Examination of Independent Prognostic Power of Gene Expressions and Histopathological Imaging Features in Cancer

Abstract
:1. Introduction
2. Data
2.1. mRNA Gene Expression Measurements
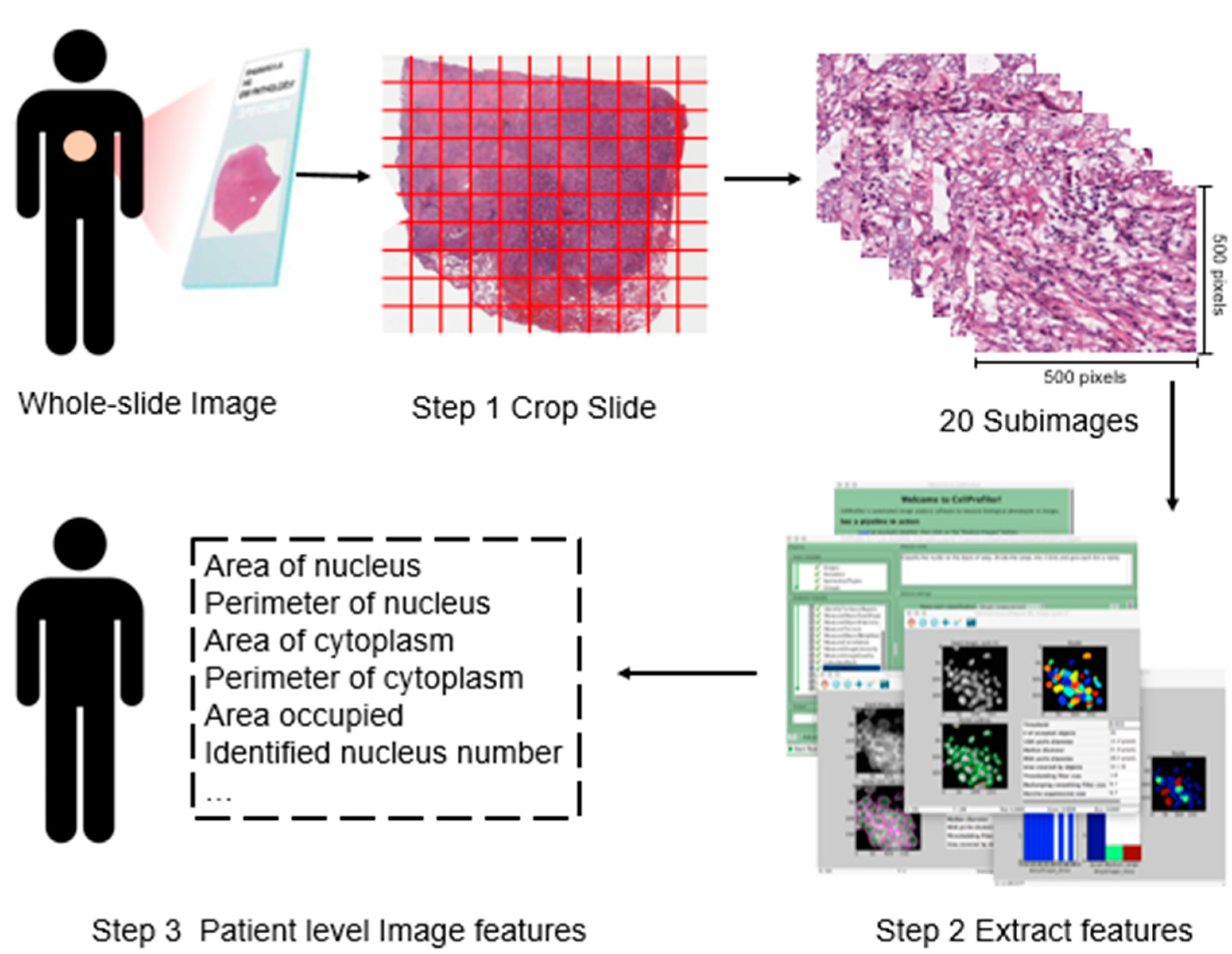
2.2. Histopathological Imaging Features
2.3. Available Data
3. Methods
4. Results
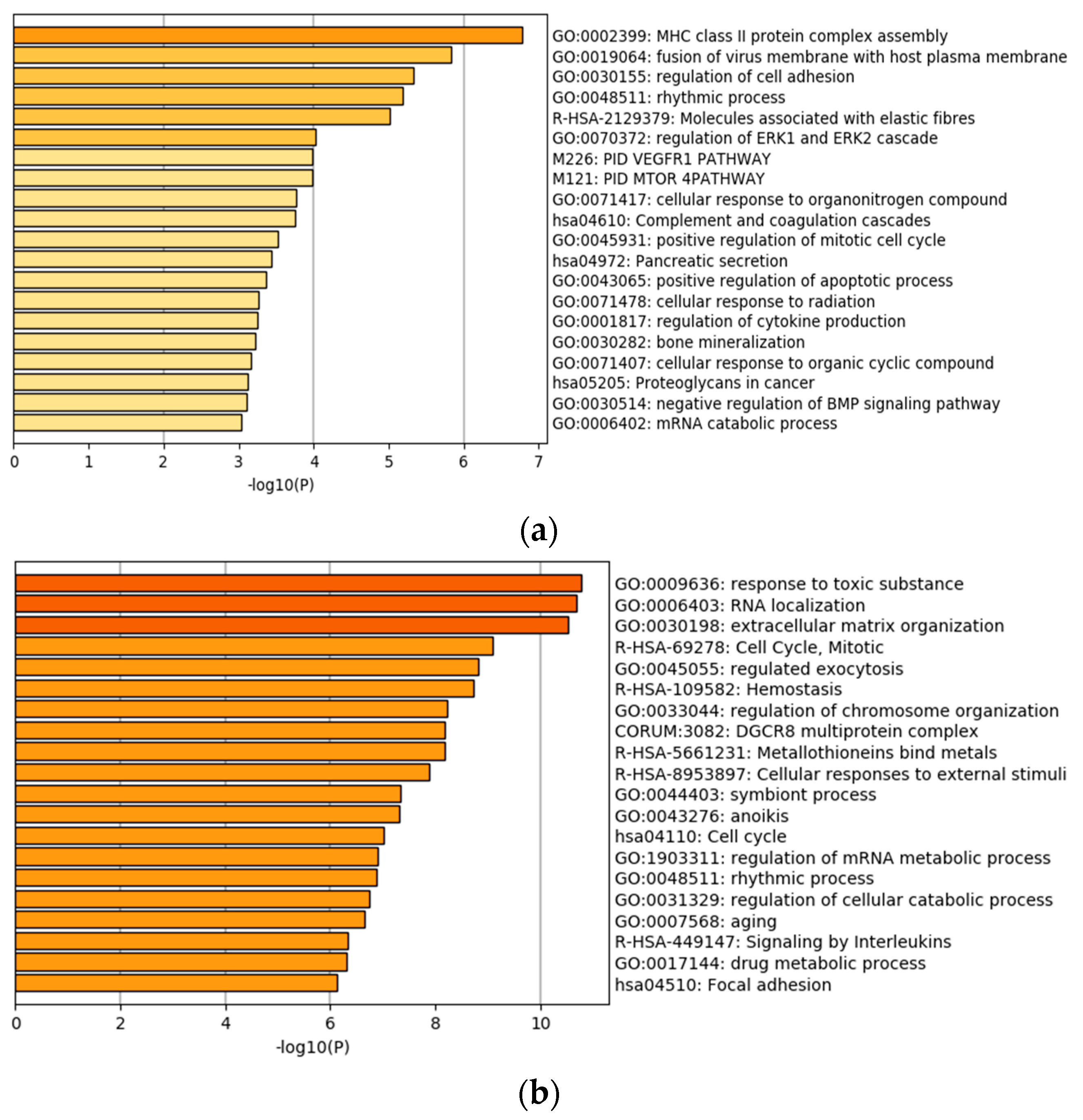
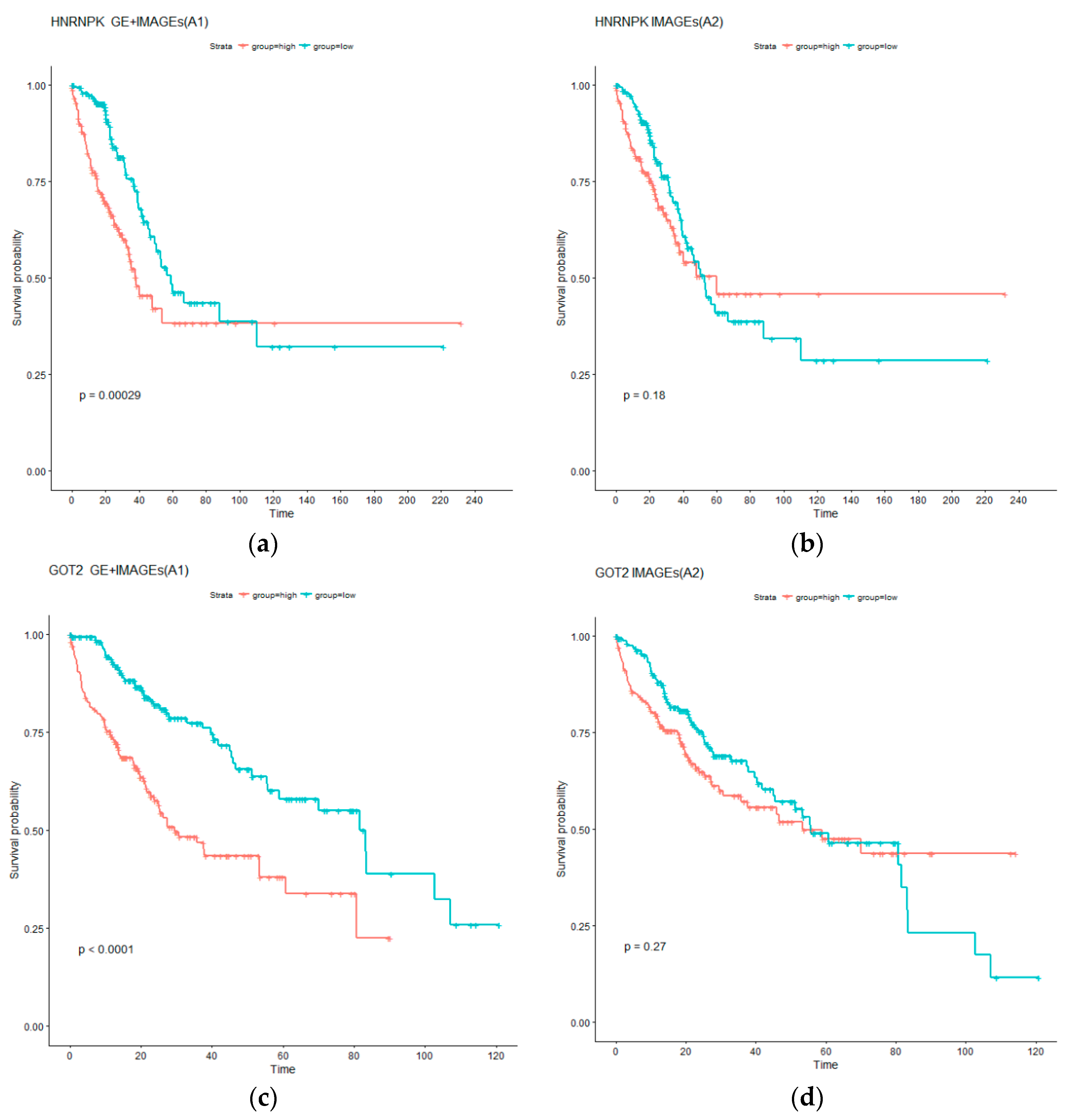
4.1. Identification of Gene Expressions with Independent Prognostic Power Conditional on Imaging Features
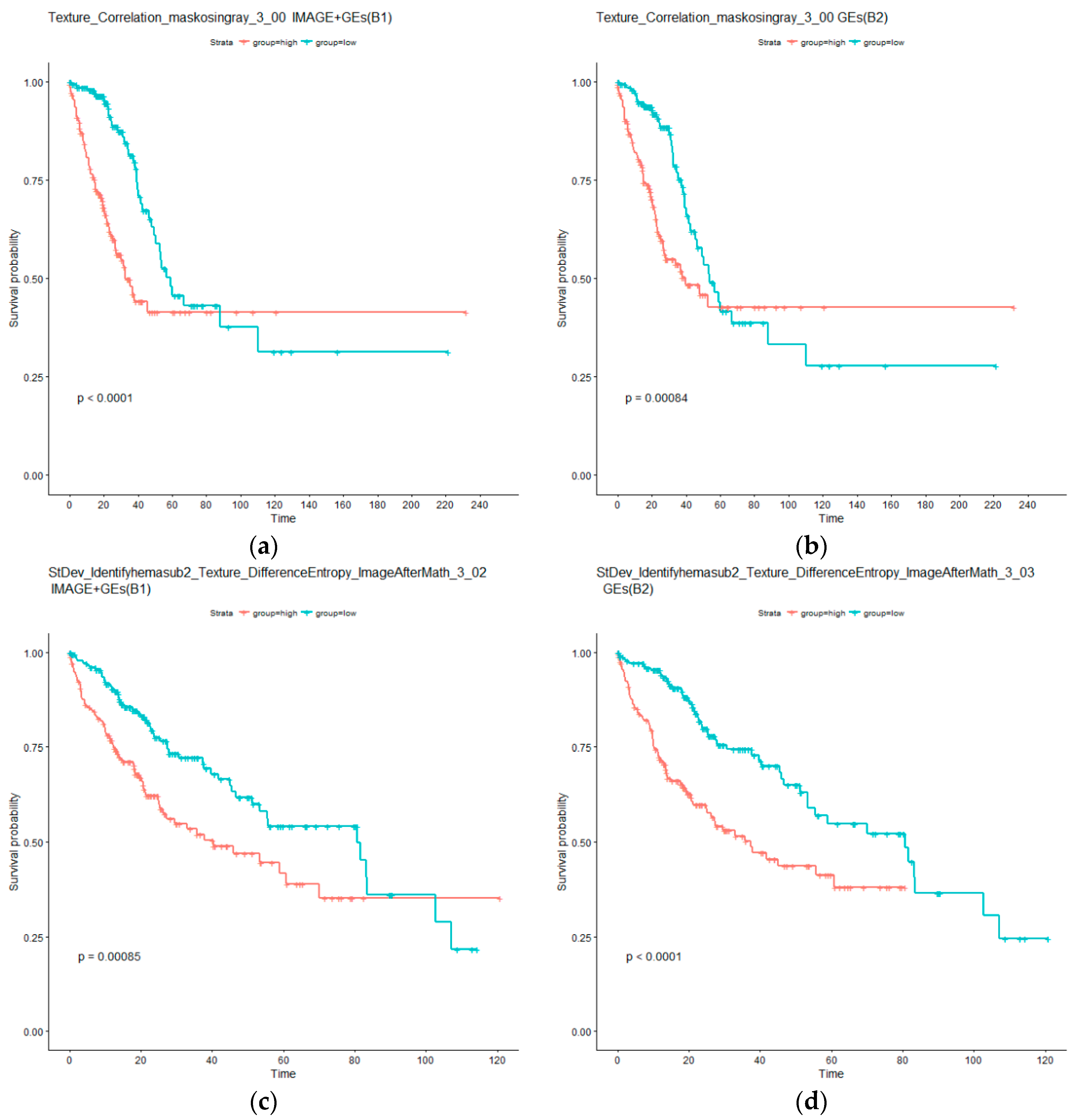
4.2. Identification of Imaging Features with Independent Prognostic Power Conditional on Gene Expressions
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mallett, S.; Royston, P.; Dutton, S.; Waters, R.; Altman, D.G. Reporting methods in studies developing prognostic models in cancer: A review. BMC Med. 2010, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Kourou, K.; Exarchos, T.P.; Exarchos, K.P.; Karamouzis, M.V.; Fotiadis, D.I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 2015, 13, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Rath, M.G.; Uhlmann, L.; Fiedler, M.; Heil, J.; Golatta, M.; Dinkic, C.; Hennigs, A.; Schott, S.; Ernst, V.; Koch, T.; et al. Oncotype DX((R)) in breast cancer patients: Clinical experience, outcome and follow-up-a case-control study. Arch. Gynecol. Obstet. 2018, 297, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, H.M.; Widschwendter, A.; Fiegl, H.; Ivarsson, L.; Goebel, G.; Perkmann, E.; Marth, C.; Widschwendter, M. DNA methylation in serum of breast cancer patients: An independent prognostic marker. Cancer Res. 2003, 63, 7641–7645. [Google Scholar] [PubMed]
- Harpole, D.H., Jr.; Herndon, J.E., 2nd; Wolfe, W.G.; Iglehart, J.D.; Marks, J.R. A prognostic model of recurrence and death in stage I non-small cell lung cancer utilizing presentation, histopathology, and oncoprotein expression. Cancer Res. 1995, 55, 51–56. [Google Scholar] [PubMed]
- Romo-Bucheli, D.; Janowczyk, A.; Gilmore, H.; Romero, E.; Madabhushi, A. Automated tubulenuclei quantification and correlation with Oncotype DX risk categories in ER+ breast cancer whole slide Images. Sci. Rep. 2016, 6, 32706. [Google Scholar] [CrossRef] [PubMed]
- Sertel, O.; Kong, J.; Shimada, H.; Catalyurek, U.V.; Saltz, J.H.; Gurcan, M.N. Computer-aided prognosis of neuroblastoma on whole-slide images: Classification of stromal development. Pattern Recognit. 2009, 42, 1093–1103. [Google Scholar] [CrossRef] [Green Version]
