Tailoring Ovarian Cancer Treatment: Implications of BRCA1/2 Mutations

Abstract
:1. Introduction
1.1. BRCA1/2 Mutations, HR Genes and High Grade Serous Ovarian Cancer
1.2. Ovarian Cancer Screening and Prevention in BRCA1/2 Mutation Carriers
2. Treatment of Ovarian Cancer & Implications of BRCA1/2 Status
2.1. Surgery
2.2. Intraperitoneal (IP) Chemotherapy
2.3. Intravenous (IV) Chemotherapy
2.3.1. Platinum
2.3.2. Non Platinum Chemotherapy
Taxanes
Trabectedin
Pegylated Liposomal Doxorubicin (PLD)
2.4. Targeted Treatment
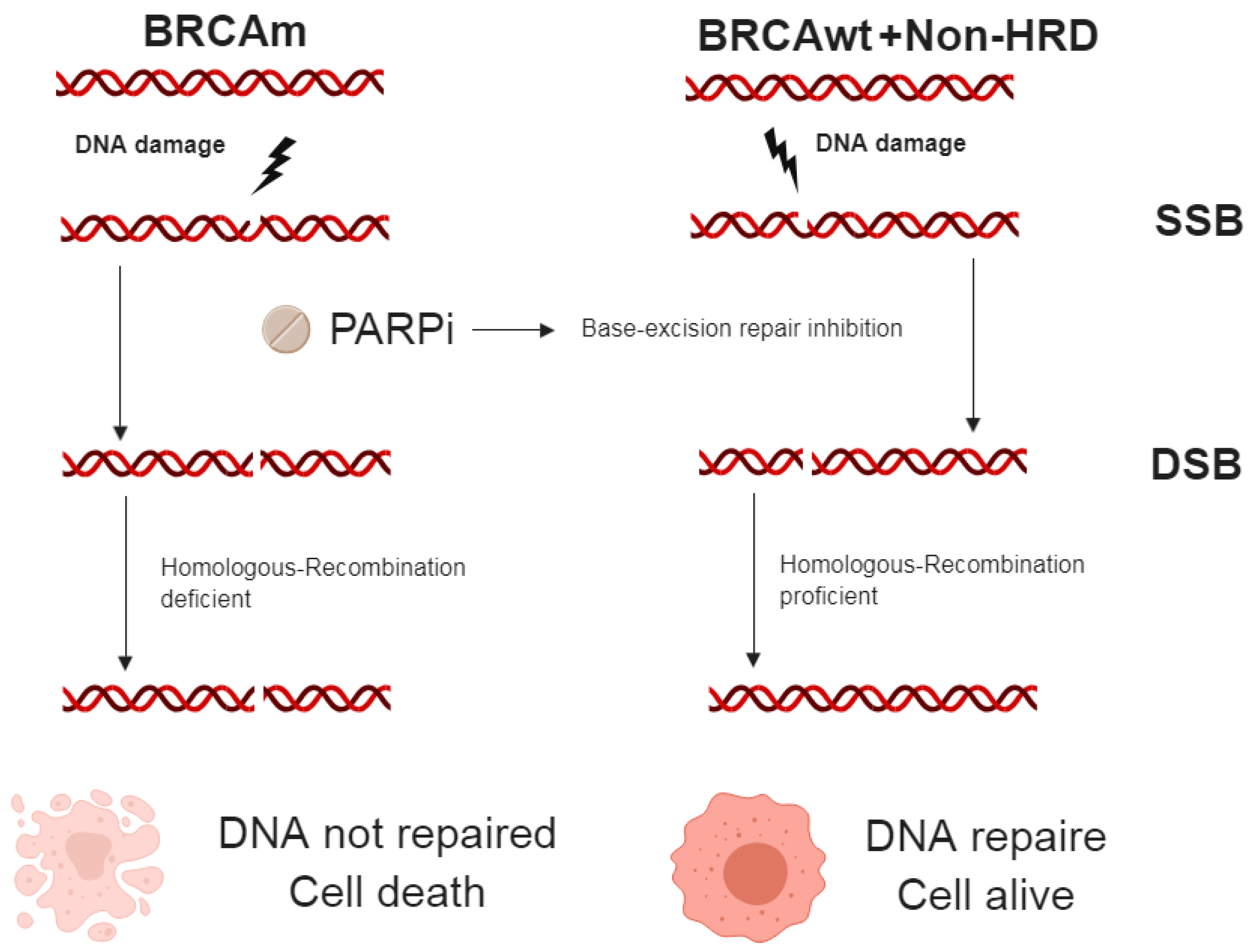
2.4.1. PARP inhibitor (PARPi)
Single Agent Treatment
Maintenance Treatment
First Line
Platinum Sensitive Relapse
Combination Treatments
2.4.2. Antiangiogenic Treatment
2.4.3. Immunotherapy
2.4.4. Other Agents Under Investigation
Cell Cycle Modulators
3. BRCA Mutations and Treatment Resistance
3.1. Increased HR Activity
3.2. Non-Homologous End Joining (NHEJ) Factors and Shieldin Complex
3.3. Drug Efflux Pump
3.4. Stromal Reactions
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Noone, A.M.; Howlader, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2015; National Cancer Institute: Bethesda, MD, USA, 2015. Available online: https://seer.cancer.gov/csr/1975_2015/ (accessed on 1 June 2019).
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Ramalingam, P. Morphologic, Immunophenotypic, and Molecular Features of Epithelial Ovarian Cancer. Oncology 2016, 30, 166–176. [Google Scholar] [PubMed]
- Kurman, R.J.; Carcangiu, M.L.; Herrington, C.S. World Health Organisation Classification of Tumours of the Female Reproductive Organs; International Agency for Research on Cancer: Lyon, France, 2014. [Google Scholar]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA A Cancer J. Clin. 2018. [Google Scholar] [CrossRef]
- Friedlander, M.; Trimble, E.; Tinker, A.; Alberts, D.; Avall-Lundqvist, E.; Brady, M.; Harter, P.; Pignata, S.; Pujade-Lauraine, E.; Sehouli, J.; et al. Clinical trials in recurrent ovarian cancer. Int. J. Gynecol. Cancer 2011, 21, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Colombo, P.E.; Fabbro, M.; Theillet, C.; Bibeau, F.; Rouanet, P.; Ray-Coquard, I. Sensitivity and resistance to treatment in the primary management of epithelial ovarian cancer. Crit. Rev. Oncol./Hematol. 2014, 89, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Gore, M. Treatment of recurrent ovarian cancer relapsing 6-12 months post platinum-based chemotherapy. Crit. Rev. Oncol./Hematol. 2007, 64, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.K.; Pujade-Lauraine, E.; Aoki, D.; Mirza, M.R.; Lorusso, D.; Oza, A.M.; du Bois, A.; Vergote, I.; Reuss, A.; Bacon, M.; et al. Fifth Ovarian Cancer Consensus Conference of the Gynecologic Cancer InterGroup: Recurrent disease. Ann. Oncol. 2017, 28, 727–732. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Kurman, R.J.; Nakayama, K.; Velculescu, V.E.; Vogelstein, B.; Kinzler, K.W.; Papadopoulos, N.; Shih Ie, M. Low-grade serous carcinomas of the ovary contain very few point mutations. J. Pathol. 2012, 226, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Shanmughapriya, S.; Nachiappan, V.; Natarajaseenivasan, K. BRCA1 and BRCA2 mutations in the ovarian cancer population across race and ethnicity: Special reference to Asia. Oncology 2013, 84, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Struewing, J.P.; Hartge, P.; Wacholder, S.; Baker, S.M.; Berlin, M.; McAdams, M.; Timmerman, M.M.; Brody, L.C.; Tucker, M.A. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N. Engl. J. Med. 1997, 336, 1401–1408. [Google Scholar] [CrossRef]
- Bayraktar, S.; Jackson, M.; Gutierrez-Barrera, A.M.; Liu, D.; Meric-Bernstam, F.; Brandt, A.; Woodson, A.; Litton, J.; Lu, K.H.; Valero, V.; et al. Genotype-Phenotype Correlations by Ethnicity and Mutation Location in BRCA Mutation Carriers. Breast J. 2015, 21, 260–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welcsh, P.L.; King, M.C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Miki, Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004, 95, 866–871. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Lu, Y.; Jin, K.; Cheng, H.; Guo, M.; Liu, Z.; Long, J.; Liu, C.; Ni, Q.; Yu, X. Pancreatic cancer: BRCA mutation and personalized treatment. Expert Rev. Anticancer Ther. 2015, 15, 1223–1231. [Google Scholar] [CrossRef]
- Taneja, S.S. Re: Germline Mutations in ATM and BRCA1/2 Distinguish Risk for Lethal and Indolent Prostate Cancer and are Associated with Early Age at Death. J. Urol. 2017, 198, 742–743. [Google Scholar] [CrossRef] [PubMed]
- Friedenson, B. BRCA1 and BRCA2 pathways and the risk of cancers other than breast or ovarian. MedGenMed 2005, 7, 60. [Google Scholar] [PubMed]
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Aarnio, M.; Sankila, R.; Pukkala, E.; Salovaara, R.; Aaltonen, L.A.; de la Chapelle, A.; Peltomaki, P.; Mecklin, J.P.; Jarvinen, H.J. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int. J. Cancer 1999, 81, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Woolderink, J.M.; De Bock, G.H.; de Hullu, J.A.; Hollema, H.; Zweemer, R.P.; Slangen, B.F.M.; Gaarenstroom, K.N.; van Beurden, M.; van Doorn, H.C.; Sijmons, R.H.; et al. Characteristics of Lynch syndrome associated ovarian cancer. Gynecol. Oncol. 2018, 150, 324–330. [Google Scholar] [CrossRef]
- Bewtra, C.; Watson, P.; Conway, T.; Read-Hippee, C.; Lynch, H.T. Hereditary ovarian cancer: A clinicopathological study. Int. J. Gynecol. Pathol. 1992, 11, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Ready, K.; Chen, H.; Gutierrez-Barrera, A.; Etzel, C.J.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Le-Petross, H.; Lu, K.; Hortobagyi, G.N.; et al. Earlier age of onset of BRCA mutation-related cancers in subsequent generations. Cancer 2012, 118, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Peng, H.L.; Zhao, X.; Zhang, L.; Hwang, W.T. Effects of BRCA1- and BRCA2-related mutations on ovarian and breast cancer survival: A meta-analysis. Clin. Cancer Res. 2015, 21, 211–220. [Google Scholar] [CrossRef]
- Kotsopoulos, J.; Rosen, B.; Fan, I.; Moody, J.; McLaughlin, J.R.; Risch, H.; May, T.; Sun, P.; Narod, S.A. Ten-year survival after epithelial ovarian cancer is not associated with BRCA mutation status. Gynecol. Oncol. 2016, 140, 42–47. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef]
- Candido-dos-Reis, F.J.; Song, H.; Goode, E.L.; Cunningham, J.M.; Fridley, B.L.; Larson, M.C.; Alsop, K.; Dicks, E.; Harrington, P.; Ramus, S.J.; et al. Germline mutation in BRCA1 or BRCA2 and ten-year survival for women diagnosed with epithelial ovarian cancer. Clin. Cancer Res. 2015, 21, 652–657. [Google Scholar] [CrossRef]
- Hyman, D.M.; Zhou, Q.; Iasonos, A.; Grisham, R.N.; Arnold, A.G.; Phillips, M.F.; Bhatia, J.; Levine, D.A.; Aghajanian, C.; Offit, K.; et al. Improved survival for BRCA2-associated serous ovarian cancer compared with both BRCA-negative and BRCA1-associated serous ovarian cancer. Cancer 2012, 118, 3703–3709. [Google Scholar] [CrossRef]
