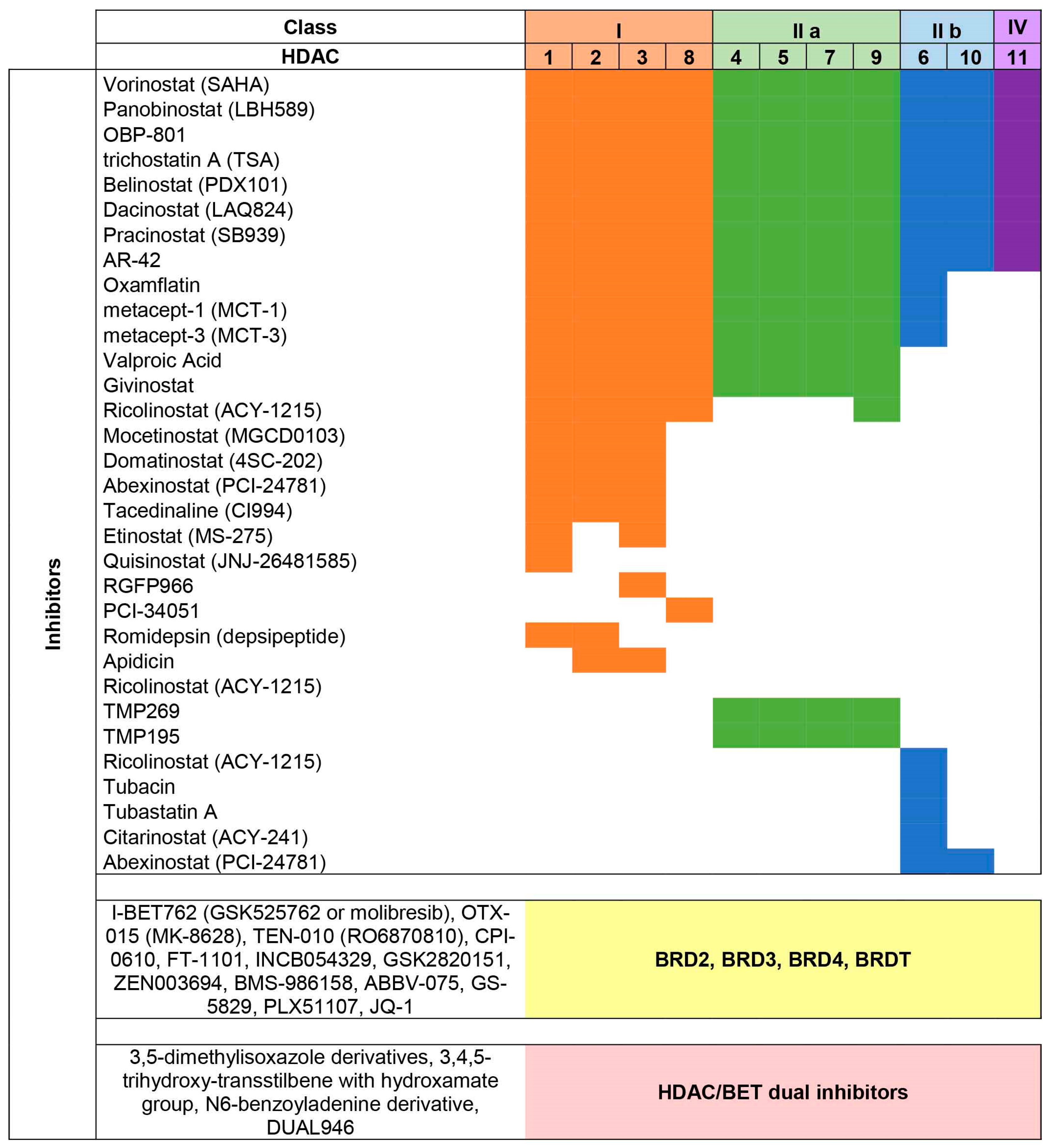{kind=link}
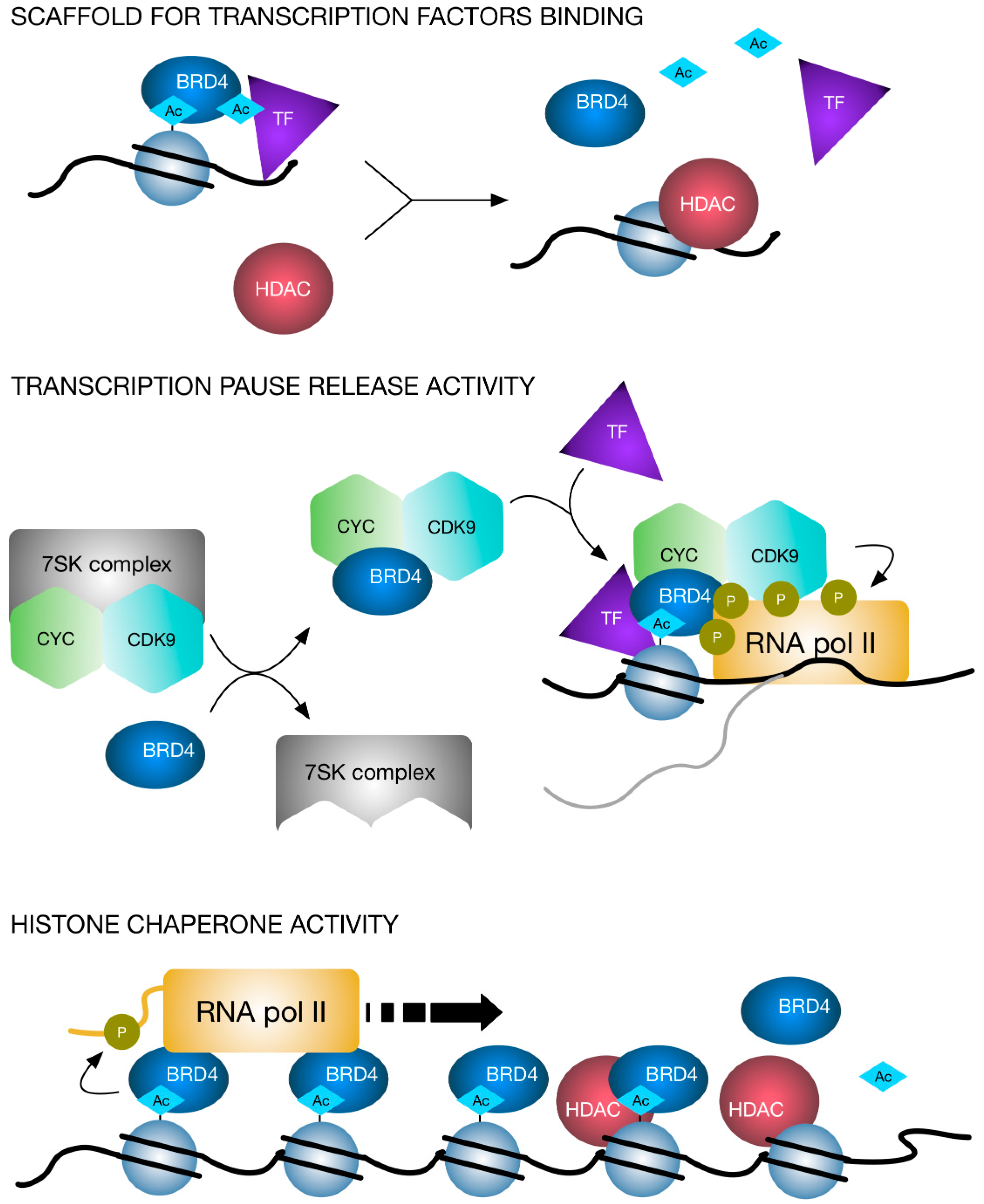{kind=link}
| HDAC Inhibitor | Class of Combination Drug | Intervention | Condition | Phase | NCT Number | Reference |
|---|---|---|---|---|---|---|
| Vorinostat | Chemotherapy | Vorinostat, paclitaxel, carboplatin | NSCLC | II | NCT00481078 | [106] |
| Chemotherapy | Vorinostat, paclitaxel, carboplatin | Adult Solid Tumor | I | NCT00287937 | [105] | |
| Chemotherapy | Vorinostat, doxorubicin | Adult Solid Tumor | I | NCT00331955 | [108] | |
| Radiotherapy | Vorinostat, radiotherapy | Pelvic Cancer | I | NCT00455351 | [109] | |
| Chemotherapy, radiotherapy | Vorinostat, capecitabine, radiotherapy | Pancreatic Cancer, Periampullary Adenocarcinoma | I | NCT00983268 | [112] | |
| Proteosome inhibitor | Vorinostat, marizomib | NSCLC, Pancreatic Cancer, Melanoma, Lymphoma, MM | I | NCT00667082 | [113] | |
| Proteosome inhibitor | Vorinostat, bortezomib | MM | I | NCT00858234 | [114] | |
| Proteosome inhibitor | Vorinostat, bortezomib | Glioblastoma, Gliosarcoma, Recurrent Adult Brain Tumor | II | NCT00641706 | [115] | |
| RTKi | Vorinostat, ridaforolimus | Lymphoma, Unspecified Adult Solid Tumor | I | NCT01169532 | [116] | |
| RTKi | Vorinostat, sirolimus, everolimus, temsirolimus | Advanced Cancer | I | NCT01087554 | [117] | |
| Hormone therapy | Vorinostat, tamoxifen | Breast Cancer | II | NCT00365599 | [118] | |
| Valproic Acid | Chemotherapy | Valproic acid, epirubicin,5-fluorouracil, cyclophosphamide | Advanced neoplasms | I | NCT00246103 | [107] |
| Chemotherapy, radiotherapy | Valproic Acid, temozolomide, radiation therapy, adjuvant therapy | High Grade Gliomas, Brain Tumors | II | NCT00302159 | [101] | |