- Sertel, O.; Kong, J.; Catalyurek, U.V.; Lozanski, G.; Saltz, J.H.; Gurcan, M.N. Histopathological Image Analysis Using Model-Based Intermediate Representations and Color Texture: Follicular Lymphoma Grading. J. Signal Process. Syst. 2009, 55, 169–183. [Google Scholar] [CrossRef]
- Yu, K.H.; Berry, G.J.; Rubin, D.L.; Re, C.; Altman, R.B.; Snyder, M. Association of omics features with histopathology patterns in lung adenocarcinoma. Cell Syst. 2017, 5, 620–627.e3. [Google Scholar] [CrossRef]
- Sabo, E.; Beck, A.H.; Montgomery, E.A.; Bhattacharya, B.; Meitner, P.; Wang, J.Y.; Resnick, M.B. Computerized morphometry as an aid in determining the grade of dysplasia and progression to adenocarcinoma in Barrett’s esophagus. Lab. Investig. 2006, 86, 1261–1271. [Google Scholar] [CrossRef]
- Cooper, L.A.; Kong, J.; Gutman, D.A.; Dunn, W.D.; Nalisnik, M.; Brat, D.J. Novel genotype-phenotype associations in human cancers enabled by advanced molecular platforms and computational analysis of whole slide images. Lab. Investig. 2015, 95, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Li, A.; Tang, B.; Wang, M. Integrating genomic data and pathological images to effectively predict breast cancer clinical outcome. Comput. Methods Programs Biomed. 2018, 161, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Mobadersany, P.; Yousefi, S.; Amgad, M.; Gutman, D.A.; Barnholtz-Sloan, J.S.; Vega, J.V.; Brat, D.J.; Cooper, L.A. Predicting cancer outcomes from histology and genomics using convolutional networks. Proc. Natl. Acad. Sci. USA 2018, 115, E2970–E2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.L.; Yao, J.W.; Luo, X.; Xiao, G.H.; Xie, Y.; Gazdar, A.; Huang, J.Z. Lung cancer survival prediction from pathological images and genetic data—An integration study. In Proceedings of the 2016 IEEE 13th International Symposium on Biomedical Imaging (ISBI), Prague, Czech Republic, 13–16 April 2016; pp. 1173–1176. [Google Scholar]
- Hutter, C.; Zenklusen, J.C. The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Soliman, K. CellProfiler: Novel Automated Image Segmentation Procedure for Super-Resolution Microscopy. Biol. Proced. Online 2015, 17, 11. [Google Scholar] [CrossRef]
- Goode, A.; Gilbert, B.; Harkes, J.; Jukic, D.; Satyanarayanan, M. OpenSlide: A vendor-neutral software foundation for digital pathology. J. Pathol. Inform. 2013, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zang, X.; Yang, L.; Huang, J.; Liang, F.; Rodriguez-Canales, J.; Wistuba, I.I.; Gazdar, A.; Xie, Y.; Xiao, G. Comprehensive Computational Pathological Image Analysis Predicts Lung Cancer Prognosis. J. Thorac. Oncol. 