- Liu, G.; Yang, D.; Sun, Y.; Shmulevich, I.; Xue, F.; Sood, A.K.; Zhang, W. Differing clinical impact of BRCA1 and BRCA2 mutations in serous ovarian cancer. Pharmacogenomics 2012, 13, 1523–1535. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shmulevich, I.; Sood, A.K.; Zhang, W. Association between BRCA2 but not BRCA1 Mutations and Beneficial Survival, Chemotherapy Sensitivity, and Gene Mutator Phenotype in Patients with Ovarian Cancer. JAMA J. Am. Med. Assoc. 2011, 306, 1557–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buys, S.S.; Partridge, E.; Black, A.; Johnson, C.C.; Lamerato, L.; Isaacs, C.; Reding, D.J.; Greenlee, R.T.; Yokochi, L.A.; Kessel, B.; et al. Effect of screening on ovarian cancer mortality: The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA 2011, 305, 2295–2303. [Google Scholar] [CrossRef]
- Lewis, K.E.; Lu, K.H.; Klimczak, A.M.; Mok, S.C. Recommendations and Choices for BRCA Mutation Carriers at Risk for Ovarian Cancer: A Complicated Decision. Cancers 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, J.R.; Risch, H.A.; Lubinski, J.; Moller, P.; Ghadirian, P.; Lynch, H.; Karlan, B.; Fishman, D.; Rosen, B.; Neuhausen, S.L.; et al. Reproductive risk factors for ovarian cancer in carriers of BRCA1 or BRCA2 mutations: A case-control study. Lancet. Oncol. 2007, 8, 26–34. [Google Scholar] [CrossRef]
- Finch, A.P.; Lubinski, J.; Moller, P.; Singer, C.F.; Karlan, B.; Senter, L.; Rosen, B.; Maehle, L.; Ghadirian, P.; Cybulski, C.; et al. Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. J. Clin. Oncol. 2014, 32, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Eleje, G.U.; Eke, A.C.; Ezebialu, I.U.; Ikechebelu, J.I.; Ugwu, E.O.; Okonkwo, O.O. Risk-reducing bilateral salpingo-oophorectomy in women with BRCA1 or BRCA2 mutations. Cochrane Database Syst. Rev. 2018, 8, CD012464. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Rookus, M.; Andrieu, N.; Brohet, R.; Chang-Claude, J.; Peock, S.; Cook, M.; Evans, D.G.; Eeles, R.; Nogues, C.; et al. Reproductive and hormonal factors, and ovarian cancer risk for BRCA1 and BRCA2 mutation carriers: Results from the International BRCA1/2 Carrier Cohort Study. Cancer Epidemiol. Biomark. Prev. 2009, 18, 601–610. [Google Scholar] [CrossRef]
- Cibula, D.; Widschwendter, M.; Majek, O.; Dusek, L. Tubal ligation and the risk of ovarian cancer: Review and meta-analysis. Hum. Reprod. Update 2011, 17, 55–67. [Google Scholar] [CrossRef]
- Bristow, R.E.; Tomacruz, R.S.; Armstrong, D.K.; Trimble, E.L.; Montz, F.J. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: A meta-analysis. J. Clin. Oncol. 2002, 20, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Long, K.C.; Tanner, E.J.; Grisham, R.N.; Arnold, A.G.; Bhatia, J.; Phillips, M.F.; Spriggs, D.R.; Soslow, R.A.; Kauff, N.D.; et al. Outcomes of primary surgical cytoreduction in patients with BRCA-associated high-grade serous ovarian carcinoma. Gynecol. Oncol. 2012, 126, 224–228. [Google Scholar] [CrossRef]
- Petrillo, M.; Marchetti, C.; De Leo, R.; Musella, A.; Capoluongo, E.; Paris, I.; Benedetti Panici, P.; Scambia, G.; Fagotti, A. BRCA mutational status, initial disease presentation, and clinical outcome in high-grade serous advanced ovarian cancer: A multicenter study. Am. J. Obstet. Gynecol. 2017, 217, 334.e331–334.e339. [Google Scholar] [CrossRef]
- Reyes, M.C.; Arnold, A.G.; Kauff, N.D.; Levine, D.A.; Soslow, R.A. Invasion patterns of metastatic high-grade serous carcinoma of ovary or fallopian tube associated with BRCA deficiency. Mod. Pathol. 2014, 27, 1405. [Google Scholar] [CrossRef] [PubMed]
- Bois, A.D.; Vergote, I.; Ferron, G.; Reuss, A.; Meier, W.; Greggi, S.; Jensen, P.T.; Selle, F.; Guyon, F.; Pomel, C.; et al. Randomized controlled phase III study evaluating the impact of secondary cytoreductive surgery in recurrent ovarian cancer: AGO DESKTOP III/ENGOT ov20. J. Clin. Oncol. 2017, 35, 5501. [Google Scholar] [CrossRef]
- Coleman, R.L.; Enserro, D.; Spirtos, N.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Kim, B.; Fujiwara, K.; Walker, J.L.; Flynn, P.J.; et al. A phase III randomized controlled trial of secondary surgical cytoreduction (SSC) followed by platinum-based combination chemotherapy (PBC), with or without bevacizumab (B) in platinum-sensitive, recurrent ovarian cancer (PSOC): A NRG Oncology/Gynecol. Oncol. Group (GOG) study. J. Clin. Oncol. 2018, 36, 5501. [Google Scholar]
- Harter, P.; du Bois, A.; Hahmann, M.; Hasenburg, A.; Burges, A.; Loibl, S.; Gropp, M.; Huober, J.; Fink, D.; Schroder, W.; et al. Surgery in recurrent ovarian cancer: The Arbeitsgemeinschaft Gynaekologische Onkologie (AGO) DESKTOP OVAR trial. Ann. Surg. Oncol. 2006, 13, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Harter, P.; Sehouli, J.; Reuss, A.; Hasenburg, A.; Scambia, G.; Cibula, D.; Mahner, S.; Vergote, I.; Reinthaller, A.; Burges, A.; et al. Prospective validation study of a predictive score for operability of recurrent ovarian cancer: The Multicenter Intergroup Study DESKTOP II. A project of the AGO Kommission OVAR, AGO Study Group, NOGGO, AGO-Austria, and MITO. Int. J. Gynecol. Cancer 2011, 21, 289–295. [Google Scholar] [CrossRef]
- Chi, D.S.; McCaughty, K.; Diaz, J.P.; Huh, J.; Schwabenbauer, S.; Hummer, A.J.; Venkatraman, E.S.; Aghajanian, C.; Sonoda, Y.; Abu-Rustum, N.R.; et al. Guidelines and selection criteria for secondary cytoreductive surgery in patients with recurrent, platinum-sensitive epithelial ovarian carcinoma. Cancer 2006, 106, 1933–1939. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.J.; Chi, D.S.; Sehouli, J.; Trope, C.G.; Jiang, R.; Ayhan, A.; Cormio, G.; Xing, Y.; Breitbach, G.P.; Braicu, E.I.; et al. A risk model for secondary cytoreductive surgery in recurrent ovarian cancer: An evidence-based proposal for patient selection. Ann. Surg. Oncol. 2012, 19, 597–604. [Google Scholar] [CrossRef]
- Gourley, C.; Michie, C.O.; Roxburgh, P.; Yap, T.A.; Harden, S.; Paul, J.; Ragupathy, K.; Todd, R.; Petty, R.; Reed, N.; et al. Increased incidence of visceral metastases in scottish patients with BRCA1/2-defective ovarian cancer: An extension of the ovarian BRCAness phenotype. J. Clin. Oncol. 2010, 28, 2505–2511. [Google Scholar] [CrossRef]
- Howell, S.B.; Pfeifle, C.L.; Wung, W.E.; Olshen, R.A.; Lucas, W.E.; Yon, J.L.; Green, M. Intraperitoneal cisplatin with systemic thiosulfate protection. Ann. Intern. Med. 1982, 97, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Alberts, D.S.; Liu, P.Y.; Hannigan, E.V.; O’Toole, R.; Williams, S.D.; Young, J.A.; Franklin, E.W.; Clarke-Pearson, D.L.; Malviya, V.K.; DuBeshter, B. Intraperitoneal cisplatin plus intravenous cyclophosphamide versus intravenous cisplatin plus intravenous cyclophosphamide for stage III ovarian cancer. N. Engl. J. Med. 1996, 335, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Markman, M.; Bundy, B.N.; Alberts, D.S.; Fowler, J.M.; Clark-Pearson, D.L.; Carson, L.F.; Wadler, S.; Sickel, J. Phase III trial of standard-dose intravenous cisplatin plus paclitaxel versus moderately high-dose carboplatin followed by intravenous paclitaxel and intraperitoneal cisplatin in small-volume stage III ovarian carcinoma: An intergroup study of the Gynecol. Oncol. Group, Southwestern Oncology Group, and Eastern Cooperative Oncology Group. J. Clin. Oncol. 2001, 19, 1001–1007. [Google Scholar] [PubMed]