| Chemotherapy, radiotherapy | Valproic acid, capecitabine, radiotherapy | Colorectal Cancer | I/II | NCT01898104 | [111] | |
| Panobinostat | Radiotherapy | Panobinostat, radiotherapy | Recurrent Glioma, High-grade Meningioma, Brain Metastasis | I | NCT01324635 | [110] |
| RTKi | Panobinostat, erlotinib | Lung Cancer, Head and Neck Cancer | I | NCT00738751 | [119] | |
| Hormone therapy | Panobinostat, bicalutamide | Prostate Cancer | I/II | NCT00878436 | [120] | |
| Romidepsin | RTKi | Erlotinib, romidepsin | Lung Cancer, Metastatic Cancer | I/II | NCT01302808 | [121] |
| Etinostat | RTKi | Etinostat, sorafenib | Advanced or Metastatic Solid Tumors, refractory or relapsed AML | I | NCT01159301 | [122] |
| Immunotherapy | Etinostat, aldesleukin (IL-2), radiotherapy | Clear Cell Renal Cell Carcinoma | I/II | NCT01038778 | [123] | |
| Ricolinostat | Proteosome inhibitor | Ricolinostat, bortezomib, dexamethasone | MM | I/II | NCT01323751 | [124] |
| BET Inhibitor | Class of Combination Drug | Intervention | Condition | Phase | NCT number | Reference |
|---|---|---|---|---|---|---|
| I-BET762 (GSK525762 or molibresib) | Monotherapy | GSK525762 | Relapsed refractory hematological malignancies | I/II | NCT01943851 | |
| Monotherapy | GSK525762 | NUT Midline Carcinoma and other Solid Cancers | I/II | NCT01587703 | ||
| Monotherapy | Molibresib | Compassionate use in NUT Midline Carcinoma | NCT03702036 | |||
| Hormone therapy | GSK525762; fulvestrant | HR+/HER2− advanced or metastatic breast cancer | I/II | NCT02964507 | ||
| Hormone therapy | GSK525762; abiraterone; enzalutamide; prednisone | Castrate-resistant prostate cancer | I | NCT03150056 | ||
| MEK inhibitors | GSK525762; trametinib | SCLC and RAS-mutated solid tumors | I/II | NCT03266159 | ||
