ADRB2-Targeting Therapies for Prostate Cancer

{kind=link}
{kind=link}
Abstract
:1. Introduction
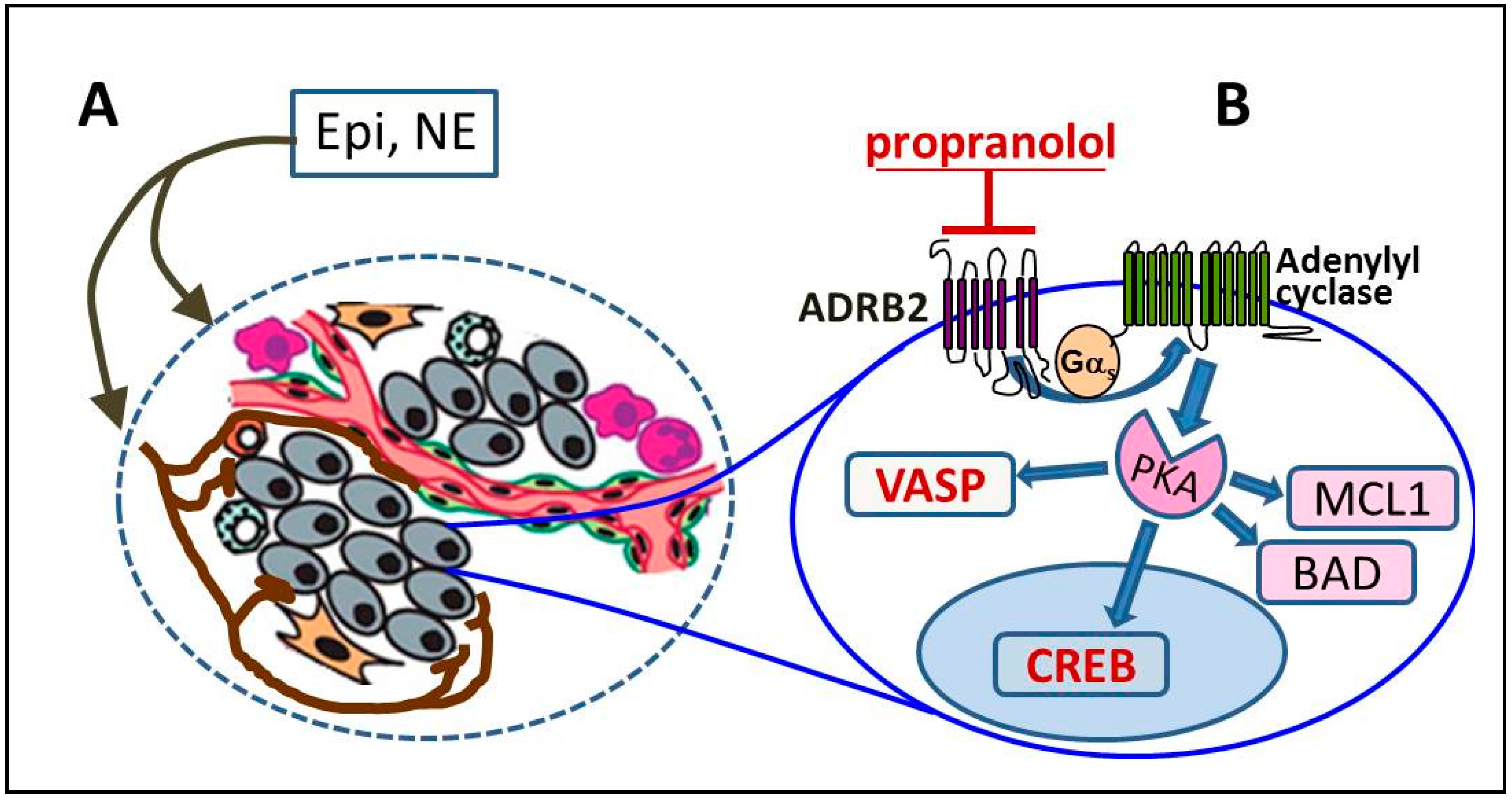
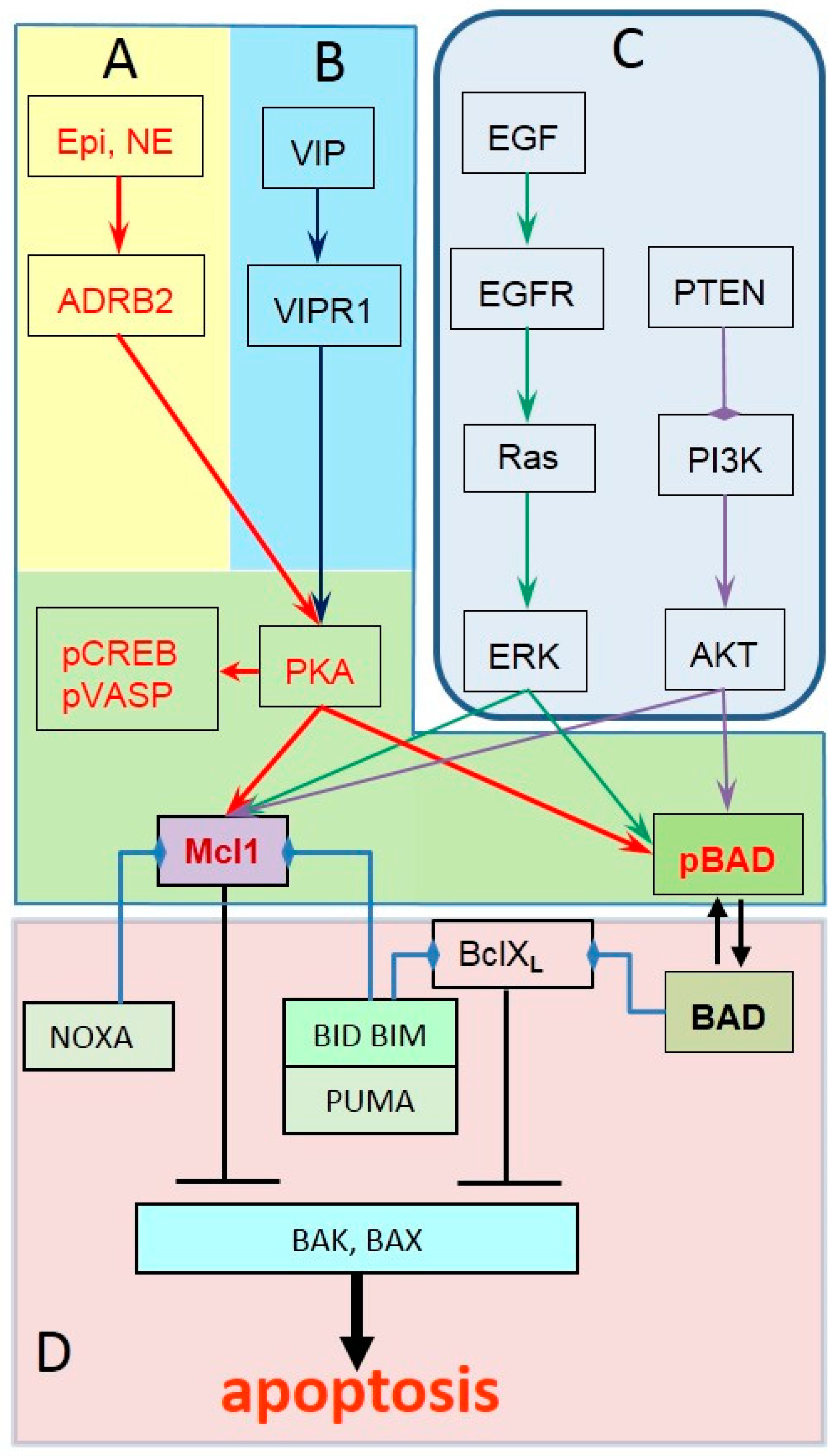
2. ADRB2 Signaling in Prostate Cancer Progression
3. Identifying Tumors with Active ADRB2 Signaling
4. Identifying Prostate Tumors Unresponsive to Propranolol
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Yap, T.A.; Smith, A.D.; Ferraldeschi, R.; Al-Lazikani, B.; Workman, P.; de Bono, J.S. Drug discovery in advanced prostate cancer: Translating biology into therapy. Nat. Rev. Drug Discov. 2016, 15, 699–718. [Google Scholar] [CrossRef]
- Collins, D.C.; Sundar, R.; Lim, J.S.; Yap, T.A. Towards Precision Medicine in the Clinic: From Biomarker Discovery to Novel Therapeutics. Trends Pharmacol. Sci. 2017, 38, 25–40. [Google Scholar] [CrossRef]
- Singh, P.; Uzgare, A.; Litvinov, I.; Denmeade, S.R.; Isaacs, J.T. Combinatorial androgen receptor targeted therapy for prostate cancer. Endocr. Relat. Cancer 2006, 13, 653–666. [Google Scholar] [CrossRef] [Green Version]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Tasken, K.A. Beta-Adrenergic Receptor Signaling in Prostate Cancer. Front. Oncol. 2014, 4, 375. [Google Scholar] [CrossRef]
- Philipp, M.; Hein, L. Adrenergic receptor knockout mice: Distinct functions of 9 receptor subtypes. Pharmacol. Ther. 2004, 101, 65–74. [Google Scholar] [CrossRef]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Gudermann, T.; Schoneberg, T.; Schultz, G. Functional and structural complexity of signal transduction via G-protein-coupled receptors. Annu. Rev. Neurosci. 1997, 20, 399–427. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J. Seven transmembrane receptors: Something old, something new. Acta Physiol. (Oxf.) 2007, 190, 9–19. [Google Scholar] [CrossRef]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef]
- Prichard, B.N.; Cruickshank, J.M.; Graham, B.R. Beta-Adrenergic blocking drugs in the treatment of hypertension. Blood Press 2001, 10, 366–386. [Google Scholar] [CrossRef]
- Baker, J.G. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br. J. Pharm. 2005, 144, 317–322. [Google Scholar] [CrossRef]
- Hoffmann, C.; Leitz, M.R.; Oberdorf-Maass, S.; Lohse, M.J.; Klotz, K.N. Comparative pharmacology of human beta-adrenergic receptor subtypes--characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedebergs Arch. Pharm. 2004, 369, 151–159. [Google Scholar] [CrossRef]
- Nagmani, R.; Pasco, D.S.; Salas, R.D.; Feller, D.R. Evaluation of beta-adrenergic receptor subtypes in the human prostate cancer cell line-LNCaP. Biochem. Pharm. 2003, 65, 1489–1494. [Google Scholar] [CrossRef]