- Tewari, D.; Java, J.J.; Salani, R.; Armstrong, D.K.; Markman, M.; Herzog, T.; Monk, B.J.; Chan, J.K. Long-term survival advantage and prognostic factors associated with intraperitoneal chemotherapy treatment in advanced ovarian cancer: A Gynecol. Oncol. group study. J. Clin. Oncol. 2015, 33, 1460–1466. [Google Scholar] [CrossRef]
- Elit, L.; Oliver, T.K.; Covens, A.; Kwon, J.; Fung, M.F.; Hirte, H.W.; Oza, A.M. Intraperitoneal chemotherapy in the first-line treatment of women with stage III epithelial ovarian cancer: A systematic review with metaanalyses. Cancer 2007, 109, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Jaaback, K.; Johnson, N.; Lawrie, T.A. Intraperitoneal chemotherapy for the initial management of primary epithelial ovarian cancer. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, L.M.; Benham-Hutchins, M.; Herzog, T.J.; Hsu, C.H.; Malone, D.C.; Skrepnek, G.H.; Slack, M.K.; Alberts, D.S. A meta-analysis of the efficacy of intraperitoneal cisplatin for the front-line treatment of ovarian cancer. Int. J. Gynecol. Cancer 2007, 17, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Lesnock, J.L.; Darcy, K.M.; Tian, C.; Deloia, J.A.; Thrall, M.M.; Zahn, C.; Armstrong, D.K.; Birrer, M.J.; Krivak, T.C. BRCA1 expression and improved survival in ovarian cancer patients treated with intraperitoneal cisplatin and paclitaxel: A Gynecol. Oncol. Group Study. Br. J. Cancer 2013, 108, 1231–1237. [Google Scholar] [CrossRef]
- van Driel, W.J.; Koole, S.N.; Sikorska, K.; Schagen van Leeuwen, J.H.; Schreuder, H.W.R.; Hermans, R.H.M.; de Hingh, I.; van der Velden, J.; Arts, H.J.; Massuger, L.; et al. Hyperthermic Intraperitoneal Chemotherapy in Ovarian Cancer. N. Engl. J. Med. 2018, 378, 230–240. [Google Scholar] [CrossRef]
- Safra, T.; Grisaru, D.; Inbar, M.; Abu-Abeid, S.; Dayan, D.; Matceyevsky, D.; Weizman, A.; Klausner, J.M. Cytoreduction surgery with hyperthermic intraperitoneal chemotherapy in recurrent ovarian cancer improves progression-free survival, especially in BRCA-positive patients—A case-control study. J. Surg. Oncol. 2014, 110, 661–665. [Google Scholar] [CrossRef]
- van den Tempel, N.; Odijk, H.; van Holthe, N.; Naipal, K.; Raams, A.; Eppink, B.; van Gent, D.C.; Hardillo, J.; Verduijn, G.M.; Drooger, J.C.; et al. Heat-induced BRCA2 degradation in human tumours provides rationale for hyperthermia-PARP-inhibitor combination therapies. Int. J. Hyperther. 2018, 34, 407–414. [Google Scholar] [CrossRef]
- Krawczyk, P.M.; Eppink, B.; Essers, J.; Stap, J.; Rodermond, H.; Odijk, H.; Zelensky, A.; van Bree, C.; Stalpers, L.J.; Buist, M.R.; et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc. Natl. Acad. Sci. USA 2011, 108, 9851–9856. [Google Scholar] [CrossRef] [Green Version]
- Schaaf, L.; van der Kuip, H.; Zopf, W.; Winter, S.; Munch, M.; Murdter, T.E.; Thon, K.P.; Steurer, W.; Aulitzky, W.E.; Ulmer, C. A Temperature of 40 degrees C Appears to be a Critical Threshold for Potentiating Cytotoxic Chemotherapy In Vitro and in Peritoneal Carcinomatosis Patients Undergoing HIPEC. Ann. Surg. Oncol. 2015, 22, S758–S765. [Google Scholar] [CrossRef]
- Poklar, N.; Pilch, D.S.; Lippard, S.J.; Redding, E.A.; Dunham, S.U.; Breslauer, K.J. Influence of cisplatin intrastrand crosslinking on the conformation, thermal stability, and energetics of a 20-mer DNA duplex. Proc. Natl. Acad. Sci. USA 1996, 93, 7606–7611. [Google Scholar] [CrossRef]
- Todd, R.C.; Lippard, S.J. Inhibition of transcription by platinum antitumor compounds. Metallomics 2009, 1, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A Gynecol. Oncol. Group study. J. Clin. Oncol. 2003, 21, 3194–3200. [Google Scholar] [CrossRef]
- du Bois, A.; Luck, H.J.; Meier, W.; Adams, H.P.; Mobus, V.; Costa, S.; Bauknecht, T.; Richter, B.; Warm, M.; Schroder, W.; et al. A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first-line treatment of ovarian cancer. J. Nat. Cancer Inst. 2003, 95, 1320–1329. [Google Scholar] [CrossRef]
- Pfisterer, J.; Plante, M.; Vergote, I.; du Bois, A.; Hirte, H.; Lacave, A.J.; Wagner, U.; Stahle, A.; Stuart, G.; Kimmig, R.; et al. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: An intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J. Clin. Oncol. 2006, 24, 4699–4707. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Wagner, U.; Aavall-Lundqvist, E.; Gebski, V.; Heywood, M.; Vasey, P.A.; Volgger, B.; Vergote, I.; Pignata, S.; Ferrero, A.; et al. Pegylated liposomal Doxorubicin and Carboplatin compared with Paclitaxel and Carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J. Clin. Oncol. 2010, 28, 3323–3329. [Google Scholar] [CrossRef] [PubMed]
- Parmar, M.K.; Ledermann, J.A.; Colombo, N.; du Bois, A.; Delaloye, J.F.; Kristensen, G.B.; Wheeler, S.; Swart, A.M.; Qian, W.; Torri, V.; et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: The ICON4/AGO-OVAR-2.2 trial. Lancet 2003, 361, 2099–2106. [Google Scholar] [PubMed]
- Quinn, J.E.; Carser, J.E.; James, C.R.; Kennedy, R.D.; Harkin, D.P. BRCA1 and implications for response to chemotherapy in ovarian cancer. Gynecol. Oncol. 2009, 113, 134–142. [Google Scholar] [CrossRef]
- Quinn, J.E.; James, C.R.; Stewart, G.E.; Mulligan, J.M.; White, P.; Chang, G.K.; Mullan, P.B.; Johnston, P.G.; Wilson, R.H.; Harkin, D.P. BRCA1 mRNA expression levels predict for overall survival in ovarian cancer after chemotherapy. Clin. Cancer Res. 2007, 13, 7413–7420. [Google Scholar] [CrossRef] [PubMed]
- Samouelian, V.; Maugard, C.M.; Jolicoeur, M.; Bertrand, R.; Arcand, S.L.; Tonin, P.N.; Provencher, D.M.; Mes-Masson, A.M. Chemosensitivity and radiosensitivity profiles of four new human epithelial ovarian cancer cell lines exhibiting genetic alterations in BRCA2, TGFbeta-RII, KRAS2, TP53 and/or CDNK2A. Cancer Chemother. Pharmacol. 2004, 54, 497–504. [Google Scholar] [CrossRef]
- Bartz, S.R.; Zhang, Z.; Burchard, J.; Imakura, M.; Martin, M.; Palmieri, A.; Needham, R.; Guo, J.; Gordon, M.; Chung, N.; et al. Small interfering RNA screens reveal enhanced cisplatin cytotoxicity in tumor cells having both BRCA network and TP53 disruptions. Mol. Cell. Biol. 2006, 26, 9377–9386. [Google Scholar] [CrossRef]
- Tassone, P.; Di Martino, M.T.; Ventura, M.; Pietragalla, A.; Cucinotto, I.; Calimeri, T.; Bulotta, A.; Neri, P.; Caraglia, M.; Tagliaferri, P. Loss of BRCA1 function increases the antitumor activity of cisplatin against human breast cancer xenografts in vivo. Cancer Biol. Ther. 2009, 8, 648–653. [Google Scholar] [CrossRef] [Green Version]
- Xing, D.; Orsulic, S. A mouse model for the molecular characterization of brca1-associated ovarian carcinoma. Cancer Res. 2006, 66, 8949–8953. [Google Scholar] [CrossRef]
- Tan, D.S.; Rothermundt, C.; Thomas, K.; Bancroft, E.; Eeles, R.; Shanley, S.; Ardern-Jones, A.; Norman, A.; Kaye, S.B.; Gore, M.E. “BRCAness” syndrome in ovarian cancer: A case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J. Clin. Oncol. 2008, 26, 5530–5536. [Google Scholar] [CrossRef] [PubMed]
