Store-Operated Calcium Entry Contributes to Cisplatin-Induced Cell Death in Non-Small Cell Lung Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
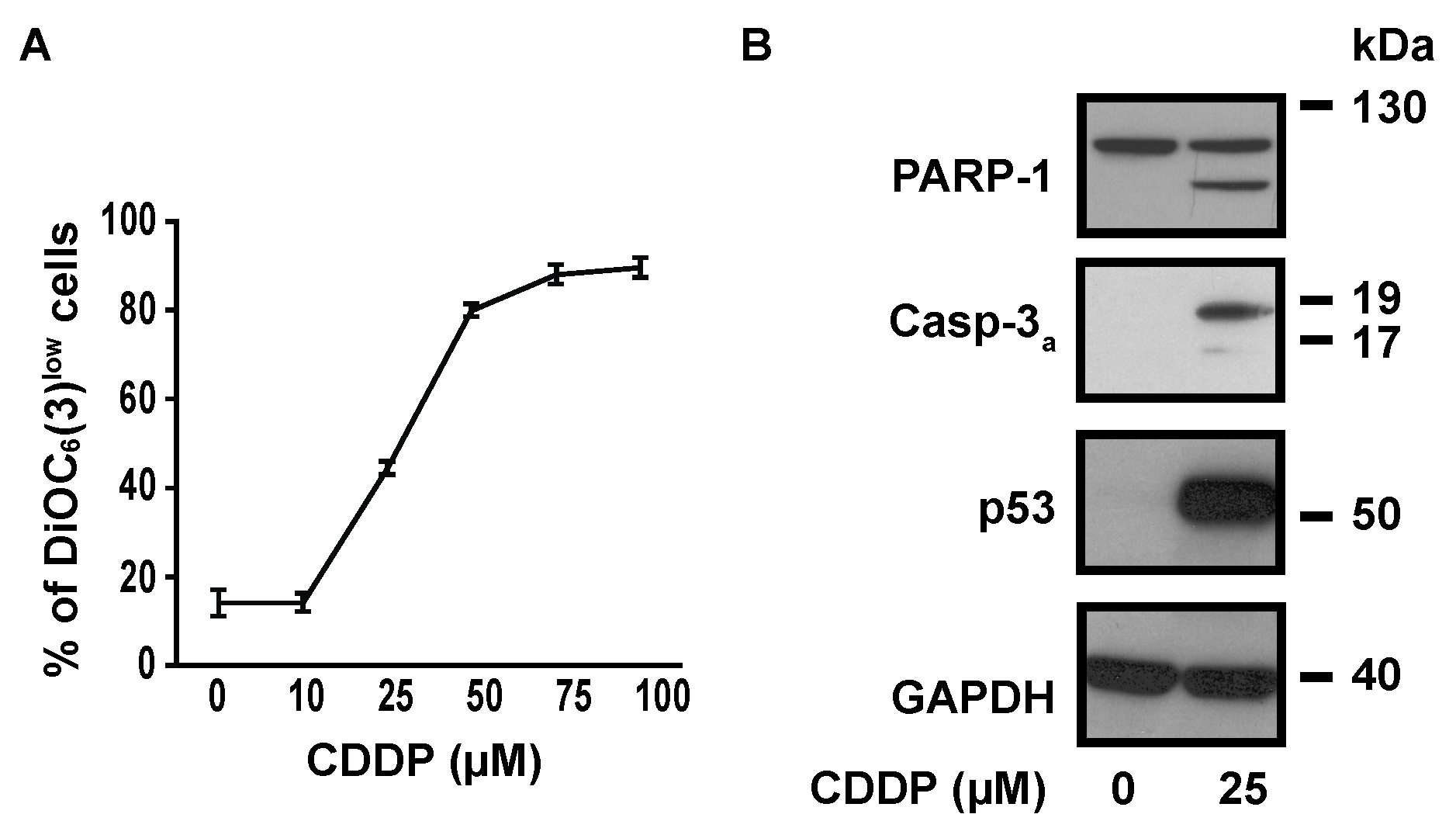
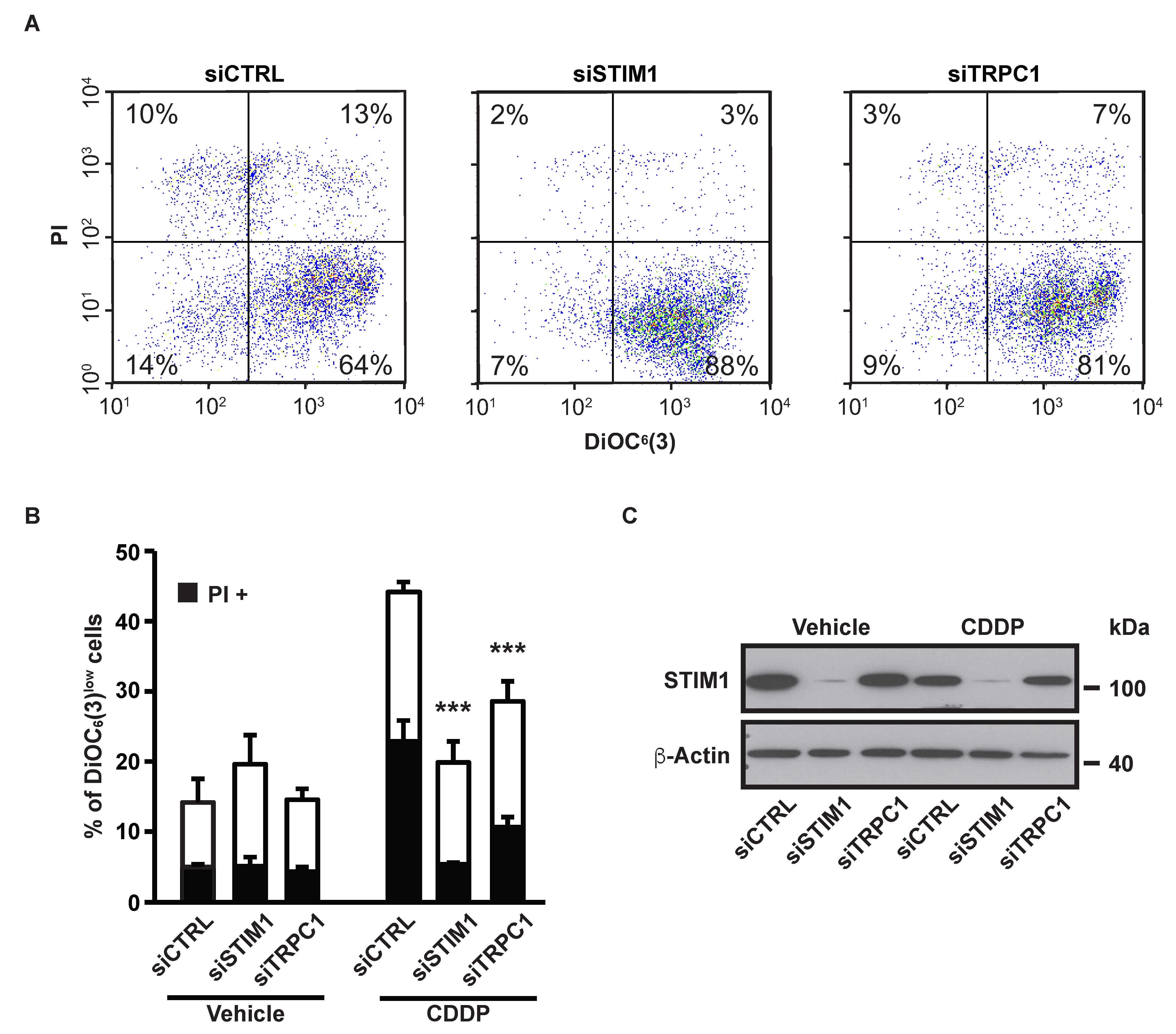
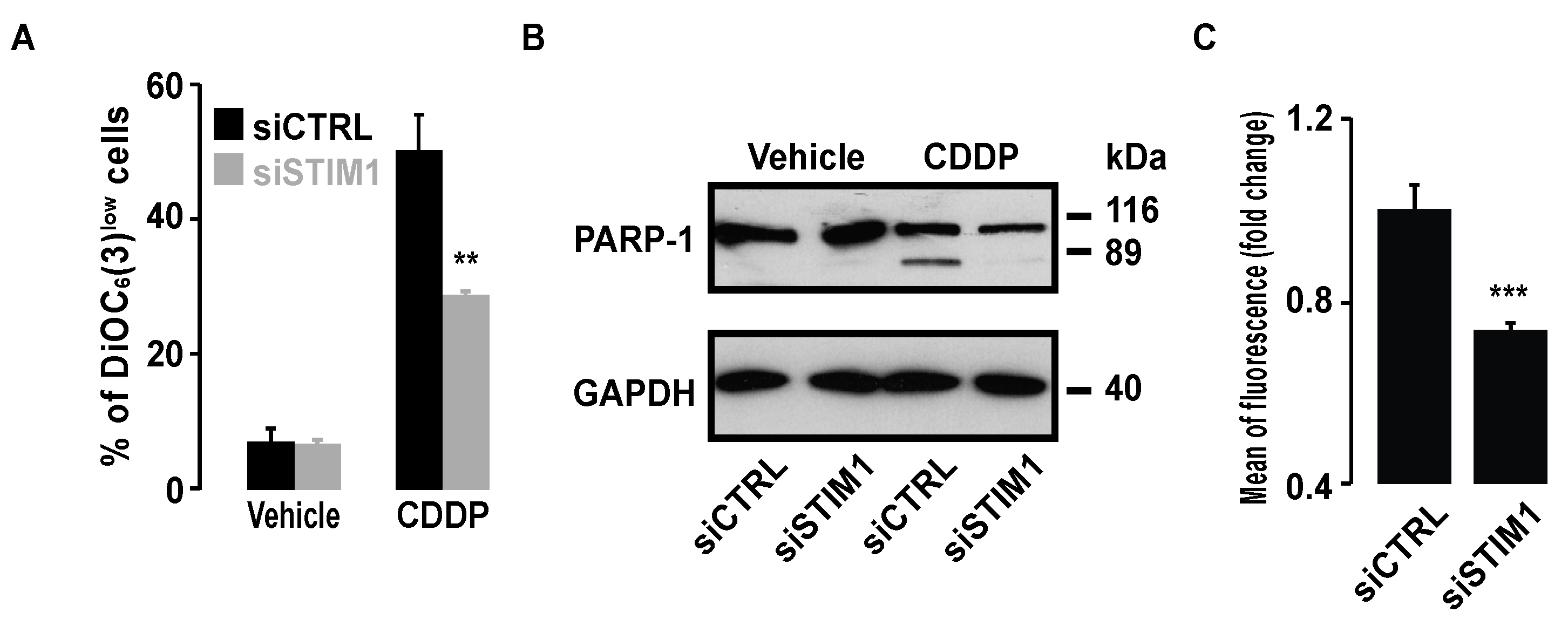
2.1. SOCE Inhibition Reduced CDDP-Dependent Cell Death
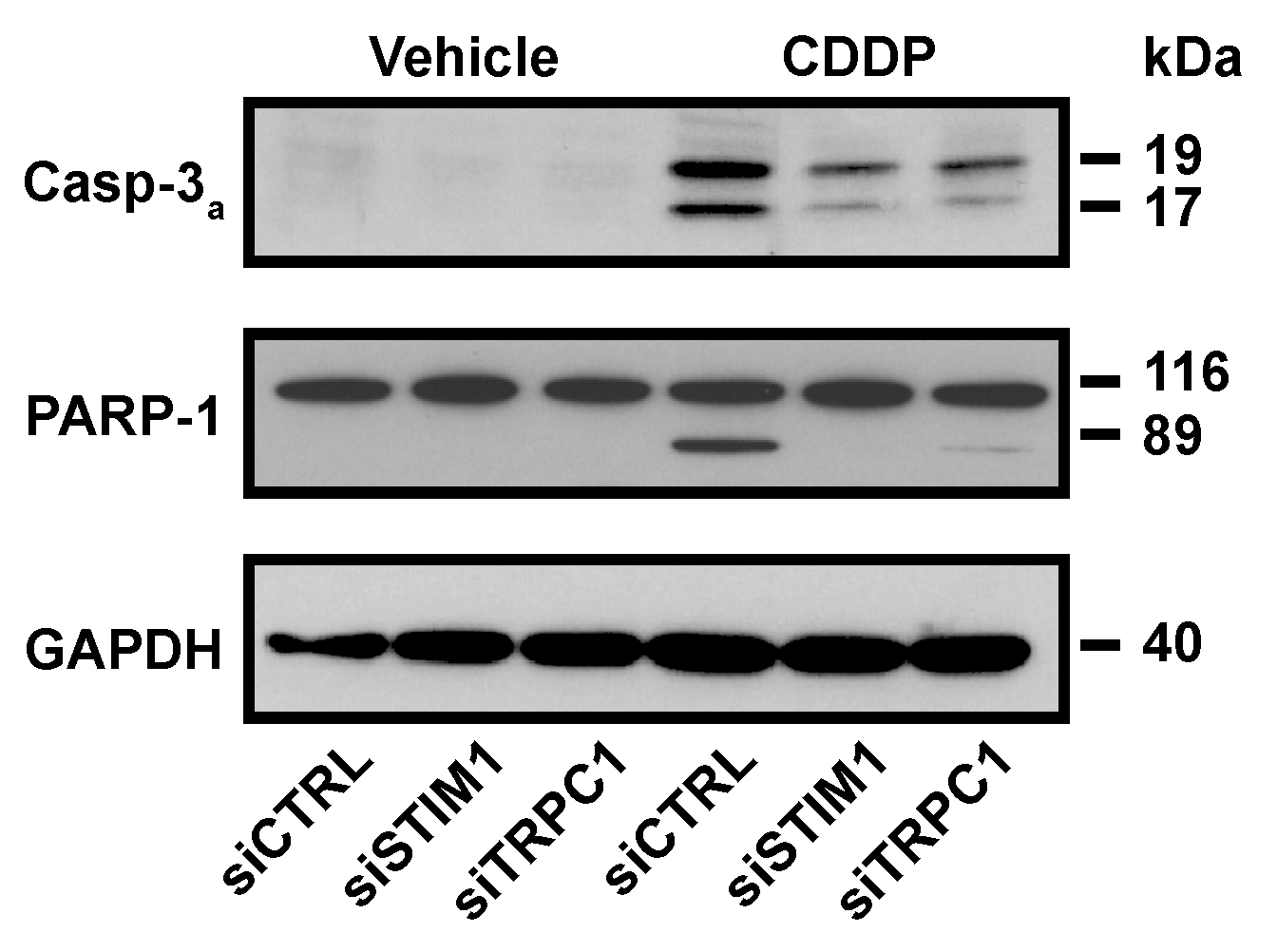
2.2. SOCE Inhibition Reduced Expression of Specific Markers of Apoptosis Induced by CDDP
2.3. SOCE Was Not Altered by CDDP
2.4. STIM1 Depletion Inhibited CDDP-Dependent ERK Activation
2.5. STIM1 Depletion Inhibited DDR Induced by CDDP
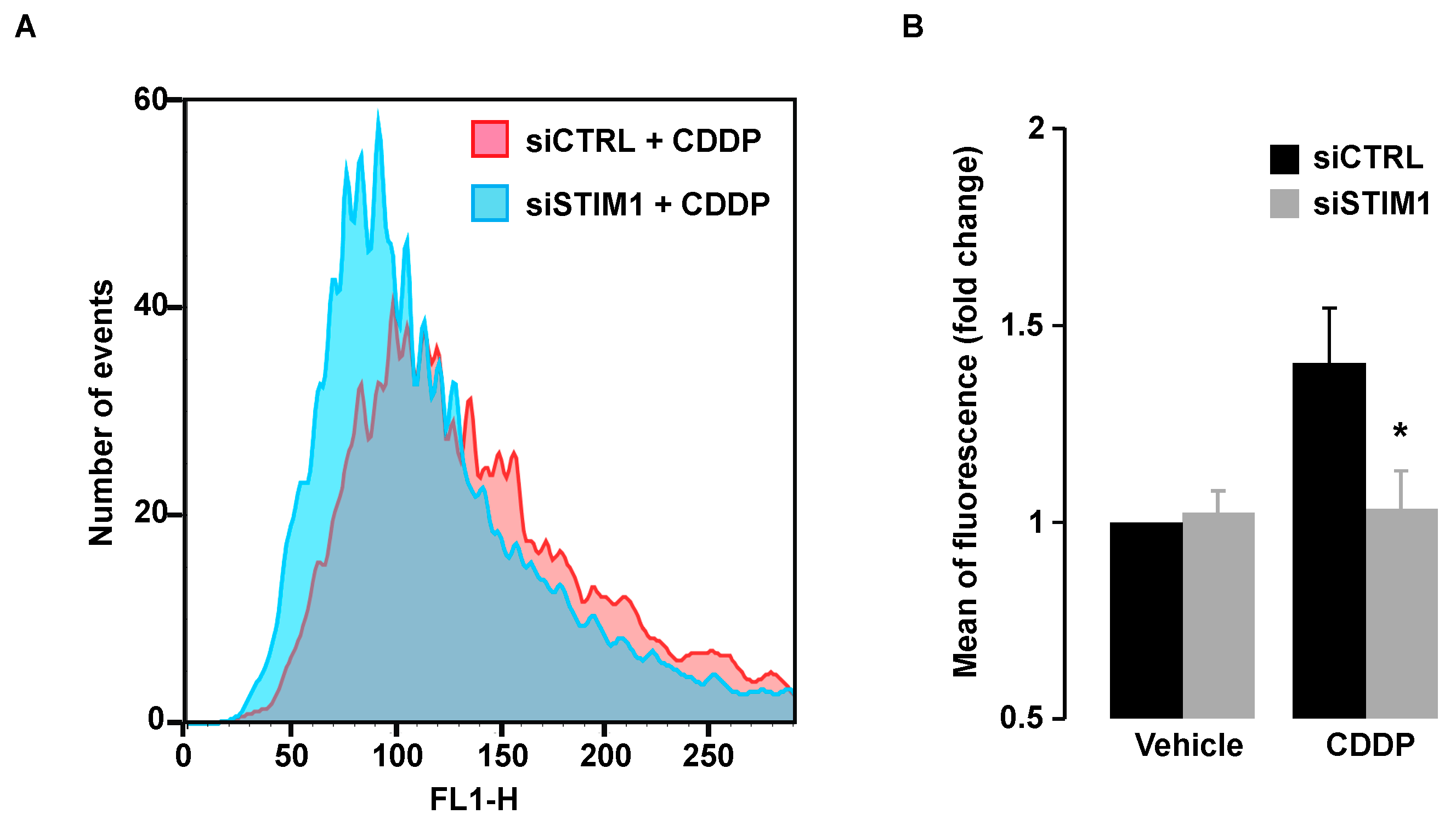
2.6. STIM1 Depletion Reduced CDDP-Induced ROS Production
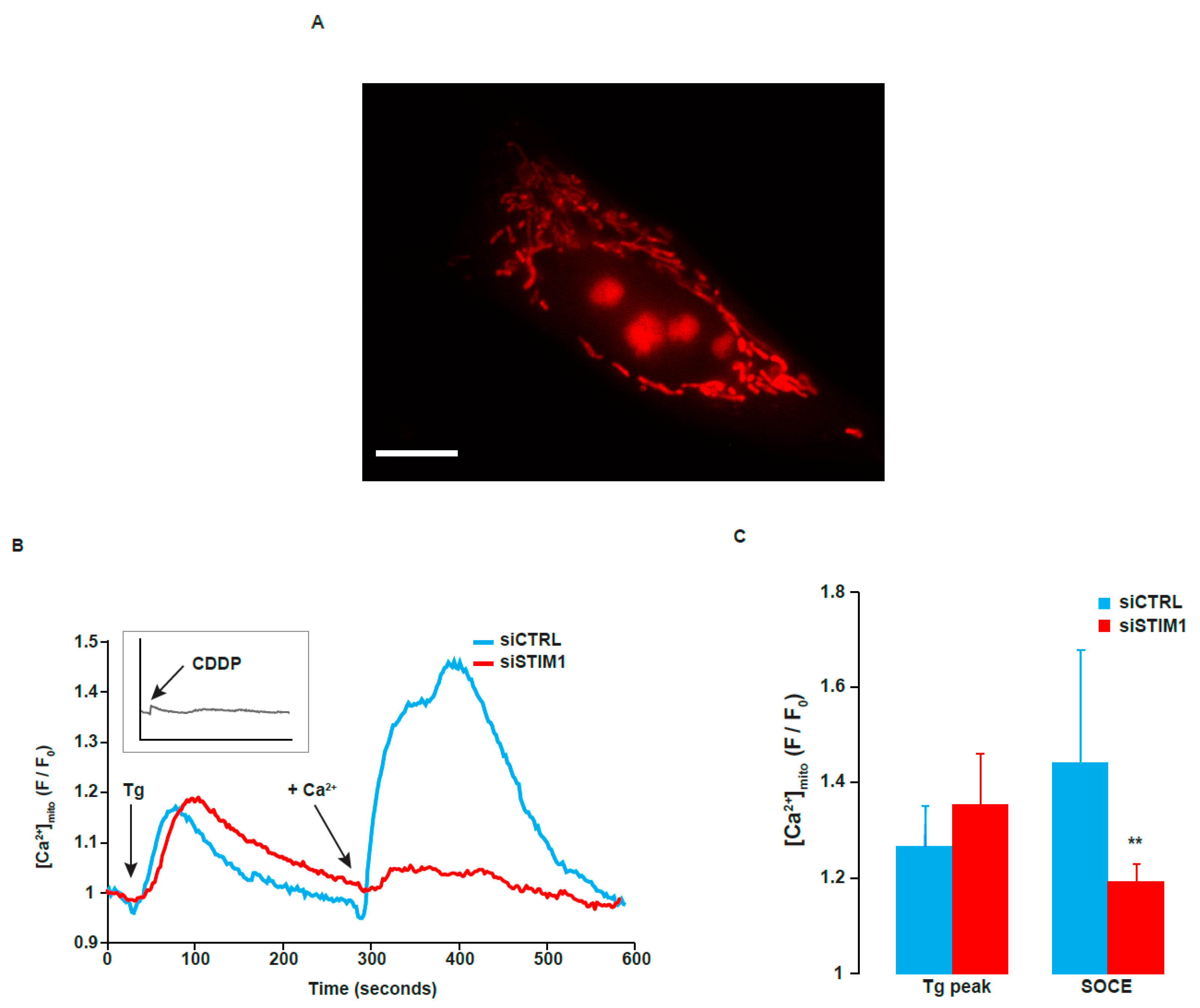
2.7. STIM1 Depletion Reduced SOCE-Dependent Entry of Ca2+ into the Mitochondria
2.8. SOCE Is Also Involved in CDDP-Induced Cell Death in Cervix Carcinoma Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. siRNA Transfection
4.3. Immunoblotting
4.4. Cytosolic and Mitochondrial Free Ca2+ Measurements
4.5. Cytofluorometric Assessment of Apoptosis-Associated Parameters and ROS Production