- Poyet, P.; Gagne, B.; Lavoie, M.; Labrie, F. Characteristics of the beta-adrenergic receptor in the rat ventral prostate using [125I]cyanopindolol. Mol. Cell. Endocrinol. 1986, 48, 59–67. [Google Scholar] [CrossRef]
- Yu, J.; Cao, Q.; Mehra, R.; Laxman, B.; Yu, J.; Tomlins, S.A.; Creighton, C.J.; Dhanasekaran, S.M.; Shen, R.; Chen, G.; et al. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell 2007, 12, 419–431. [Google Scholar] [CrossRef]
- Ramberg, H.; Eide, T.; Krobert, K.A.; Levy, F.O.; Dizeyi, N.; Bjartell, A.S.; Abrahamsson, P.A.; Tasken, K.A. Hormonal regulation of beta2-adrenergic receptor level in prostate cancer. Prostate 2008, 68, 1133–1142. [Google Scholar] [CrossRef]
- Flierl, M.A.; Rittirsch, D.; Nadeau, B.A.; Chen, A.J.; Sarma, J.V.; Zetoune, F.S.; McGuire, S.R.; List, R.P.; Day, D.E.; Hoesel, L.M.; et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature 2007, 449, 721–725. [Google Scholar] [CrossRef]
- Marino, F.; Cosentino, M. Adrenergic modulation of immune cells: An update. Amino Acids 2013, 45, 55–71. [Google Scholar] [CrossRef]
- Dantzer, R. Neuroimmune Interactions: From the Brain to the Immune System and Vice Versa. Physiol. Rev. 2018, 98, 477–504. [Google Scholar] [CrossRef]
- Palm, D.; Lang, K.; Niggemann, B.; Drell, T.L.; Masur, K.; Zaenker, K.S.; Entschladen, F. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int. J. Cancer 2006, 118, 2744–2749. [Google Scholar] [CrossRef]
- Hassan, S.; Karpova, Y.; Baiz, D.; Yancey, D.; Pullikuth, A.; Flores, A.; Register, T.; Cline, J.M.; D’Agostino, R., Jr.; Danial, N.; et al. Behavioral stress accelerates prostate cancer development in mice. J. Clin. Investig. 2013, 123, 874–886. [Google Scholar] [CrossRef] [Green Version]
- Kulik, G. Personalized prostate cancer therapy based on systems analysis of the apoptosis regulatory network. Asian J. Androl. 2015, 17, 471–474. [Google Scholar]
- Sun, X.; Bao, J.; Nelson, K.C.; Li, K.C.; Kulik, G.; Zhou, X. Systems modeling of anti-apoptotic pathways in prostate cancer: Psychological stress triggers a synergism pattern switch in drug combination therapy. PLoS Comput. Biol. 2013, 9, e1003358. [Google Scholar] [CrossRef]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar] [CrossRef]
- Datta, S.R.; Katsov, A.; Hu, L.; Petros, A.; Fesik, S.W.; Yaffe, M.B.; Greenberg, M.E. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell 2000, 6, 41–51. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Holmgreen, S.P.; Huang, D.C.; Adams, J.M.; Cory, S. Survival activity of Bcl-2 homologs Bcl-w and A1 only partially correlates with their ability to bind pro-apoptotic family members. Cell Death Differ. 1999, 6, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Gulla, S.; Chen, T.S.; Fire, E.; Grant, R.A.; Keating, A.E. Determinants of BH3 binding specificity for Mcl-1 versus Bcl-xL. J. Mol. Biol. 2010, 398, 747–762. [Google Scholar] [CrossRef]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef] [Green Version]