- Vencken, P.M.; Kriege, M.; Hoogwerf, D.; Beugelink, S.; van der Burg, M.E.; Hooning, M.J.; Berns, E.M.; Jager, A.; Collee, M.; Burger, C.W.; et al. Chemosensitivity and outcome of BRCA1- and BRCA2-associated ovarian cancer patients after first-line chemotherapy compared with sporadic ovarian cancer patients. Ann. Oncol. 2011, 22, 1346–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsop, K.; Fereday, S.; Meldrum, C.; deFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Gorodnova, T.V.; Sokolenko, A.P.; Ivantsov, A.O.; Iyevleva, A.G.; Suspitsin, E.N.; Aleksakhina, S.N.; Yanus, G.A.; Togo, A.V.; Maximov, S.Y.; Imyanitov, E.N. High response rates to neoadjuvant platinum-based therapy in ovarian cancer patients carrying germ-line BRCA mutation. Cancer Lett 2015, 369, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.A.; Masson, J.Y.; McIlwraith, M.J.; Stasiak, A.Z.; Stasiak, A.; Venkitaraman, A.R.; West, S.C. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol. Cell 2001, 7, 273–282. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef]
- Barbuti, A.M.; Chen, Z.S. Paclitaxel Through the Ages of Anticancer Therapy: Exploring Its Role in Chemoresistance and Radiation Therapy. Cancers 2015, 7, 2360–2371. [Google Scholar] [CrossRef] [Green Version]
- Piccart, M.J.; Bertelsen, K.; James, K.; Cassidy, J.; Mangioni, C.; Simonsen, E.; Stuart, G.; Kaye, S.; Vergote, I.; Blom, R.; et al. Randomized intergroup trial of cisplatin-paclitaxel versus cisplatin-cyclophosphamide in women with advanced epithelial ovarian cancer: Three-year results. J. Nat. Cancer Inst. 2000, 92, 699–708. [Google Scholar] [CrossRef]
- Ten Bokkel Huinink, W.; Gore, M.; Carmichael, J.; Gordon, A.; Malfetano, J.; Hudson, I.; Broom, C.; Scarabelli, C.; Davidson, N.; Spanczynski, M.; et al. Topotecan versus paclitaxel for the treatment of recurrent epithelial ovarian cancer. J. Clin. Oncol. 1997, 15, 2183–2193. [Google Scholar] [CrossRef] [PubMed]
- Markman, M.; Blessing, J.; Rubin, S.C.; Connor, J.; Hanjani, P.; Waggoner, S. Phase II trial of weekly paclitaxel (80 mg/m2) in platinum and paclitaxel-resistant ovarian and primary peritoneal cancers: A Gynecol. Oncol. Group study. Gynecol. Oncol. 2006, 101, 436–440. [Google Scholar] [CrossRef]
- Luvero, D.; Milani, A.; Ledermann, J.A. Treatment options in recurrent ovarian cancer: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2014, 6, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S.; Kamen, B.A. The anti-angiogenic basis of metronomic chemotherapy. Nat. Rev. Cancer 2004, 4, 423–436. [Google Scholar] [CrossRef]
- Katsumata, N.; Yasuda, M.; Isonishi, S.; Takahashi, F.; Michimae, H.; Kimura, E.; Aoki, D.; Jobo, T.; Kodama, S.; Terauchi, F.; et al. Long-term results of dose-dense paclitaxel and carboplatin versus conventional paclitaxel and carboplatin for treatment of advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer (JGOG 3016): A randomised, controlled, open-label trial. Lancet Oncol. 2013, 14, 1020–1026. [Google Scholar] [CrossRef]
- Clamp, A.R.; McNeish, I.; Dean, A.; Gallardo, D.; Weon-Kim, J.; O’Donnell, D.; Hook, J.; Coyle, C.; Blagden, S.P.; Brenton, J.; et al. 929O_PRICON8: A GCIG phase III randomised trial evaluating weekly dose- dense chemotherapy integration in first-line epithelial ovarian/fallopian tube/primary peritoneal carcinoma (EOC) treatment: Results of primary progression- free survival (PFS) analysis. Ann. Oncol. 2017, 28, mdx440.039. [Google Scholar]
- Pignata, S.; Scambia, G.; Katsaros, D.; Gallo, C.; Pujade-Lauraine, E.; De Placido, S.; Bologna, A.; Weber, B.; Raspagliesi, F.; Panici, P.B.; et al. Carboplatin plus paclitaxel once a week versus every 3 weeks in patients with advanced ovarian cancer (MITO-7): A randomised, multicentre, open-label, phase 3 trial. Lancet. Oncol. 2014, 15, 396–405. [Google Scholar] [CrossRef]
- Rosenberg, P.; Andersson, H.; Boman, K.; Ridderheim, M.; Sorbe, B.; Puistola, U.; Paro, G. Randomized trial of single agent paclitaxel given weekly versus every three weeks and with peroral versus intravenous steroid premedication to patients with ovarian cancer previously treated with platinum. Acta Oncol. 2002, 41, 418–424. [Google Scholar] [CrossRef]
- Safra, T.; Rogowski, O.; Muggia, F.M. The effect of germ-line BRCA mutations on response to chemotherapy and outcome of recurrent ovarian cancer. Int. J. Gynecol. Cancer 2014, 24, 488–495. [Google Scholar] [CrossRef]
- Nientiedt, C.; Heller, M.; Endris, V.; Volckmar, A.L.; Zschabitz, S.; Tapia-Laliena, M.A.; Duensing, A.; Jager, D.; Schirmacher, P.; Sultmann, H.; et al. Mutations in BRCA2 and taxane resistance in prostate cancer. Sci. Rep. 2017, 7, 4574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriege, M.; Jager, A.; Hooning, M.J.; Huijskens, E.; Blom, J.; van Deurzen, C.H.; Bontenbal, M.; Collee, J.M.; Menke-Pluijmers, M.B.; Martens, J.W.; et al. The efficacy of taxane chemotherapy for metastatic breast cancer in BRCA1 and BRCA2 mutation carriers. Cancer 2012, 118, 899–907. [Google Scholar] [CrossRef]
- Soares, D.G.; Escargueil, A.E.; Poindessous, V.; Sarasin, A.; de Gramont, A.; Bonatto, D.; Henriques, J.A.; Larsen, A.K. Replication and homologous recombination repair regulate DNA double-strand break formation by the antitumor alkylator ecteinascidin 743. Proc. Natl. Acad. Sci. USA 2007, 104, 13062–13067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monk, B.J.; Lorusso, D.; Italiano, A.; Kaye, S.B.; Aracil, M.; Tanovic, A.; D’Incalci, M. Trabectedin as a chemotherapy option for patients with BRCA deficiency. Cancer Treat. Rev. 2016, 50, 175–182. [Google Scholar] [CrossRef]
- Monk, B.J.; Herzog, T.J.; Kaye, S.B.; Krasner, C.N.; Vermorken, J.B.; Muggia, F.M.; Pujade-Lauraine, E.; Lisyanskaya, A.S.; Makhson, A.N.; Rolski, J.; et al. Trabectedin plus pegylated liposomal Doxorubicin in recurrent ovarian cancer. J. Clin. Oncol. 2010, 28, 3107–3114. [Google Scholar] [CrossRef]
- Poveda, A.; Vergote, I.; Tjulandin, S.; Kong, B.; Roy, M.; Chan, S.; Filipczyk-Cisarz, E.; Hagberg, H.; Kaye, S.B.; Colombo, N.; et al. Trabectedin plus pegylated liposomal doxorubicin in relapsed ovarian cancer: Outcomes in the partially platinum-sensitive (platinum-free interval 6-12 months) subpopulation of OVA-301 phase III randomized trial. Ann. Oncol. 2011, 22, 39–48. [Google Scholar] [CrossRef]
- Monk, B.J.; Herzog, T.J.; Kaye, S.B.; Krasner, C.N.; Vermorken, J.B.; Muggia, F.M.; Pujade-Lauraine, E.; Park, Y.C.; Parekh, T.V.; Poveda, A.M. Trabectedin plus pegylated liposomal doxorubicin (PLD) versus PLD in recurrent ovarian cancer: Overall survival analysis. Eur. J. Cancer 2012, 48, 2361–2368. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Ghatage, P.; Parekh, T.; Henitz, E.; Knoblauch, R.; Matos-Pita, A.S.; Nieto, A.; Park, Y.C.; Cheng, P.S.; Li, W.; et al. Effect of BRCA1 and XPG mutations on treatment response to trabectedin and pegylated liposomal doxorubicin in patients with advanced ovarian cancer: Exploratory analysis of the phase 3 OVA-301 study. Ann. Oncol. 2015, 26, 914–920. [Google Scholar] [CrossRef]
