1. Introduction
Ovarian cancer (OC) has the highest fatality rate of any gynecologic cancer [
1]. Although OC is considered a chemo-responsive tumor with very high initial response rates to standard platinum-based therapy [
2], most women eventually develop recurrence, which rapidly evolves into resistant disease and is incurable [
2]. One of the critical factors contributing to relapse is persistence of quiescent cancer cells that are not eliminated by chemotherapy. It has been speculated that these residual cells are cancer stem cells (CSCs), which can subsequently differentiate and generate recurrent tumors. CSCs have been isolated from OC cell lines, malignant ascites, and primary tumors [
3,
4,
5]. CSCs are characterized by the expression of specific cell surface markers and the ability to self-renew, differentiate, and generate tumors when injected in small numbers into NOD/SCID mice. In culture, CSCs grow as spheres, are able to differentiate into cell subtypes with different phenotypes, and have been implicated in tumor heterogeneity, tumor dormancy, and resistance to chemotherapy and radiation [
6]. Several surface markers have been proposed for ovarian CSC identification, including CD44+/CD117+, CD44+/MyoD, CD133+, CD133/ALDH+, or ALDH+. CD44/CD117+ cells are resistant to chemotherapy, generate tumors in immunodeficient mice and display activation of recognized stem cell pathways (e.g.,
nanog,
nestin,
notch) [
5]. CD44+/MyoD+ cells generate tumors, grow as spheroids and are highly resistant to chemotherapy [
7]. CD133+ cells are highly resistant to platinum [
8], while ALDH+ cells are tumorigenic in vivo, express stem cell restricted transcription factors, form spheres in non-adherent cultures, and are chemoresistant [
4,
9,
10]. Double positive ALDH+/CD133+ cells are highly tumorigenic and chemo-resistant and represent a rare cell subpopulation within tumors [
4]. Here, we focused on the properties of ALDH+ cells and new modalities to target them.
The highly conserved aldehyde dehydrogenase (ALDH) family includes 19 enzymes involved in the metabolism of reactive aldehydes [
11]. Through their detoxification functions, ALDHs exert cytoprotective roles in various tissues [
12]. In addition, the enzymes catalyze retinol oxidation to retinal, a limiting step during the synthesis of retinoic acid, which activates an important cellular differentiation pathway. Recent reports have linked ALDHs, and particularly ALDH1A1, to stem cells, both in normal tissues, such as the hematopoietic milieu [
13], as well as in malignancy [
14]. It remains unknown whether the enzyme is only a marker for stem cells or whether it is functionally implicated in maintaining their characteristics. While several other markers have been proposed over the years for identifying ovarian CSCs [
5], ALDH1A1 activity detectable through the Aldefluor assay has been validated by several groups, and appears to be a robust phenotype [
4,
10,
15]. The percentage of ALDH+ cells in high grade serous ovarian cancer (HGSOC) cell lines varies between 0.2% to 10%, and ALDH expression is higher in mucinous and endometrioid cancers compared to serous carcinomas [
16]. Furthermore, ALDH positivity in OC has been correlated with worse patient survival [
16,
17]. An enrichment in ALDH+ cells was observed after treatment with platinum both in cells lines and xenografts [
18,
19], supporting the hypothesis that post-therapy tumors are enriched in cells with progenitor characteristics.
In recent years, emerging efforts have focused on developing new therapies targeting CSCs [
20]. Various strategies have been pursued, including agents which block pathways selectively activated in stem cells [
21] or which target specific markers expressed on the surface of CSCs. As high ALDH activity appears to be a hallmark of ovarian CSCs [
22], development of inhibitors for this family of enzymes is garnering interest [
23,
24]. We have previously reported that CM37, a small molecule inhibitor for ALDH1A1 discovered from a high-throughput screen of a diverse chemical library, was a potent inhibitor of OC cell proliferation as spheres but had modest effects on differentiated cells [
25]. Here, we describe in further detail the effects of this selective inhibitor in ovarian cancer models and the significance of the ALDH1A1 enzyme to the ovarian cancer stemness phenotype.