- Perciavalle, R.M.; Opferman, J.T. Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol. 2013, 23, 22–29. [Google Scholar] [CrossRef]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, Y.; Kato, T.; Kitagawa, M.; Fujita, H.; Kitagawa, S. Calpain inhibition delays neutrophil apoptosis via cyclic AMP-independent activation of protein kinase A and protein kinase A-mediated stabilization of Mcl-1 and X-linked inhibitor of apoptosis (XIAP). Arch. Biochem. Biophys. 2008, 477, 227–231. [Google Scholar] [CrossRef]
- Yu, M.; Liu, T.; Chen, Y.; Li, Y.; Li, W. Combination therapy with protein kinase inhibitor H89 and Tetrandrine elicits enhanced synergistic antitumor efficacy. J. Exp. Clin. Cancer Res. 2018, 37, 114. [Google Scholar] [CrossRef]
- Sastry, K.S.; Karpova, Y.; Prokopovich, S.; Smith, A.J.; Essau, B.; Gersappe, A.; Carson, J.P.; Weber, M.J.; Register, T.C.; Chen, Y.Q.; et al. Epinephrine protects cancer cells from apoptosis via activation of cAMP-dependent protein kinase and BAD phosphorylation. J. Biol. Chem. 2007, 282, 14094–14100. [Google Scholar] [CrossRef]
- Hulsurkar, M.; Li, Z.; Zhang, Y.; Li, X.; Zheng, D.; Li, W. Beta-adrenergic signaling promotes tumor angiogenesis and prostate cancer progression through HDAC2-mediated suppression of thrombospondin-1. Oncogene 2017, 36, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic nerve development contributes to prostate cancer progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [PubMed]
- Zahalka, A.H.; Arnal-Estape, A.; Maryanovich, M.; Nakahara, F.; Cruz, C.D.; Finley, L.W.S.; Frenette, P.S. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 2017, 358, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perron, L.; Bairati, I.; Harel, F.; Meyer, F. Antihypertensive drug use and the risk of prostate cancer (Canada). Cancer Causes Control 2004, 15, 535–541. [Google Scholar] [CrossRef]
- Grytli, H.H.; Fagerland, M.W.; Fossa, S.D.; Tasken, K.A. Association between use of beta-blockers and prostate cancer-specific survival: A cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur. Urol. 2014, 65, 635–641. [Google Scholar] [CrossRef]