- Lorusso, D.; Scambia, G.; Pignata, S.; Sorio, R.; Amadio, G.; Lepori, S.; Mosconi, A.; Pisano, C.; Mangili, G.; Maltese, G.; et al. Prospective phase II trial of trabectedin in BRCA-mutated and/or BRCAness phenotype recurrent ovarian cancer patients: The MITO 15 trial. Ann. Oncol. 2016, 27, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.A.; Patil, Y.; La-Beck, N.M. New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy. Drug Resist. Updat 2016, 29, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Wagner, U.; Marth, C.; Largillier, R.; Kaern, J.; Brown, C.; Heywood, M.; Bonaventura, T.; Vergote, I.; Piccirillo, M.C.; Fossati, R.; et al. Final overall survival results of phase III GCIG CALYPSO trial of pegylated liposomal doxorubicin and carboplatin vs paclitaxel and carboplatin in platinum-sensitive ovarian cancer patients. Br. J. Cancer 2012, 107, 588–591. [Google Scholar] [CrossRef] [Green Version]
- Gordon, A.N.; Fleagle, J.T.; Guthrie, D.; Parkin, D.E.; Gore, M.E.; Lacave, A.J. Recurrent epithelial ovarian carcinoma: A randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J. Clin. Oncol. 2001, 19, 3312–3322. [Google Scholar] [CrossRef]
- Gordon, A.N.; Tonda, M.; Sun, S.; Rackoff, W. Long-term survival advantage for women treated with pegylated liposomal doxorubicin compared with topotecan in a phase 3 randomized study of recurrent and refractory epithelial ovarian cancer. Gynecol. Oncol. 2004, 95, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pignata, S.; Scambia, G.; Ferrandina, G.; Savarese, A.; Sorio, R.; Breda, E.; Gebbia, V.; Musso, P.; Frigerio, L.; Del Medico, P.; et al. Carboplatin plus paclitaxel versus carboplatin plus pegylated liposomal doxorubicin as first-line treatment for patients with ovarian cancer: The MITO-2 randomized phase III trial. J. Clin. Oncol. 2011, 29, 3628–3635. [Google Scholar] [CrossRef] [PubMed]
- Mahner, S.; Meier, W.; du Bois, A.; Brown, C.; Lorusso, D.; Dell’Anna, T.; Cretin, J.; Havsteen, H.; Bessette, P.; Zeimet, A.G.; et al. Carboplatin and pegylated liposomal doxorubicin versus carboplatin and paclitaxel in very platinum-sensitive ovarian cancer patients: Results from a subset analysis of the CALYPSO phase III trial. Eur. J. Cancer 2015, 51, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.F.; Marsh, E.B.; Elmasri, W.; Halberstadt, S.; Vandecker, S.; Sammel, M.D.; Bradbury, A.R.; Daly, M.; Karlan, B.; Rubin, S.C. A high response rate to liposomal doxorubicin is seen among women with BRCA mutations treated for recurrent epithelial ovarian cancer. Gynecol. Oncol. 2011, 123, 486–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safra, T.; Borgato, L.; Nicoletto, M.O.; Rolnitzky, L.; Pelles-Avraham, S.; Geva, R.; Donach, M.E.; Curtin, J.; Novetsky, A.; Grenader, T.; et al. BRCA mutation status and determinant of outcome in women with recurrent epithelial ovarian cancer treated with pegylated liposomal doxorubicin. Mol. Cancer Ther. 2011, 10, 2000–2007. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Lubinski, J.; Matulonis, U.; Ang, J.E.; Gourley, C.; Karlan, B.Y.; Amnon, A.; Bell-McGuinn, K.M.; Chen, L.M.; Friedlander, M.; et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J. Clin. Oncol. 2012, 30, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Ame, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malanga, M.; Althaus, F.R. The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem. Cell Biol. 2005, 83, 354–364. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Noel, G.; Giocanti, N.; Fernet, M.; Megnin-Chanet, F.; Favaudon, V. Poly(ADP-ribose) polymerase (PARP-1) is not involved in DNA double-strand break recovery. BMC Cell Biol. 2003, 4, 7. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Domchek, S.M.; Aghajanian, C.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol. Oncol. 2016, 140, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet. Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet. Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Schelman, W.R.; Wilding, G.; Moreno, V.; Baird, R.D.; Miranda, S.; Hylands, L.; Riisnaes, R.; Forster, M.; Omlin, A.; et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. Lancet. Oncol. 2013, 14, 882–892. [Google Scholar] [CrossRef]
- Oza, A.M.; Tinker, A.V.; Oaknin, A.; Shapira-Frommer, R.; McNeish, I.A.; Swisher, E.M.; Ray-Coquard, I.; Bell-McGuinn, K.; Coleman, R.L.; O’Malley, D.M.; et al. Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: Integrated analysis of data from Study 10 and ARIEL2. Gynecol. Oncol. 2017, 147, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, K.D.; Adimi, P.; Jakobsen, A. Veliparib Monotherapy to Patients With BRCA Germ Line Mutation and Platinum-Resistant or Partially Platinum-Sensitive Relapse of Epithelial Ovarian Cancer: A Phase I/II Study. Int. J. Gynecol. Cancer 2017, 27, 1842–1849. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, J.; Moreno, V.; Gupta, A.; Kaye, S.B.; Dean, E.; Middleton, M.R.; Friedlander, M.; Gourley, C.; Plummer, R.; Rustin, G.; et al. An Adaptive Study to Determine the Optimal Dose of the Tablet Formulation of the PARP Inhibitor Olaparib. Target. Oncol. 2016, 11, 401–415. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.G.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. QUADRA: A phase 2, open-label, single-arm study to evaluate niraparib in patients (pts) with relapsed ovarian cancer (ROC) who have received ≥3 prior chemotherapy regimens. J. Clin. Oncol. 2018, 36, 5514. [Google Scholar] [CrossRef]
- Drew, Y.; Ledermann, J.; Hall, G.; Rea, D.; Glasspool, R.; Highley, M.; Jayson, G.; Sludden, J.; Murray, J.; Jamieson, D.; et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br. J. Cancer 2016, 114, 723–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; LoRusso, P.; Patel, M.R.; Domchek, S.M.; Balmana, J.; Drew, Y.; Chen, L.M.; et al. A Phase I-II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef]
- Coleman, R.L.; Sill, M.W.; Bell-McGuinn, K.; Aghajanian, C.; Gray, H.J.; Tewari, K.S.; Rubin, S.C.; Rutherford, T.J.; Chan, J.K.; Chen, A.; et al. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation—An NRG Oncology/Gynecol. Oncol. Group study. Gynecol. Oncol. 2015, 137, 386–391. [Google Scholar]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
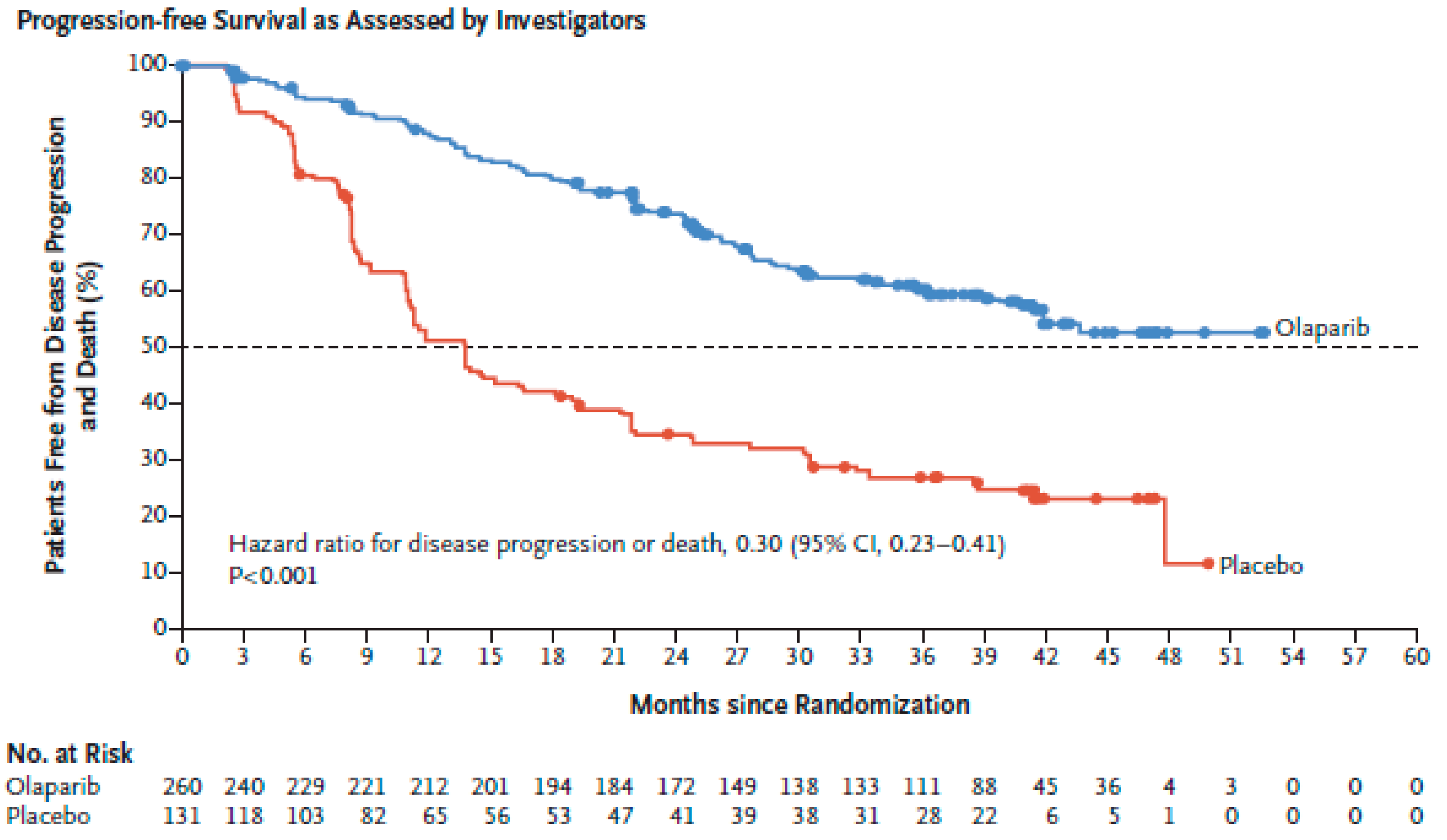
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: An updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet. Oncol. 2016, 17, 1579–1589. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Travis, L.B.; Holowaty, E.J.; Bergfeldt, K.; Lynch, C.F.; Kohler, B.A.; Wiklund, T.; Curtis, R.E.; Hall, P.; Andersson, M.; Pukkala, E.; et al. Risk of leukemia after platinum-based chemotherapy for ovarian cancer. N. Engl. J. Med. 1999, 340, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Korach, J.; Turner, S.; Milenkova, T.; Alecu, I.; McMurtry, E.; Bloomfield, R.; Pujade-Lauraine, E. Incidence of myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) in patients (pts) with a germline (g) BRCA mutation (m) and platinum-sensitive relapsed ovarian cancer (PSR OC) receiving maintenance olaparib in SOLO2: Impact of prior lines of platinum therapy. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Zhong, L.; Tran, A.T.; Tomasino, T.; Nugent, E.; Smith, J.A. Cost-Effectiveness of Niraparib and Olaparib as Maintenance Therapy for Patients with Platinum-Sensitive Recurrent Ovarian Cancer. J. Manag. Care Spec. Pharm. 