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rosenberg, B.; VanCamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Berndtsson, M.; Hagg, M.; Panaretakis, T.; Havelka, A.M.; Shoshan, M.C.; Linder, S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int. J. Cancer 2007, 120, 175–180. [Google Scholar] [CrossRef]
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 2003, 278, 9100–9106. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Wang, X.; Martindale, J.L.; Holbrook, N.J. Requirement for ERK activation in cisplatin-induced apoptosis. J. Biol. Chem. 2000, 275, 39435–39443. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.L.; Jin, B.J.; Cheng, C.G.; Zhang, F.X.; Wang, S.W.; Wang, Y.; Wu, B. Apatinib resensitizes cisplatin-resistant non-small cell lung carcinoma A549 cell through reversing multidrug resistance and suppressing ERK signaling pathway. Eur. Rev. Med Pharmacol. Sci. 2017, 21, 5370–5377. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.W.; Wu, T.C.; Wu, J.Y.; Cheng, Y.W.; Chen, Y.C.; Lee, M.C.; Chen, C.Y.; Lee, H. Phosphorylation of paxillin confers cisplatin resistance in non-small cell lung cancer via activating ERK-mediated Bcl-2 expression. Oncogene 2014, 33, 4385–4395. [Google Scholar] [CrossRef]
- Yeh, P.Y.; Chuang, S.E.; Yeh, K.H.; Song, Y.C.; Ea, C.K.; Cheng, A.L. Increase of the resistance of human cervical carcinoma cells to cisplatin by inhibition of the MEK to ERK signaling pathway partly via enhancement of anticancer drug-induced NF kappa B activation. Biochem. Pharmacol. 2002, 63, 1423–1430. [Google Scholar] [CrossRef]
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [Green Version]
- Bong, A.H.L.; Monteith, G.R. Calcium signaling and the therapeutic targeting of cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1786–1794. [Google Scholar] [CrossRef] [PubMed]
- Busselberg, D.; Florea, A.M. Targeting Intracellular Calcium Signaling ([Ca2+]i) to Overcome Acquired Multidrug Resistance of Cancer Cells: A Mini-Overview. Cancers 2017, 9, 48. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, X.; Li, H.; Chen, Z.; Yao, X.; Jin, J.; Ma, X. TRPC5-induced autophagy promotes drug resistance in breast carcinoma via CaMKKbeta/AMPKalpha/mTOR pathway. Sci. Rep. 2017, 7, 3158. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, Z.; Hua, D.; He, D.; Wang, L.; Zhang, P.; Wang, J.; Cai, Y.; Gao, C.; Zhang, X.; et al. Essential role for TrpC5-containing extracellular vesicles in breast cancer with chemotherapeutic resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 6389–6394. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Chen, I.; Shimoda, L.A.; Park, Y.; Zhang, C.; Tran, L.; Zhang, H.; Semenza, G.L. Chemotherapy-Induced Ca2+ Release Stimulates Breast Cancer Stem Cell Enrichment. Cell Rep. 2017, 18, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Splettstoesser, F.; Florea, A.M.; Busselberg, D. IP(3) receptor antagonist, 2-APB, attenuates cisplatin induced Ca2+-influx in HeLa-S3 cells and prevents activation of calpain and induction of apoptosis. Br. J. Pharmacol. 2007, 151, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Bourguignon, L.Y. Hyaluronan-CD44 promotes phospholipase C-mediated Ca2+ signaling and cisplatin resistance in head and neck cancer. Arch. Otolaryngol. Head Neck Surg. 2006, 132, 19–24. [Google Scholar] [CrossRef]
- Al-Bahlani, S.; Fraser, M.; Wong, A.Y.; Sayan, B.S.; Bergeron, R.; Melino, G.; Tsang, B.K. P73 regulates cisplatin-induced apoptosis in ovarian cancer cells via a calcium/calpain-dependent mechanism. Oncogene 2011, 30, 4219–4230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Taweel, N.; Varghese, E.; Florea, A.M.; Busselberg, D. Cisplatin (CDDP) triggers cell death of MCF-7 cells following disruption of intracellular calcium ([Ca2+]i) homeostasis. J. Toxicol. Sci. 2014, 39, 765–774. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathopulos, P.B.; Ikura, M. Store operated calcium entry: From concept to structural mechanisms. Cell Calcium 2017, 63, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Trebak, M.; Putney, J.W., Jr. ORAI Calcium Channels. Physiology 2017, 32, 332–342. [Google Scholar] [CrossRef]
- Sun, X.; Wei, Q.; Cheng, J.; Bian, Y.; Tian, C.; Hu, Y.; Li, H. Enhanced Stim1 expression is associated with acquired chemo-resistance of cisplatin in osteosarcoma cells. Hum. Cell 2017, 30, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.D.; Xia, X.; Lv, X.F.; Yu, B.X.; Yuan, J.N.; Mai, X.Y.; Shang, J.Y.; Zhou, J.G.; Liang, S.J.; Pang, R.P. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell. Mol. Med. 2017, 21, 904–915. [Google Scholar] [CrossRef]
- Tajeddine, N.; Gailly, P. TRPC1 protein channel is major regulator of epidermal growth factor receptor signaling. J. Biol. Chem. 2012, 287, 16146–16157. [Google Scholar] [CrossRef] [PubMed]
- Emeriau, N.; de Clippele, M.; Gailly, P.; Tajeddine, N. Store operated calcium entry is altered by the inhibition of receptors tyrosine kinase. Oncotarget 2018, 9, 16059–16073. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhang, M.; Xu, L.; Lin, D.; Cai, S.; Zou, F. The apoptosis of non-small cell lung cancer induced by cisplatin through modulation of STIM1. Exp. Toxicol. Pathol. Off. J. Ges. Fur Toxikol. Pathol. 2013, 65, 1073–1081. [Google Scholar] [CrossRef]
- Vandebrouck, A.; Ducret, T.; Basset, O.; Sebille, S.; Raymond, G.; Ruegg, U.; Gailly, P.; Cognard, C.; Constantin, B. Regulation of store-operated calcium entries and mitochondrial uptake by minidystrophin expression in cultured myotubes. FASEB J. 2006, 20, 136–138. [Google Scholar] [CrossRef]
- Yu, W.; Chen, Y.; Dubrulle, J.; Stossi, F.; Putluri, V.; Sreekumar, A.; Putluri, N.; Baluya, D.; Lai, S.Y.; Sandulache, V.C. Cisplatin generates oxidative stress which is accompanied by rapid shifts in central carbon metabolism. Sci. Rep. 2018, 8, 4306. [Google Scholar] [CrossRef] [PubMed]
- Tajeddine, N.; Galluzzi, L.; Kepp, O.; Hangen, E.; Morselli, E.; Senovilla, L.; Araujo, N.; Pinna, G.; Larochette, N.; Zamzami, N.; et al. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene 2008, 27, 4221–4232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Zeng, Q.; Zhang, B.; Liu, H.; Wang, W. Promotion of p53 expression and reactive oxidative stress production is involved in zerumbone-induced cisplatin sensitization of non-small cell lung cancer cells. Biochimie 2014, 107 Pt B, 257–262. [Google Scholar] [CrossRef]