- Armaiz-Pena, G.N.; Allen, J.K.; Cruz, A.; Stone, R.L.; Nick, A.M.; Lin, Y.G.; Han, L.Y.; Mangala, L.S.; Villares, G.J.; Vivas-Mejia, P.; et al. Src activation by beta-adrenoreceptors is a key switch for tumour metastasis. Nat. Commun. 2013, 4, 1403. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Jacobs, E.J.; Deka, A.; Patel, A.V.; Bain, E.B.; Thun, M.J.; Calle, E.E. Use of blood-pressure-lowering medication and risk of prostate cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Causes. Control 2009, 20, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.M.; Carey, I.M.; Owen, C.G.; Harris, T.; Dewilde, S.; Cook, D.G. Does beta-adrenoceptor blocker therapy improve cancer survival? Findings from a population-based retrospective cohort study. Br. J. Clin. Pharmcol. 2011, 72, 157–161. [Google Scholar] [CrossRef]
- Bhaskaran, K.; Rachet, B.; Evans, S.; Smeeth, L. Re: Helene Hartvedt Grytli, Morten Wang Fagerland, Sophie D. Fossa, Kristin Austlid Tasken. Association between use of beta-blockers and prostate cancer-specific survival: A cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur Urol. In press. http://dx.doi.org/10.1016/j.eururo.2013.01.007. Eur. Urol. 2013, 64, e86–e87. [Google Scholar] [CrossRef]
- Chang, P.Y.; Huang, W.Y.; Lin, C.L.; Huang, T.C.; Wu, Y.Y.; Chen, J.H.; Kao, C.H. Propranolol Reduces Cancer Risk: A Population-Based Cohort Study. Medicine (Baltimore) 2015, 94, e1097. [Google Scholar] [CrossRef]
- Emilien, G.; Maloteaux, J.M. Current therapeutic uses and potential of beta-adrenoceptor agonists and antagonists. Eur. J. Clin. Pharmcol. 1998, 53, 389–404. [Google Scholar] [CrossRef]
- Ellison, K.E.; Gandhi, G. Optimising the use of beta-adrenoceptor antagonists in coronary artery disease. Drugs 2005, 65, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.R.; Goldstein, D.S. Sympathoneural and adrenomedullary responses to mental stress. Compr. Physiol. 2015, 5, 119–146. [Google Scholar] [PubMed]
- Ullrich, P.M.; Carson, M.R.; Lutgendorf, S.K.; Williams, R.D. Cancer fear and mood disturbance after radical prostatectomy: Consequences of biochemical evidence of recurrence. J. Urol. 2003, 169, 1449–1452. [Google Scholar] [CrossRef]
- Stone, A.A.; Mezzacappa, E.S.; Donatone, B.A.; Gonder, M. Psychosocial stress and social support are associated with prostate-specific antigen levels in men: Results from a community screening program. Health Psychol. 1999, 18, 482–486. [Google Scholar] [CrossRef]
- Turner, E.L.; Lane, J.A.; Metcalfe, C.; Down, L.; Donovan, J.L.; Hamdy, F.; Neal, D.; Vedhara, K. Psychological distress and prostate specific antigen levels in men with and without prostate cancer. Brain Behav. Immun. 2009, 23, 1073–1078. [Google Scholar] [CrossRef]
- Saxe, G.A.; Major, J.M.; Nguyen, J.Y.; Freeman, K.M.; Downs, T.M.; Salem, C.E. Potential attenuation of disease progression in recurrent prostate cancer with plant-based diet and stress reduction. Integr. Cancer 2006, 5, 206–213. [Google Scholar] [CrossRef]