2017, 12, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.H.; Zhang, C.; Berry, G.J.; Altman, R.B.; Re, C.; Rubin, D.L.; Snyder, M. Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features. Nat. Commun. 2016, 7, 12474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, J.; James, I. Linear regression with censored data. Biometrika 1979, 66, 429–436. [Google Scholar] [CrossRef]
- Chai, H.; Zhang, Q.Z.; Huang, J.; Ma, S.G. Inference for low-dimensional covariates in a high-dimensional accelerated failure time model. Stat. Sin. 2019. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Chen, J.H.; Chen, Z.H. Extended Bayesian information criteria for model selection with large model spaces. Biometrika 2008, 95, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.H.; Fan, P.; Chang, S.W.; Tsao, Y.P.; Huang, H.P.; Chen, S.L. NRIP/DCAF6 stabilizes the androgen receptor protein by displacing DDB2 from the CUL4A-DDB1 E3 ligase complex in prostate cancer. Oncotarget 2017, 8, 21501–21515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Yang, Z.; Tsuji, T.; Gong, J.; Xie, J.; Chen, C.; Li, W.; Amar, S.; Luo, Z. LITAF and TNFSF15, two downstream targets of AMPK, exert inhibitory effects on tumor growth. Oncogene 2011, 30, 1892–1900. [Google Scholar] [CrossRef] [Green Version]
- Kopajtich, R.; Murayama, K.; Janecke, A.R.; Haack, T.B.; Breuer, M.; Knisely, A.S.; Harting, I.; Ohashi, T.; Okazaki, Y.; Watanabe, D.; et al. Biallelic IARS Mutations Cause Growth Retardation with Prenatal Onset, Intellectual Disability, Muscular Hypotonia, and Infantile Hepatopathy. Am. J. Hum. Genet. 2016, 99, 414–422. [Google Scholar] [CrossRef]
- Tian, T.; Ikeda, J.; Wang, Y.; Mamat, S.; Luo, W.; Aozasa, K.; Morii, E. Role of leucine-rich pentatricopeptide repeat motif-containing protein (LRPPRC) for anti-apoptosis and tumourigenesis in cancers. Eur. J. Cancer 2012, 48, 2462–2473. [Google Scholar] [CrossRef]
- Van Beijnum, J.R.; Moerkerk, P.T.M.; Gerbers, A.J.; de Brune, A.P.; Arends, J.W.; Hoogenboom, H.R.; Hufton, S.E. Target validation for genomics using peptide-specific phage antibodies: A study of five gene products overexpressed in colorectal cancer. Int. J. Cancer 2002, 101, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Szczyrba, J.; Nolte, E.; Hart, M.; Doll, C.; Wach, S.; Taubert, H.; Keck, B.; Kremmer, E.; Stohr, R.; Hartmann, A.; et al. Identification of ZNF217, hnRNP-K, VEGF-A and IPO7 as targets for microRNAs that are downregulated in prostate carcinoma. Int. J. Cancer 2013, 132, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Losada, A. Cohesin in cancer: Chromosome segregation and beyond. Nat. Rev. Cancer 2014, 14, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Sivula, A.; Talvensaari-Mattila, A.; Lundin, J.; Joensuu, H.; Haglund, C.; Ristimaki, A.; Turpeenniemi-Hujanen, T. Association of cyclooxygenase-2 and matrix metalloproteinase-2 expression in human breast cancer. Breast Cancer Res. Treat. 2005, 89, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Anedchenko, E.A.; Dmitriev, A.A.; Krasnov, G.S.; Kondrat’eva, T.T.; Kopantsev, E.P.; Vinogradova, T.V.; Zinov’eva, M.V.; Zborovskaia, I.B.; Polotskii, B.E.; Sakharova, O.V.; et al. Down-regulation of RBSP3/CTDSPL, NPRL2/G21, RASSF1A, ITGA9, HYAL1 and HYAL2 genes in non-small cell lung cancer. Mol. Biol. (Mosk) 2008, 42, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Xavier, P.; Pros, E.; Bonastre, E.; Moran, S.; Aza, A.; Grana, O.; Gomez-Lopez, G.; Derdak, S.; Dabad, M.; Esteve-Codina, A.; et al. Genomic and Molecular Screenings Identify Different Mechanisms for Acquired Resistance to MET Inhibitors in Lung Cancer Cells. Mol. Cancer Ther. 2017, 16, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Boss, J.M.; Hu, S.X.; Xu, H.J.; Blanck, G. Apoptosis-independent retinoblastoma protein rescue of HLA class II messenger RNA IFN-gamma inducibility in non-small cell lung carcinoma cells. Lack of surface class II expression associated with a specific defect in HLA-DRA induction. J. Immunol. 1996, 156, 2495–2502. [Google Scholar] [PubMed]
- Pino, I.; Pio, R.; Toledo, G.; Zabalegui, N.; Vicent, S.; Rey, N.; Lozano, M.D.; Torre, W.; Garcia-Foncillas, J.; Montuenga, L.M. Altered patterns of expression of members of the heterogeneous nuclear ribonucleoprotein (hnRNP) family in lung cancer. Lung Cancer 2003, 41, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Oyewumi, M.O.; Manickavasagam, D.; Novak, K.; Wehrung, D.; Paulic, N.; Moussa, F.M.; Sondag, G.R.; Safadi, F.F. Osteoactivin (GPNMB) ectodomain protein promotes growth and invasive behavior of human lung cancer cells. Oncotarget 2016, 7, 13932–13944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieniasz, M.; Oszajca, K.; Eusebio, M.; Kordiak, J.; Bartkowiak, J.; Szemraj, J. The positive correlation between gene expression of the two angiogenic factors: VEGF and BMP-2 in lung cancer patients. Lung Cancer 2009, 66, 319–326. [Google Scholar] [CrossRef]