| OTX-015 (MK-8628) | Monotherapy | OTX-015 | Hematological malignancies | I | NCT01713582 | [67,68] |
| Monotherapy | MK-8628 | Glioblastoma multiforme | II | NCT02296476 | ||
| Monotherapy | MK-8628 | Advanced solid tumors | I | NCT02698176 | ||
| Monotherapy | MK-8628 | Advanced solid tumors | I | NCT02259114 | ||
| Monotherapy | MK-8628 | Hematological malignancies | I | NCT02698189 | ||
| Chemotherapy | OTX-015; azacitidine | AML | I/II | NCT02303782 | ||
| TEN-010 (RO6870810) | Monotherapy | RO6870810 | Advanced solid tumors | I | NCT01987362 | |
| Monotherapy | RO6870810 | AML and myelodysplastic syndrome (MDS) | I | NCT02308761 | ||
| Immune checkpoint inhibitors | RO6870810; atezolizumab | TNBC and/or ovarian cancer | I | NCT03292172 | ||
| Anti CD38 | RO6870810; daratumumab | Advanced MM | I | NCT03068351 | ||
| BCL2 inhibitor | RO6870810; venetoclax; rituximab | Diffuse large B-cell lymphoma and/or high-grade B-cell lymphoma | I | NCT03255096 | ||
| CPI-0610 | Monotherapy | CPI-0610 | Malignant Peripheral Nerve Sheath Tumors | II | NCT02986919 | |
| Monotherapy | CPI-0610 | MM | I | NCT02157636 | ||
| Monotherapy | CPI-0610 | Progressive lymphoma | I | NCT01949883 | ||
| JAK inhibitor | CPI-0610; ruxolitinib | Acute Leukemia, Myelodysplastic Syndrome, Myelodysplastic/Myeloproliferative Neoplasms, and Myelofibrosis | I/II | NCT02158858 | ||
| FT-1101 | Chemotherapy | FT-1101; azacitidine | AML or non-Hodgkin Lymphoma | I | NCT02543879 | |
| INCB054329 | Monotherapy | INCB054329 | Advanced Solid Tumors and Hematologic Malignancy | I/II | NCT02431260 | |
| GSK2820151 | Monotherapy | GSK2820151 | Advanced or recurrent solid tumors | I | NCT02630251 | |
| ZEN003694 | Monotherapy | ZEN003694 | Metastatic Castration-Resistant Prostate Cancer | I | NCT02705469 | |