- Hassan, S.; Karpova, Y.; Flores, A.; D’Agostino, R., Jr.; Danhauer, S.C.; Hemal, A.; Kulik, G. A pilot study of blood epinephrine levels and CREB phosphorylation in men undergoing prostate biopsies. Int. Urol. Nephrol. 2014, 46, 505–510. [Google Scholar] [CrossRef]
- White, C.W.; Xie, J.H.; Ventura, S. Age-related changes in the innervation of the prostate gland: Implications for prostate cancer initiation and progression. Organogenesis 2013, 9, 206–215. [Google Scholar] [CrossRef]
- Goepel, M.; Wittmann, A.; Rubben, H.; Michel, M.C. Comparison of adrenoceptor subtype expression in porcine and human bladder and prostate. Urol. Res. 1997, 25, 199–206. [Google Scholar] [CrossRef]
- Thaker, P.H.; Han, L.Y.; Kamat, A.A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; Jennings, N.B.; Armaiz-Pena, G.; Bankson, J.A.; Ravoori, M.; et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 2006, 12, 939–944. [Google Scholar] [CrossRef]
- Cohen, L.; de Moor, C.; Devine, D.; Baum, A.; Amato, R.J. Endocrine levels at the start of treatment are associated with subsequent psychological adjustment in cancer patients with metastatic disease. Psychosom. Med. 2001, 63, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Palmgren, J.S.; Karavadia, S.S.; Wakefield, M.R. Unusual and underappreciated: Small cell carcinoma of the prostate. Semin. Oncol. 2007, 34, 22–29. [Google Scholar] [CrossRef]
- Lutgendorf, S.K.; DeGeest, K.; Sung, C.Y.; Arevalo, J.M.; Penedo, F.; Lucci, J., III; Goodheart, M.; Lubaroff, D.; Farley, D.M.; Sood, A.K.; et al. Depression, social support, and beta-adrenergic transcription control in human ovarian cancer. Brain Behav. Immun. 2009, 23, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Lutgendorf, S.K.; DeGeest, K.; Dahmoush, L.; Farley, D.; Penedo, F.; Bender, D.; Goodheart, M.; Buekers, T.E.; Mendez, L.; Krueger, G.; et al. Social isolation is associated with elevated tumor norepinephrine in ovarian carcinoma patients. Brain Behav. Immun. 2011, 25, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingenfeld, K.; Whooley, M.A.; Neylan, T.C.; Otte, C.; Cohen, B.E. Effect of current and lifetime posttraumatic stress disorder on 24-h urinary catecholamines and cortisol: Results from the Mind Your Heart Study. Psychoneuroendocrinology 2015, 52, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Gandubert, C.; Scali, J.; Ancelin, M.L.; Carriere, I.; Dupuy, A.M.; Bagnolini, G.; Ritchie, K.; Sebanne, M.; Martrille, L.; Baccino, E.; et al. Biological and psychological predictors of posttraumatic stress disorder onset and chronicity. A one-year prospective study. Neurobiol. Stress 2016, 3, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Kjeldsen, S.E.; Flaaten, B.; Eide, I.; Helgeland, A.; Leren, P. Evidence of increased peripheral catecholamine release in patients with long-standing, untreated essential hypertension. Scand. J. Clin. Lab. Investig. 