- Zhan, W.; Wang, W.; Han, T.; Xie, C.; Zhang, T.; Gan, M.; Wang, J.B. COMMD9 promotes TFDP1/E2F1 transcriptional activity via interaction with TFDP1 in non-small cell lung cancer. Cell Signal. 2017, 30, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Tokumoto, H. Analysis of HLA-DRB1-related alleles in Japanese patients with lung cancer—relationship to genetic susceptibility and resistance to lung cancer. J. Cancer Res. Clin. Oncol. 1998, 124, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Mura, M.; Hopkins, T.G.; Michael, T.; Abd-Latip, N.; Weir, J.; Aboagye, E.; Mauri, F.; Jameson, C.; Sturge, J.; Gabra, H.; et al. LARP1 post-transcriptionally regulates mTOR and contributes to cancer progression. Oncogene 2015, 34, 5025–5036. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Lee, Y.J.; Hung, H.H.; Tseng, W.P.; Tu, C.C.; Lee, H.; Wu, W.J. ZAK inhibits human lung cancer cell growth via ERK and JNK activation in an AP-1-dependent manner. Cancer Sci. 2010, 101, 1374–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, G.Z.; Zhou, R.L.; Zhang, Q.Y.; Zhang, Y.; Liu, J.J.; Rui, J.A.; Wei, X.; Ye, D.X. Molecular cloning and characterization of LAPTM4B, a novel gene upregulated in hepatocellular carcinoma. Oncogene 2003, 22, 5060–5069. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Cao, L.; Zheng, S. CAPZA1 modulates EMT by regulating actin cytoskeleton remodelling in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 13. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Yamamoto, H.; Takemasa, I.; Yamada, D.; Uemura, M.; Wada, H.; Kobayashi, S.; Marubashi, S.; Eguchi, H.; Tanemura, M.; et al. PLOD2 induced under hypoxia is a novel prognostic factor for hepatocellular carcinoma after curative resection. Liver Int. 2012, 32, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xu, L.; Su, T.; Ke, Z.; Peng, Z.; Zhang, N.; Peng, S.; Zhang, Q.; Liu, G.; Wei, G.; et al. Autocrine STIP1 signaling promotes tumor growth and is associated with disease outcome in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2017, 493, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, M.; Huether, A.; Sutter, A.P.; Baradari, V.; Schuppan, D.; Scherubl, H. Blockade of IGF-1 receptor tyrosine kinase has antineoplastic effects in hepatocellular carcinoma cells. Biochem. Pharmacol. 2006, 71, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sun, H.C.; Wang, W.Q.; Zhang, Q.B.; Zhuang, P.Y.; Xiong, Y.Q.; Zhu, X.D.; Xu, H.X.; Kong, L.Q.; Wu, W.Z.; et al. Sorafenib down-regulates expression of HTATIP2 to promote invasiveness and metastasis of orthotopic hepatocellular carcinoma tumors in mice. Gastroenterology 2012, 143, 1641–1649. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, J.; Huan, L.; Lian, J.; Bao, C.; Li, Y.; Ge, C.; Li, J.; Yao, M.; Liang, L.; et al. GNAI3 inhibits tumor cell migration and invasion and is post-transcriptionally regulated by miR-222 in hepatocellular carcinoma. Cancer Lett. 2015, 356, 978–984. [Google Scholar] [CrossRef]