| Hormone therapy | ZEN003694; Enzalutamide | Metastatic Castration-Resistant Prostate Cancer | I/II | NCT02711956 | ||
| BMS-986158 | Immune checkpoint inhibitors | BMS-986158; Nivolumab | Selected advanced cancers | I/II | NCT02419417 | |
| ABBV-075 | BCL2 inhibitor | ABBV-075; Venetoclax | Selected hematological and solid cancers | I | NCT02391480 | |
| GS-5829 | Hormone therapy | GS-5829; Exemestane; Fulvestrant | Advanced Estrogen Receptor Positive, HER2 Negative-Breast Cancer | I/II | NCT02983604 | |
| Hormone therapy | GS-5829; Exemestane; Fulvestrant | Advanced solid tumors and lymphomas | I | NCT02392611 | ||
| Hormone therapy | GS-5829; Enzatulamide | Metastatic Castrate-Resistant Prostate Cancer | I/II | NCT02607228 | ||
| PLX51107 | Monotherapy | PLX51107 | Advanced solid and hematologic malignancies | I/II | NCT02683395 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manzotti, G.; Ciarrocchi, A.; Sancisi, V. Inhibition of BET Proteins and Histone Deacetylase (HDACs): Crossing Roads in Cancer Therapy. Cancers 2019, 11, 304. https://doi.org/10.3390/cancers11030304
Manzotti G, Ciarrocchi A, Sancisi V. Inhibition of BET Proteins and Histone Deacetylase (HDACs): Crossing Roads in Cancer Therapy. Cancers. 2019; 11(3):304. https://doi.org/10.3390/cancers11030304
Chicago/Turabian StyleManzotti, Gloria, Alessia Ciarrocchi, and Valentina Sancisi. 2019. "Inhibition of BET Proteins and Histone Deacetylase (HDACs): Crossing Roads in Cancer Therapy" Cancers 11, no. 3: 304. https://doi.org/10.3390/cancers11030304
APA StyleManzotti, G., Ciarrocchi, A., & Sancisi, V. (2019). Inhibition of BET Proteins and Histone Deacetylase (HDACs): Crossing Roads in Cancer Therapy. Cancers, 11(3), 304. https://doi.org/10.3390/cancers11030304