3. Discussion
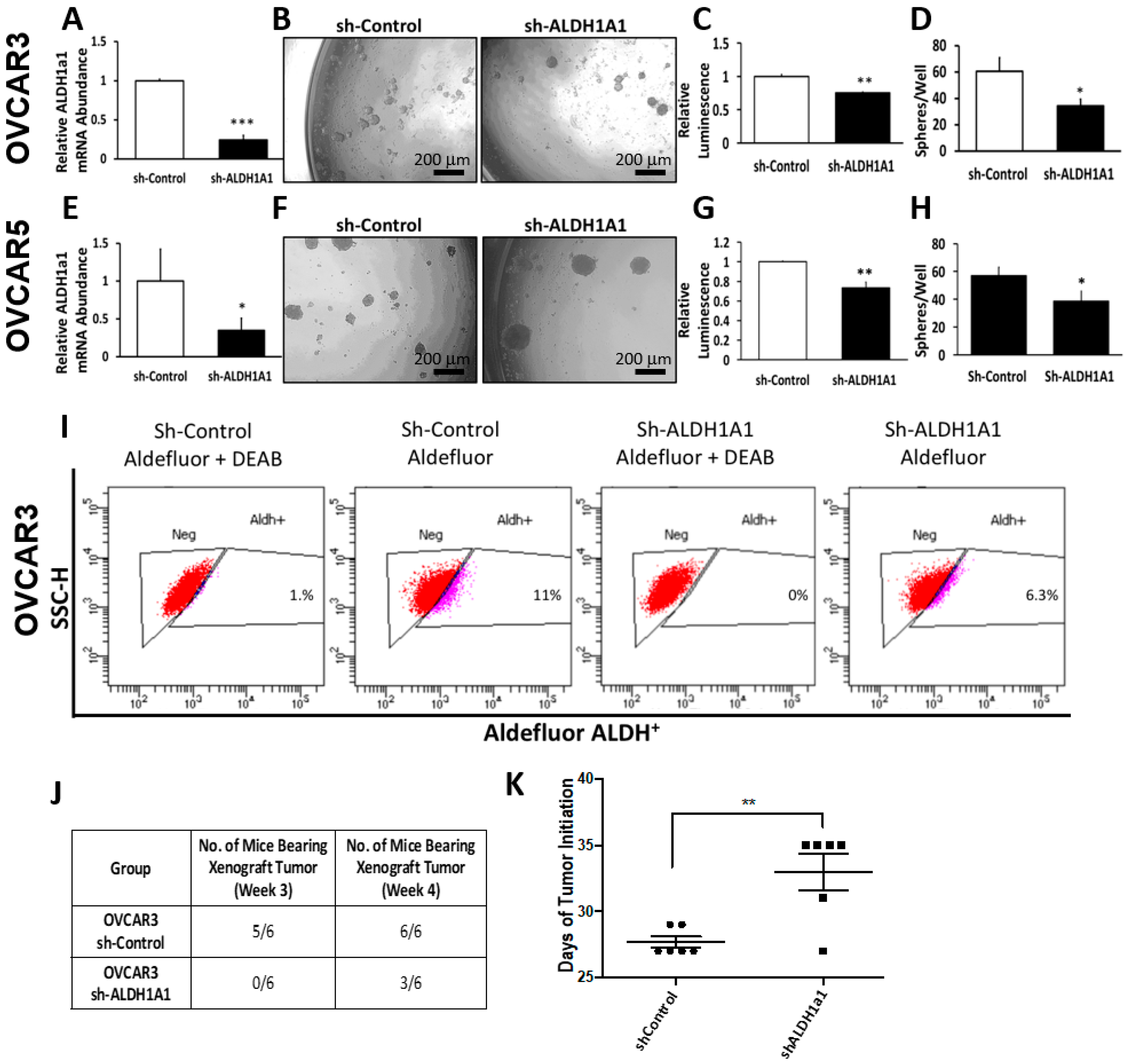
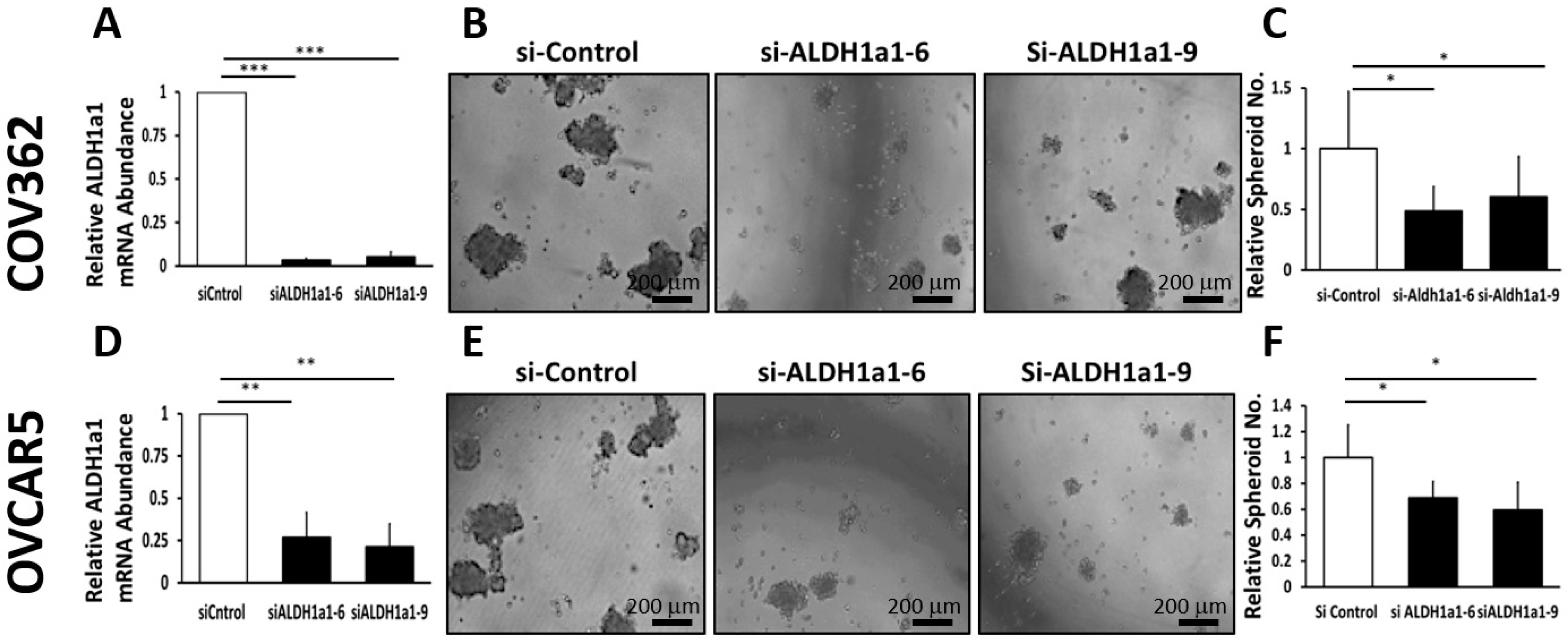
Our data provide proof of principle support for the concept that ALDH inhibitors block stemness and cell proliferation in OC models, and back further development of such small molecules for targeting and eradicating CSCs. We show that CM37, a selective and potent ALDH1A1 inhibitor [
25], effectively inhibits OC cell proliferation as spheres, induces accumulation of intracellular ROS, and inflicts DNA damage. These effects were consistent between cell lines and primary cancer cells sorted from malignant ascites. Similar effects were observed following knockdown of the
ALDH1A1 isoform, supporting its significance to the maintenance of stemness characteristics. However, functional redundancy among isoforms in this family and the distinct patterns of expression of isoforms in various models or cell lines may limit the activity of highly selective inhibitors, suggesting that drugs with a broader spectrum of inhibitory activity might induce more potent anti-CSCs results. Our findings have several implications.
Aldefluor activity has been increasingly recognized as a marker of cells with progenitor/stemness characteristics in OC [
22], but also in other cancer models [
13,
24]. There are 19 genes encoding various aldehyde dehydrogenases with similar functions and tissue specific patterns of expression. The main catalytic activity of ALDHs is to oxidize aldehydes generated from cellular metabolism through an NAPD(P)-dependent reaction. However, ALDHs participate in other cellular processes, including the regulation of retinoid signaling by conversion of retinal into retinoic acid. This role places a spotlight on ALDH as a key regulatory node in the process of cellular differentiation. Indeed, ALDH inhibition by DEAB was shown to delay cytokine-induced differentiation of hematopoietic progenitor cells through direct inhibition of retinoic signaling in these progenitors [
27].