- Zheng, Y.; Gery, S.; Sun, H.; Shacham, S.; Kauffman, M.; Koeffler, H.P. KPT-330 inhibitor of XPO1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2014, 74, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.H.; Cheng, T.Y.; Chen, T.Y.; Chang, K.M.; Chuang, V.P.; Kao, K.J. Plasmalemmal Vesicle Associated Protein (PLVAP) as a therapeutic target for treatment of hepatocellular carcinoma. BMC Cancer 2014, 14, 815. [Google Scholar] [CrossRef] [PubMed]
- Bangoura, G.; Liu, Z.S.; Qian, Q.; Jiang, C.Q.; Yang, G.F.; Jing, S. Prognostic significance of HIF-2alpha/EPAS1 expression in hepatocellular carcinoma. World J. Gastroenterol. 2007, 13, 3176–3182. [Google Scholar] [CrossRef] [PubMed]
- Bejnordi, B.E.; Veta, M.; van Diest, P.J.; van Ginneken, B.; Karssemeijer, N.; Litjens, G.; van der Laak, J.; the CAMELYON16 Consortium; Hermsen, M.; Manson, Q.F.; et al. Diagnostic Assessment of Deep Learning Algorithms for Detection of Lymph Node Metastases in Women With Breast Cancer. JAMA 2017, 318, 2199–2210. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | LUAD | LIHC |
|---|---|---|
| Sample size | 316 | 358 |
| Age at diagnosis: median (range) | 66 (39–88) | 61 (16–90) |
| Follow-up: median (range) | 6.03 (0–214.77) | 19.25 (0–120.73) |
| Vital status: n (%) | ||
| Alive | 213 (67.4%) | 233 (65.0%) |
| Deceased | 103 (32.6%) | 125 (35.0%) |
| Sex: n (%) | ||
| Male | 144 (45.6%) | 242 (67.6%) |
| Female | 172 (54.4%) | 116 (32.4%) |
| Cancer stage: n (%) | ||
| I | 180 (57.0%) | 166 (46.4%) |
| II | 69 (18.7%) | 82 (22.9%) |
| III | 41 (13%) | 83 (23.2%) |
| IV | 21 (6.6%) | 5 (1.4%) |
| NA | 5 (1.6%) | 22 (6.1%) |
| Gene | Evidence | PMID |
|---|---|---|
| HYAL2 | Real-time PCR studies showed that HYAL2 genes were down regulated in non-small cell lung cancer [35]. | 19140316 |
| MAPK1IP1L | MAPK1IP1L gene was found to be related with acquired resistance to MET inhibitors in lung cancer cells [36]. | 28396363 |
| HLA-DRA | Lack of surface class II expression was found to be associated with a specific defect in HLA-DRA induction in non-small cell lung carcinoma cells [37]. | 8786310 |
| HNRNPK | Higher levels of hnRNP mRNAs were found in SCLC as compared to NSCLC. hnRNP K protein localization varied with cellular confluence [38]. | 12871776 |