1982, 42, 217–223. [Google Scholar] [CrossRef]
- Tsao, P.; von Zastrow, M. Downregulation of G protein-coupled receptors. Curr. Opin. Neurobiol. 2000, 10, 365–369. [Google Scholar] [CrossRef]
- Collins, S.; Caron, M.G.; Lefkowitz, R.J. Regulation of adrenergic receptor responsiveness through modulation of receptor gene expression. Annu. Rev. Physiol. 1991, 53, 497–508. [Google Scholar] [CrossRef]
- Prowatke, I.; Devens, F.; Benner, A.; Grone, E.F.; Mertens, D.; Grone, H.J.; Lichter, P.; Joos, S. Expression analysis of imbalanced genes in prostate carcinoma using tissue microarrays. Br. J. Cancer 2007, 96, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Aljameeli, A.; Thakkar, A.; Shah, G. Calcitonin receptor increases invasion of prostate cancer cells by recruiting zonula occludens-1 and promoting PKA-mediated TJ disassembly. Cell. Signal. 2017, 36, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Chigurupati, S.; Anbalagan, M.; Shah, G. Calcitonin increases tumorigenicity of prostate cancer cells: Evidence for the role of protein kinase A and urokinase-type plasminogen receptor. Mol. Endocrinol. 2006, 20, 1894–1911. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martinez, A.B.; Carmena, M.J.; Arenas, M.I.; Bajo, A.M.; Prieto, J.C.; Sanchez-Chapado, M. Overexpression of vasoactive intestinal peptide receptors and cyclooxygenase-2 in human prostate cancer. Analysis of potential prognostic relevance. Histol. Histopathol. 2012, 27, 1093–1101. [Google Scholar] [PubMed]
- Nelson, J.; Bagnato, A.; Battistini, B.; Nisen, P. The endothelin axis: Emerging role in cancer. Nat. Rev. Cancer 2003, 3, 110–116. [Google Scholar] [CrossRef]
- Taub, J.S.; Guo, R.; Leeb-Lundberg, L.M.; Madden, J.F.; Daaka, Y. Bradykinin receptor subtype 1 expression and function in prostate cancer. Cancer Res. 2003, 63, 2037–2041. [Google Scholar] [PubMed]
- Xu, L.L.; Stackhouse, B.G.; Florence, K.; Zhang, W.; Shanmugam, N.; Sesterhenn, I.A.; Zou, Z.; Srikantan, V.; Augustus, M.; Roschke, V.; et al. PSGR, a novel prostate-specific gene with homology to a G protein-coupled receptor, is overexpressed in prostate cancer. Cancer Res. 2000, 60, 6568–6572. [Google Scholar] [PubMed]