2018, 24, 1219–1228. [Google Scholar] [PubMed]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.; Sonke, G.S.; Colombo, N.; Spacek, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Lheureux, S.; Lai, Z.; Dougherty, B.A.; Runswick, S.; Hodgson, D.R.; Timms, K.M.; Lanchbury, J.S.; Kaye, S.; Gourley, C.; Bowtell, D.; et al. Long-Term Responders on Olaparib Maintenance in High-Grade Serous Ovarian Cancer: Clinical and Molecular Characterization. Clin. Cancer Res. 2017, 23, 4086–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lheureux, S.; Bruce, J.P.; Burnier, J.V.; Karakasis, K.; Shaw, P.A.; Clarke, B.A.; Yang, S.Y.; Quevedo, R.; Li, T.; Dowar, M.; et al. Somatic BRCA1/2 Recovery as a Resistance Mechanism After Exceptional Response to Poly (ADP-ribose) Polymerase Inhibition. J. Clin. Oncol. 2017, 35, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Del Conte, G.; Sessa, C.; von Moos, R.; Vigano, L.; Digena, T.; Locatelli, A.; Gallerani, E.; Fasolo, A.; Tessari, A.; Cathomas, R.; et al. Phase I study of olaparib in combination with liposomal doxorubicin in patients with advanced solid tumours. Br. J. Cancer 2014, 111, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Bindra, R.S.; Gibson, S.L.; Meng, A.; Westermark, U.; Jasin, M.; Pierce, A.J.; Bristow, R.G.; Classon, M.K.; Glazer, P.M. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res. 2005, 65, 11597–11604. [Google Scholar] [CrossRef] [PubMed]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Maseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol. Cell. Biol. 2004, 24, 8504–8518. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.J.; Yang, K.; Taylor-Harding, B.; Wiedemeyer, W.R.; Buckanovich, R.J. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia 2014, 16, 343–353.e341-342. [Google Scholar] [CrossRef]
- Chan, N.; Bristow, R.G. “Contextual” synthetic lethality and/or loss of heterozygosity: Tumor hypoxia and modification of DNA repair. Clin. Cancer Res. 2010, 16, 4553–4560. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Birrer, M.J.; Lee, J.-m.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.R.; Hurteau, J.; et al. Overall survival and updated progression-free survival results from a randomized phase 2 trial comparing the combination of olaparib and cediranib against olaparib alone in recurrent platinum-sensitive ovarian cancer. J. Clin. Oncol. 2017, 35, 5535. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Wenham, R.M.; Wahner Hendrickson, A.E.; Armstrong, D.K.; Chan, N.; Cohn, D.E.; Lee, J.M.; Penson, R.T.; Cristea, M.C.; et al. A phase 2 biomarker trial of combination cediranib and olaparib in relapsed platinum (plat) sensitive and plat resistant ovarian cancer (ovca). J. Clin. Oncol. 2018, 36, 5519. [Google Scholar] [CrossRef]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmana, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, Y.H.; Garcia-Garcia, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzman, M.; Grueso, J.; et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Cardnell, R.J.; Feng, Y.; Diao, L.; Fan, Y.H.; Masrorpour, F.; Wang, J.; Shen, Y.; Mills, G.B.; Minna, J.D.; Heymach, J.V.; et al. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin. Cancer Res. 2013, 19, 6322–6328. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann. Oncol. 2017, 28, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Westin, S.N.; Litton, J.K.; Williams, R.A.; Shepherd, C.J.; Brugger, W.; Pease, E.J.; Soliman, P.T.; Frumovitz, M.M.; Levenback, C.F.; Sood, A.; et al. Phase I trial of olaparib (PARP inhibitor) and vistusertib (mTORC1/2 inhibitor) in recurrent endometrial, ovarian and triple negative breast cancer. J. Clin. Oncol. 2018, 36, 5504. [Google Scholar] [CrossRef]
- Westin, S.; Litton, J.; Williams, R.; Soliman, P.; Frumovitz, M.; Schmeler, K.; Jazaeri, A.; Sood, A.; Lu, K.; Moulder, S.; et al. 391PPhase I expansion of olaparib (PARP inhibitor) and AZD5363 (AKT inhibitor) in recurrent ovarian, endometrial and triple negative breast cancer. Ann. Oncol. 2017, 28, mdx367.025. [Google Scholar] [CrossRef]
- Ang, J.E.; Gourley, C.; Powell, C.B.; High, H.; Shapira-Frommer, R.; Castonguay, V.; De Greve, J.; Atkinson, T.; Yap, T.A.; Sandhu, S.; et al. Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: A multi-institutional study. Clin. Cancer Res. 2013, 19, 5485–5493. [Google Scholar] [CrossRef] [PubMed]
- Mesiano, S.; Ferrara, N.; Jaffe, R.B. Role of vascular endothelial growth factor in ovarian cancer: Inhibition of ascites formation by immunoneutralization. Am. J. Pathol. 1998, 153, 1249–1256. [Google Scholar] [CrossRef]
- Byrne, A.T.; Ross, L.; Holash, J.; Nakanishi, M.; Hu, L.; Hofmann, J.I.; Yancopoulos, G.D.; Jaffe, R.B. Vascular endothelial growth factor-trap decreases tumor burden, inhibits ascites, and causes dramatic vascular remodeling in an ovarian cancer model. Clin. Cancer Res. 2003, 9, 5721–5728. [Google Scholar] [PubMed]
- Hollingsworth, H.C.; Kohn, E.C.; Steinberg, S.M.; Rothenberg, M.L.; Merino, M.J. Tumor angiogenesis in advanced stage ovarian carcinoma. Am. J. Pathol. 1995, 147, 33–41. [Google Scholar] [PubMed]
- Duncan, T.J.; Al-Attar, A.; Rolland, P.; Scott, I.V.; Deen, S.; Liu, D.T.; Spendlove, I.; Durrant, L.G. Vascular endothelial growth factor expression in ovarian cancer: A model for targeted use of novel therapies? Clin. Cancer Res. 2008, 14, 3030–3035. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.M.; Brady, M.F.; Harrell, M.I.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Burger, R.A.; Tewari, K.S.; et al. Mutations in Homologous Recombination Genes and Outcomes in Ovarian Carcinoma Patients in GOG 218: An NRG Oncology/Gynecol. Oncol. Group Study. Clin. Cancer Res. 2018, 24, 777–783. [Google Scholar] [CrossRef]
- Harter, P.; Johnson, T.; Berton-Rigaud, D.; Park, S.Y.; Friedlander, M.; Del Campo, J.M.; Shimada, M.; Forget, F.; Mirza, M.R.; Colombo, N.; et al. BRCA1/2 mutations associated with progression-free survival in ovarian cancer patients in the AGO-OVAR 16 study. Gynecol. Oncol. 2016, 140, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef]
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936. [Google Scholar] [CrossRef]
- du Bois, A.; Floquet, A.; Kim, J.W.; Rau, J.; del Campo, J.M.; Friedlander, M.; Pignata, S.; Fujiwara, K.; Vergote, I.; Colombo, N.; et al. Incorporation of pazopanib in maintenance therapy of ovarian cancer. J. Clin. Oncol. 2014, 32, 3374–3382. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Sun, C.; Feng, Y.; Jia, Q.; Zhu, B. Potent immunogenicity in BRCA1-mutated patients with high-grade serous ovarian carcinoma. J. Cell. Mol. Med. 2018. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Nebot-Bral, L.; Brandao, D.; Verlingue, L.; Rouleau, E.; Caron, O.; Despras, E.; El-Dakdouki, Y.; Champiat, S.; Aoufouchi, S.; Leary, A.; et al. Hypermutated tumours in the era of immunotherapy: The paradigm of personalised medicine. Eur. J. Cancer 2017, 84, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAlpine, J.N.; Porter, H.; Kobel, M.; Nelson, B.H.; Prentice, L.M.; Kalloger, S.E.; Senz, J.; Milne, K.; Ding, J.; Shah, S.P.; et al. BRCA1 and BRCA2 mutations correlate with TP53 abnormalities and presence of immune cell infiltrates in ovarian high-grade serous carcinoma. Mod. Pathol. 2012, 25, 740–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, B.; Tinker, A.V.; Lee, C.H.; Subramanian, S.; van de Rijn, M.; Turbin, D.; Kalloger, S.; Han, G.; Ceballos, K.; Cadungog, M.G.; et al. Intraepithelial T cells and prognosis in ovarian carcinoma: Novel associations with stage, tumor type, and BRCA1 loss. Mod. Pathol. 2009, 22, 393–402. [Google Scholar] [CrossRef]