| GPNMB | Osteoactivin (GPNMB) ectodomain protein was shown to promote growth and invasive behavior of human lung cancer cells [39]. | 26883195 |
| BMP2 | Positive correlation was found between gene expressions of two angiogenic factors, VEGF and BMP-2, in lung cancer patients [40]. | 19324447 |
| COMMD6 | COMMD9 was demonstrated to promote TFDP1/E2F1 transcriptional activity via interaction with TFDP1 in non-small cell lung cancer [41]. | 27871936 |
| HLA-DRB1 | Lung cancer patients in Japan showed an increased frequency of HLA-DRB1*0901 and a decreased frequency of HLA-DRB1*1302 and DRB1*14-related alleles when compared to the other subjects [42]. | 9808426 |
| LARP1 | LARP1 post-transcriptionally regulates mTOR and contributes to cancer progression [43]. | 25531318 |
| ZAK | ZAK inhibits human lung cancer cell growth via ERK and JNK activation in an AP-1-dependent manner [44]. | 20331627 |
| Gene | Evidence | PMID |
|---|---|---|
| LAPTM4B | LAPTM4B is a potential proto-oncogene, whose overexpression is involved in carcinogenesis and progression of HCC [45]. | 12902989 |
| CAPZA1 | CAPZA1 expression levels were negatively correlated with the biological characteristics of primary HCC and patient prognosis [46]. | 28093067 |
| PLOD2 | PLOD2 expression was identified as a significant, independent factor of poor prognosis for HCC patients [47]. | 22098155 |
| STIP1 | STIP1 was upregulated in HCC and associated with poor clinical prognosis [48]. | 28887036 |
| IGF1 | Inhibition of IGF-1R tyrosine kinase (IGF-1R-TK) by NVP-AEW541 induces growth inhibition, apoptosis and cell cycle arrest in human HCC cell lines without accompanying cytotoxicity [49]. | 16530734 |
| HTATIP2 | HepG2 cells that expressed transgenic HTATIP2 formed more invasive tumors in mice following administration of sorafenib. Sorafenib therapy prolonged recurrence-free survival in patients who expressed lower levels of HTATIP2 compared with higher levels [50]. | 22922424 |
| GNAI3 | GNAI3 inhibits tumor cell migration and invasion and is post-transcriptionally regulated by miR-222 in hepatocellular carcinoma [51]. | 25444921 |
| XPO1 | Exportin-1 (XPO1, CRM1) mediates the nuclear export of several key growth regulatory and tumor suppressor proteins [52]. | 25030088 |
| PLVAP | PLVAP was identified as a gene specifically expressed in vascular endothelial cells of HCC but not in non-tumorous liver tissues [53]. | 25376302 |
| EPAS1 | HIF-2alpha/EPAS1 expression may play an important role in tumor progression and prognosis of HCC [54]. | 17589895 |