- Sastry, K.S.; Chouchane, A.I.; Wang, E.; Kulik, G.; Marincola, F.M.; Chouchane, L. Cytoprotective effect of neuropeptides on cancer stem cells: Vasoactive intestinal peptide-induced antiapoptotic signaling. Cell Death Dis. 2017, 8, e2844. [Google Scholar] [CrossRef]
- Sastry, K.S.; Smith, A.J.; Karpova, Y.; Datta, S.R.; Kulik, G. Diverse antiapoptotic signaling pathways activated by vasoactive intestinal polypeptide, epidermal growth factor, and phosphatidylinositol 3-kinase in prostate cancer cells converge on BAD. J. Biol. Chem. 2006, 281, 20891–20901. [Google Scholar] [CrossRef]
- Yan, J.; Xiang, J.; Lin, Y.; Ma, J.; Zhang, J.; Zhang, H.; Sun, J.; Danial, N.N.; Liu, J.; Lin, A. Inactivation of BAD by IKK inhibits TNFalpha-induced apoptosis independently of NF-kappaB activation. Cell 2013, 152, 304–315. [Google Scholar] [CrossRef]
- Yancey, D.; Nelson, K.C.; Baiz, D.; Hassan, S.; Flores, A.; Pullikuth, A.; Karpova, Y.; Axanova, L.; Moore, V.; Sui, G.; et al. BAD dephosphorylation and decreased expression of MCL-1 induce rapid apoptosis in prostate cancer cells. PLoS ONE 2013, 8, e74561. [Google Scholar] [CrossRef] [PubMed]
- Santer, F.R.; Erb, H.H.; Oh, S.J.; Handle, F.; Feiersinger, G.E.; Luef, B.; Bu, H.; Schafer, G.; Ploner, C.; Egger, M.; et al. Mechanistic rationale for MCL1 inhibition during androgen deprivation therapy. Oncotarget 2015, 6, 6105–6122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, A.; Tang, J.; Hong, Y.; Song, J.; Terranova, P.F.; Thrasher, J.B.; Svojanovsky, S.; Wang, H.G.; Li, B. Androgen receptor-dependent regulation of Bcl-xL expression: Implication in prostate cancer progression. Prostate 2008, 68, 453–461. [Google Scholar] [CrossRef]
- Krajewska, M.; Krajewski, S.; Epstein, J.I.; Shabaik, A.; Sauvageot, J.; Song, K.; Kitada, S.; Reed, J.C. Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am. J. Pathol. 1996, 148, 1567–1576. [Google Scholar] [PubMed]
- Zellweger, T.; Ninck, C.; Bloch, M.; Mirlacher, M.; Koivisto, P.A.; Helin, H.J.; Mihatsch, M.J.; Gasser, T.C.; Bubendorf, L. Expression patterns of potential therapeutic targets in prostate cancer. Int. J. Cancer 2005, 113, 619–628. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulik, G. ADRB2-Targeting Therapies for Prostate Cancer. Cancers 2019, 11, 358. https://doi.org/10.3390/cancers11030358
Kulik G. ADRB2-Targeting Therapies for Prostate Cancer. Cancers. 2019; 11(3):358. https://doi.org/10.3390/cancers11030358
Chicago/Turabian StyleKulik, George. 2019. "ADRB2-Targeting Therapies for Prostate Cancer" Cancers 11, no. 3: 358. https://doi.org/10.3390/cancers11030358
APA StyleKulik, G. (2019). ADRB2-Targeting Therapies for Prostate Cancer. Cancers, 11(3), 358. https://doi.org/10.3390/cancers11030358