- Chirino, Y.I.; Pedraza-Chaverri, J. Role of oxidative and nitrosative stress in cisplatin-induced nephrotoxicity. Exp. Toxicol. Pathol. 2009, 61, 223–242. [Google Scholar] [CrossRef]
- Manohar, S.; Leung, N. Cisplatin nephrotoxicity: A review of the literature. J. Nephrol. 2018, 31, 15–25. [Google Scholar] [CrossRef]
- Tajeddine, N. How do reactive oxygen species and calcium trigger mitochondrial membrane permeabilisation? Biochim. Biophys. Acta 2016, 1860, 1079–1088. [Google Scholar] [CrossRef]
- Hempel, N.; Trebak, M. Crosstalk between calcium and reactive oxygen species signaling in cancer. Cell Calcium 2017, 63, 70–96. [Google Scholar] [CrossRef] [PubMed]
- Prins, D.; Groenendyk, J.; Touret, N.; Michalak, M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 2011, 12, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Alansary, D.; Schmidt, B.; Dorr, K.; Bogeski, I.; Rieger, H.; Kless, A.; Niemeyer, B.A. Thiol dependent intramolecular locking of Orai1 channels. Sci. Rep. 2016, 6, 33347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Chen, W.; Zhang, L.; Liu, W.B.; Fei, Z. Inhibition of store-operated calcium entry attenuates MPP(+)-induced oxidative stress via preservation of mitochondrial function in PC12 cells: Involvement of Homer1a. PLoS ONE 2013, 8, e83638. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gualdani, R.; de Clippele, M.; Ratbi, I.; Gailly, P.; Tajeddine, N. Store-Operated Calcium Entry Contributes to Cisplatin-Induced Cell Death in Non-Small Cell Lung Carcinoma. Cancers 2019, 11, 430. https://doi.org/10.3390/cancers11030430
Gualdani R, de Clippele M, Ratbi I, Gailly P, Tajeddine N. Store-Operated Calcium Entry Contributes to Cisplatin-Induced Cell Death in Non-Small Cell Lung Carcinoma. Cancers. 2019; 11(3):430. https://doi.org/10.3390/cancers11030430
Chicago/Turabian StyleGualdani, Roberta, Marie de Clippele, Ikram Ratbi, Philippe Gailly, and Nicolas Tajeddine. 2019. "Store-Operated Calcium Entry Contributes to Cisplatin-Induced Cell Death in Non-Small Cell Lung Carcinoma" Cancers 11, no. 3: 430. https://doi.org/10.3390/cancers11030430
APA StyleGualdani, R., de Clippele, M., Ratbi, I., Gailly, P., & Tajeddine, N. (2019). Store-Operated Calcium Entry Contributes to Cisplatin-Induced Cell Death in Non-Small Cell Lung Carcinoma. Cancers, 11(3), 430. https://doi.org/10.3390/cancers11030430