| LUAD | LIHC | ||
|---|---|---|---|
| Feature Name | Adjusted p Value | Feature Name | Adjusted p Value |
| Mean_Identifyeosinprimarycytoplasm_Texture_Correlation_maskosingray_3_00 | 1.16 × 10−4 | StDev_Identifyhemasub2_Texture_DifferenceEntropy_ImageAfterMath_3_02 | 9.46 × 10−6 |
| Median_Identifyeosinprimarycytoplasm_Texture_Correlation_maskosingray_3_00 | 1.59 × 10−4 | StDev_Identifyhemasub2_Texture_SumEntropy_ImageAfterMath_3_00 | 9.46 × 10−6 |
| StDev_Identifyeosinprimarycytoplasm_Texture_Correlation_maskosingray_3_00 | 1.59 × 10−4 | StDev_Identifyhemasub2_Texture_SumEntropy_ImageAfterMath_3_02 | 1.85 × 10−5 |
| StDev_Identifyhemasub2_AreaShape_Orientation | 1.59 × 10−4 | StDev_Identifyhemasub2_Texture_DifferenceEntropy_ImageAfterMath_3_00 | 2.92 × 10−5 |
| StDev_Identifyhemasub2_AreaShape_Zernike_6_6 | 1.05 × 10−4 | StDev_Identifyhemasub2_Texture_SumEntropy_ImageAfterMath_3_01 | 3.64 × 10−5 |
| StDev_Identifyhemasub2_AreaShape_Zernike_9_1 | 1.59 × 10−4 | StDev_Identifyhemasub2_Texture_DifferenceEntropy_ImageAfterMath_3_01 | 4.08 × 10−5 |
| StDev_Identifyhemasub2_AreaShape_Zernike_9_9 | 1.59 × 10−4 | StDev_Identifyhemasub2_Texture_DifferenceEntropy_ImageAfterMath_3_03 | 4.82 × 10−5 |
| StDev_Identifyhemasub2_Texture_DifferenceEntropy_ImageAfterMath_3_03 | 1.64 × 10−4 | StDev_Identifyhemasub2_Texture_SumEntropy_ImageAfterMath_3_03 | 9.24 × 10−5 |
| StDev_Identifyhemasub2_Texture_SumEntropy_ImageAfterMath_3_01 | 1.64 × 10−4 | Granularity_2_ImageAfterMath | 1.07 × 10−4 |
| Texture_Correlation_maskosingray_3_00 | 1.59 × 10−4 | ||
| Granularity_15_ImageAfterMath | 7.66 × 10−4 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, T.; Wu, M.; Ma, S. Examination of Independent Prognostic Power of Gene Expressions and Histopathological Imaging Features in Cancer. Cancers 2019, 11, 361. https://doi.org/10.3390/cancers11030361
Zhong T, Wu M, Ma S. Examination of Independent Prognostic Power of Gene Expressions and Histopathological Imaging Features in Cancer. Cancers. 2019; 11(3):361. https://doi.org/10.3390/cancers11030361
Chicago/Turabian StyleZhong, Tingyan, Mengyun Wu, and Shuangge Ma. 2019. "Examination of Independent Prognostic Power of Gene Expressions and Histopathological Imaging Features in Cancer" Cancers 11, no. 3: 361. https://doi.org/10.3390/cancers11030361
APA StyleZhong, T., Wu, M., & Ma, S. (2019). Examination of Independent Prognostic Power of Gene Expressions and Histopathological Imaging Features in Cancer. Cancers, 11(3), 361. https://doi.org/10.3390/cancers11030361