- Disis, M.L.; Patel, M.R.; Pant, S.; Hamilton, E.P.; Lockhart, A.C.; Kelly, K.; Beck, J.T.; Gordon, M.S.; Weiss, G.J.; Taylor, M.H.; et al. Avelumab (MSB0010718C; anti-PD-L1) in patients with recurrent/refractory ovarian cancer from the JAVELIN Solid Tumor phase Ib trial: Safety and clinical activity. J. Clin. Oncol. 2016, 34, 5533. [Google Scholar] [CrossRef]
- Varga, A.; Piha-Paul, S.A.; Ott, P.A.; Mehnert, J.M.; Berton-Rigaud, D.; Morosky, A.; Zhao, G.Q.; Rangwala, R.A.; Matei, D. Pembrolizumab in patients (pts) with PD-L1−positive (PD-L1+) advanced ovarian cancer: Updated analysis of KEYNOTE-028. J. Clin. Oncol. 2017, 35, 5513. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef]
- Matulonis, U.; Shapira-Frommer, R.; Santin, A.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.; et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Interim results from the phase 2 KEYNOTE-100 study. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Kristeleit, R.; Davidenko, I.; Shirinkin, V.; El-Khouly, F.; Bondarenko, I.; Goodheart, M.J.; Gorbunova, V.; Penning, C.A.; Shi, J.G.; Liu, X.; et al. A randomised, open-label, phase 2 study of the IDO1 inhibitor epacadostat (INCB024360) versus tamoxifen as therapy for biochemically recurrent (CA-125 relapse)-only epithelial ovarian cancer, primary peritoneal carcinoma, or fallopian tube cancer. Gynecol. Oncol. 2017, 146, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A.; Fossati, M.; Buzzonetti, A.; Scambia, G.; Fattorossi, A. A robust immune system conditions the response to abagovomab (anti-idiotypic monoclonal antibody mimicking the CA125 protein) vaccination in ovarian cancer patients. Immunol. Lett. 2017, 191, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Konstantinopoulos, P.A. From checkpoint to checkpoint: DNA damage ATR/Chk1 checkpoint signalling elicits PD-L1 immune checkpoint activation. Br. J. Cancer 2018, 118, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Na. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Waggoner, S.E.; Vidal, G.A.; Mita, M.M.; Fleming, G.F.; Holloway, R.W.; Le, L.V.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. J. Clin. Oncol. 2018, 36, 106. [Google Scholar] [CrossRef]
- Drew, Y.; de Jonge, M.; Hong, S.H.; Park, Y.H.; Wolfer, A.; Brown, J.; Ferguson, M.; Gore, M.E.; Alvarez, R.H.; Gresty, C.; et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in germline BRCA-mutated (gBRCAm) platinum-sensitive relapsed (PSR) ovarian cancer (OC). Gynecol. Oncol. 2018, 149, 246–247. [Google Scholar] [CrossRef]
- Do, K.; Doroshow, J.H.; Kummar, S. Wee1 kinase as a target for cancer therapy. Cell Cycle 2013, 12, 3159–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Shi, H.; Ren, F.; Zhang, M.; Ji, P.; Wang, W.; Liu, C. The aberrant upstream pathway regulations of CDK1 protein were implicated in the proliferation and apoptosis of ovarian cancer cells. J. Ovarian Res. 2017, 10, 60. [Google Scholar] [CrossRef]
- Zhang, M.; Dominguez, D.; Chen, S.; Fan, J.; Qin, L.; Long, A.; Li, X.; Zhang, Y.; Shi, H.; Zhang, B. WEE1 inhibition by MK1775 as a single-agent therapy inhibits ovarian cancer viability. Oncol. Lett. 2017, 14, 3580–3586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J. Clin. Oncol. 2015, 33, 3409–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.; Li, Y.C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Karnak, D.; Engelke, C.G.; Parsels, L.A.; Kausar, T.; Wei, D.; Robertson, J.R.; Marsh, K.B.; Davis, M.A.; Zhao, L.; Maybaum, J.; et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin. Cancer Res. 2014, 20, 5085–5096. [Google Scholar] [CrossRef] [Green Version]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet. Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Drost, R.; Dhillon, K.K.; van der Gulden, H.; van der Heijden, I.; Brandsma, I.; Cruz, C.; Chondronasiou, D.; Castroviejo-Bermejo, M.; Boon, U.; Schut, E.; et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J. Clin. Investig. 2016, 126, 2903–2918. [Google Scholar] [CrossRef] [Green Version]
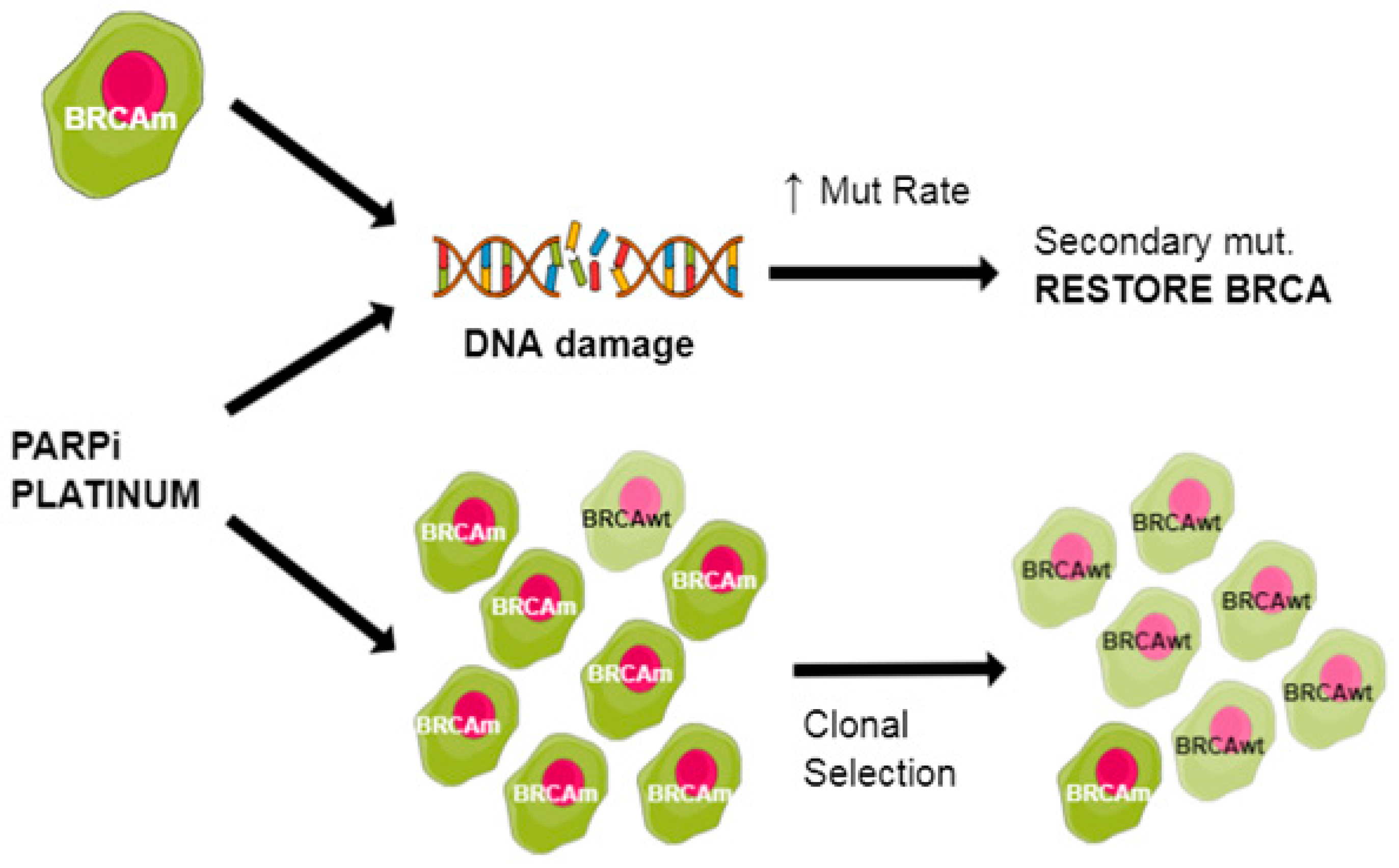
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, K.K.; Swisher, E.M.; Taniguchi, T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011, 102, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Xu, X.; Bunting, S.F.; Liu, J.; Wang, R.H.; Cao, L.L.; Wu, J.J.; Peng, T.N.; Chen, J.; Nussenzweig, A.; et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol. Cell 2009, 35, 534–541. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callen, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; Lopez-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef]
- Yang, Z.M.; Liao, X.M.; Chen, Y.; Shen, Y.Y.; Yang, X.Y.; Su, Y.; Sun, Y.M.; Gao, Y.L.; Ding, J.; Zhang, A.; et al. Combining 53BP1 with BRCA1 as a biomarker to predict the sensitivity of poly(ADP-ribose) polymerase (PARP) inhibitors. Acta Pharmacol. Sin. 2017, 38, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.P.; Wickramanayake, A.; Norquist, B.M.; Pennil, C.C.; Garcia, R.L.; Agnew, K.J.; Taniguchi, T.; Welcsh, P.; Swisher, E.M. 53BP1 expression in sporadic and inherited ovarian carcinoma: Relationship to genetic status and clinical outcomes. Gynecol. Oncol. 2013, 128, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Alvarez-Quilon, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef]
- Durmus, S.; Sparidans, R.W.; van Esch, A.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral availability and brain accumulation of the PARP inhibitor rucaparib (AG-014699). Pharm. Res. 2015, 32, 37–46. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Phase | N (Ovarian) | Prior Treatments | BRCA/HR | Platinum Sensitivity | Dosage | RR (RECIST 1.1) % CI 95% | |
|---|---|---|---|---|---|---|---|
| OLAPARIB | |||||||
| Fong et al. [127] (NCT00516373) | Phase 1 | 50 | 1: 10% 2: 22% 3: 22% ≥4: 46% | 48 gBRCAm (96%) | 13 PlatS (27%) | Escalation: 40 mg qd- 600 mg bid. Expansion: 200 mg bid. | 28% (16.2–42.5) PlatS: 46.2% (19.2–74.9) PlatR: 33.5% (15.6–55.3) Plat Refractory: 0% (0–24.7) |
| Mateo et al. [134] (NCT00777582) | Phase 1 Multitumour | Group 1 and 6 (OC): 53 | Group 1: median 6 (200 mg bid) and 3 (400 mg bid) Group 6: median 4 (300 mg bid) and 3 (400 mg bid) | Group 1 and 6: 53 gBRCAm (100%) | Group 1: NR Group 6: 15 PlatS (37%) | 200–400 mg bid. | Group 1 and 6: 30% (18.3–44.3) |
| Audeh et al. [129] (NCT00494442) | Phase 2 | 57 | Median 3 (400 mg bid) and 4 (100 mg bid) | 57 gBRCAm (100%) | 19 PlatS (33%) | 400 mg bid | 33% (20–51) PlatS: 38% PlatR: 30% |
| 100 mg bid | 13% (4–31) PlatS: 50% PltR: NR | ||||||
| Gelmon et al. [128] (NCT00679783) | Phase 2 Ovarian and breast cancer | 64 | Median 3 | 17 BRCAm (26%) | 25 PlatS (38.5%) | 400 mg bid | 29% (19–41) BRCAm: PlatS: 60% PlatR:33% BRCAwt/u: PlatS: 50% PlatR: 4% |
| Kaufman et al. [123] (NCT01078662) | Phase 2. Multitumour | 193 | Mean 4.3 | 193 BRCAm (100%) | All PlatR or intolerant. | 400 mg bid | 31.1% (24.6–38.1%) |
| Kaye et al. [115] (NCT00628251) | Phase 2. Olaparib vs. PLD | 97 | Olaparib 200 mg: ≥3 lines: 59% | 97 BRCAm (100%) | 48 PlatS | 200 mg bid | 25% |
| Olaparib 400 mg: ≥3 lines: 78% | 400 mg bid | 31% | |||||
| PLD: ≥3 lines: 51% | PLD | 18% | |||||
| NIRAPARIB | |||||||
| Sandhu et al. [130] (NCT00749502) | Phase 1 | 49 | Overall population median: 5 | 22 BRCAm (45%) | 13 PlatS (27%) | Part A: 30–400 mg qd Part B: 300 mg qd | BRCAm: 40% (19–64) PlatS: 50% (19–81) PlatR: 33% (7–70) BRCAwt: NR PlatS: 33% (1–91) PlatR: 5% (<1–26) |
| Moore et al. [135] QUADRA (NCT02354586) Abstract | Phase 2 | 463 | All patients ≥3 prior lines | NR | 162 Plat refractory 152 PlatR 118 PlatS 31 U | 300 mg qd | HRD: PlatS 27.5% (15.9–41.7) BRCAm: 38.9% (7–18) BRCAwt: 21.2% (7–33) Non-HRD: NR. |
| RUCAPARIB | |||||||
| Drew et al. [136] (NCT00664781) | Phase 2. OC and breast cancer | 51 | Overall population: median 2 | 47 BRCAm (92%) | NR | IV (4–18 mg/m2, 5 days q 3w) Oral (92 qd–600 bid). | Overall population OC + breast: 7%. IV cohort: 2% Oral cohort: 15% |
| Kristeleit et al. [137] STUDY 10 (NCT01482715) | Phase 1–2. Multitumour | Part 1: 20 OC (multi-tumour) | Overall population: Median 4 | Part 1: Overall population 64% | Part 1: Overall population 14.3% | Part 1: 40 mg qd–840 mg bid | Part 1: NR |
| Part 2A: 42 (only ovarian) | Median 2 | Part 2A: 42 BRCAm (100%) | Part 2A: 42 PlatS. (100%) | Part 2A: 600 mg bid. | Part 2A: 59.5% (43.3–74.4) | ||
| Swisher et al. [126] ARIEL 2 part 1 (NCT01891344) | Phase 2 | 206 | BRCAm: ≥2 lines 58% LOHh, BRCAwt: ≥2 lines 46% LOHl, BRCAwt: ≥2 lines 33% LOHu, BRCAwt: ≥2 lines 17% | 40 BRCAm (19%) 82 LOHh (BRCAwt) 70 LOHl (BRCAwt) 12 LOHu (BRCAwt) | 206 PlatS (100%) | 600 mg bid | BRCAm: 80% (64–91) LOHh: 29% (20–40) LOHl: 10% (4–20) LOHu: 33% (10–65) |
| Oza et al. [131] Pool data: ARIEL2+ STUDY 10 | Phase 2 | 106 (integrated efficacy population) | ≥3 prior chemotherapies, 61.3% | 106 BRCAm (100%) | 79 PlatS (74.5%) | 600 mg bid | 53.8% (43.8–63.5) PFI > 12 m: 73.9% (51.6–89.8) PFI 6–12 m: 62.5% (48.5–75.1) PFI < 6 m: 18.5% (6.3–38.1) |
| VELIPAPRIB | |||||||
| Coleman et al. [138] (NCT01540565) | Phase 2 | 50 | 1: 28% 2: 36% 3: 36% | 50 BRCAm (100%) | 20 PlatS (40%) | 400 mg bid | 26% (16–38) PlatS: 35% (18–56) PlatR: 20% (9–36) |
| Steffensen et al. [132] (NCT01472783) | Phase 1–2 | 48 | Median 4 | 48 BRCAm (100%) | 13 Plat S (with PFI 6–12 m) Phase I: 4 (12.5%) Phase II: 9 (28.1%) | Phase I: escalation from 300 mg bid | Phase I: NR |
| Phase II: 300 mg bid | Phase II:44% Plat S: 50% Plat R: 41% | ||||||
| TALAZOPARIB | |||||||
| De Bono et al. [133] (NCT01286987) | Phase 1 | 34 Part 1: 23 Part 2: 11 BRCAm | Overall median: 2.5 Part 1 median: 4 Part 2: median 2 | Part 1: NR Part 2: 11 (100%) | NR | Part 1: escalation 0.025–1.1 mg qd. Part 2: expansion. 1 mg qd. | Both cohorts: BRCAm: 48%. PlatS: 55% Plat R: 20% |
| Part 2: 41.7% | |||||||
| Trial Design | Patient Characteristics | BRCA/HRD | PFS | PFS (BRCAm) | PFS (BRCAwt/HRD) | PFS (BRCAwt/non-HRD) | OS | |
|---|---|---|---|---|---|---|---|---|
| OLAPARIB-FRONT LINE | ||||||||
| Moore et al. [139] SOLO1 (NCT01844986) | Phase III. Olaparib vs. placebo. | N: 391 Serous 96% Front line. | gBRCAm 99% | At 3 years: 60% vs. 27% HR 0.3, p < 0.001 | (Same as overall population) | NA | NA | Immature HR 0.95 (95% CI 0.6–1.53) |
| OLAPARIB-PLATINUM SENSTIVE RECURRENCE | ||||||||
| Ledermann et al. [140,141,142] STUDY19 (NCT00753545) | Phase II. Olaparib vs. placebo. | N: 265 High grade serous 91% ≤2 prior platinum 58% | g/sBRCAm 51% | 8.4 vs. 4.8 months HR 0.35, p < 0.001. | 11.2 vs. 4.3 months HR0.18, p < 0.0001 | NA | 7.4 vs. 5.5 months HR 0.54, p = 0.0075 | 29.8 vs. 27.8 months HR0.73, p = 0.025, NS. |
| Pujade-Lauraine et al. [143] SOLO2/ENGOT-OV21 (NCT01874353) | Phase III. Olaparib vs. placebo. | N: 295 Serous 91% ≤2 prior platinum 58% | gBRCAm 97% | 19.1 vs. 5.5 months HR 0.3, p < 0.0001. | (Same as overall population) | NA | NA | Immature. HR: 0.8, p = 0.43 |
| NIRAPARIB-PLATINUM SENSTIVE RECURRENCE | ||||||||
| Mirza et al. [144] ENGOT-OV16/NOVA (NCT01847274) | Phase III. Niraparib vs. placebo. | N: 553 Predominantly high grade serous. ≤2 prior chemotherapy 60% | gBRCAm: 203 (37%) gBRCAwt:350 (63%) HRD: 162 Non-HRD: 134 HRDu: 54 | NR | 21 vs. 5.5 months HR0.27, p < 0.001. | 12.9 vs. 3.8 months HR0.38, p < 0.001 | 6.9 vs. 3.8 months HR 0.58. p = 0.02. | NR |
| RUCAPARIB-PLATINUM SENSTIVE RECURRENCE | ||||||||
| Coleman et al. [145] ARIEL 3 (NCT01968213) | Phase III. Rucaparib vs. placebo. | N: 564 Serous 95% ≤2 prior chemotherapy 63% | g/sBRCAm 196 (35%) LOHh 158 (28%) LOHl 161 (29%) LOHu 49 (9%) | 10.8 vs. 5.4 months HR 0.36, p < 0.0001 | 16 vs. 5.4 months HR0.23, p < 0.0001 | 9.7 vs. 5.4 months HR0.44, p < 0.0001 | 6.7 vs. 5.4 months HR 0.58, p = 0.0049 | NR |
| Olaparib | Niraparib | Rucaparib | |
|---|---|---|---|
| Dosage | Cap: 400 mg bid Tab: 300 mg bid | 300 mg qd | 600 mg bid |
| FDA Approval | -2014: Single agent treatment. Cap formulation. gBRCAm, ≥3 prior Ch lines. -2017: Maintenance treatment. PlatS, and response to platinum Ch. Regardless of BRCAm status. -2018: First-line maintenance. Germline or somatic BRCAm carriers. | -2017: Maintenance treatment. PlatS relapse, and response to platinum Ch. Regardless of BRCAm status. | -2018: Maintenance treatment. PlatS, and response to platinum Ch. Regardless of BRCAm status. |
| EMA Approval | -2014: Maintenance treatment. Cap formulation. g/sBRCAm, PlatS relapse, and response to platinum. -2018: Maintenance treatment. Tablet formulation. PlatS relapse, and response to platinum Ch. Regardless of BRCAm status. | -2017: Maintenance treatment. PlatS relapse, and response to platinum Ch. Regardless BRCAm status. | -2018: Maintenance treatment. Germline or somatic BRCAm, PlatS and response to platinum Ch. |
| Toxicity | |||
| Drug class toxicity | GI, fatigue, myelosuppression, headache, decrease appetite, creatinine increase, dyspnea. | GI, fatigue, myelosuppression, headache, decrease appetite, creatinine increase, dyspnea. | GI, fatigue, myelosuppression, headache, decrease appetite, creatinine increase, dyspnea. |
| ≥10%, grade ≥3 | Anemia. | Anemia, neutropenia, Thrombocytopenia. | Anemia, ↑ALT/AST. |
| Drug specific | - | Hypertension | ↑ALT/AST |
| Other | Overall risk of MDS/AML 1%. | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madariaga, A.; Lheureux, S.; Oza, A.M. Tailoring Ovarian Cancer Treatment: Implications of BRCA1/2 Mutations. Cancers 2019, 11, 416. https://doi.org/10.3390/cancers11030416
Madariaga A, Lheureux S, Oza AM. Tailoring Ovarian Cancer Treatment: Implications of BRCA1/2 Mutations. Cancers. 2019; 11(3):416. https://doi.org/10.3390/cancers11030416
Chicago/Turabian StyleMadariaga, Ainhoa, Stephanie Lheureux, and Amit M. Oza. 2019. "Tailoring Ovarian Cancer Treatment: Implications of BRCA1/2 Mutations" Cancers 11, no. 3: 416. https://doi.org/10.3390/cancers11030416
APA StyleMadariaga, A., Lheureux, S., & Oza, A. M. (2019). Tailoring Ovarian Cancer Treatment: Implications of BRCA1/2 Mutations. Cancers, 11(3), 416. https://doi.org/10.3390/cancers11030416