ALDH1A enzymes are expressed abundantly in the liver, pancreas, intestine, and hypophysis and are present variably in cancer, particularly in colon, liver, pancreatic, and some ovarian and breast tumors [
17]. Among the various isoforms, ALDH1A1, 1A2, and 1A3 have been shown to be expressed and active in cancer [
28] and have been implicated in the development of chemo-resistance [
17,
25], a key characteristic of CSCs. Evaluation of specific isoforms in cancer tissue is somewhat limited by the specificity of available antibodies. Additionally, ALDH expression is considered to be restricted to progenitor cells, a rare cell population within tumors, further limiting the ability to distinguish between isoforms. The available literature supports that ALDH1A3, ALDH3A2, and ALDH7A1 isozymes are expressed more abundantly in ovarian tumors, particularly in mucinous and endometrioid type tumors [
16]. Our current results support the significance of ALHDH1A1 isoform to proliferation of OC cells as spheres and maintenance of a CSC phenotype, but also show that other ALDH1A isoforms are expressed in OC cells and could compensate for selective inhibition or knock down of ALDH1A1.
ALDH1 activity is increased in OC cells growing as spheroids compared to monolayers [
18,
25] and in OC cells and tumors that survived exposure to platinum [
18,
19]. Recent data from our group and others demonstrate that platinum activates cancer-associated fibroblasts in ovarian xenografts, thereby stimulating IL6 secretion, which in turn, transcriptionally upregulates ALDH activity [
19]. These observations suggest that a potential application of ALDH inhibitors could occur after completion of standard platinum-based therapy, with the goal of eradicating residual, chemotherapy-resistant, or tolerant cells, which are enriched for ALDH activity and responsible for giving rise to recurrent, recalcitrant tumors. Other strategies which could indirectly block ALDH1A1 include agents targeting transcriptional regulatory mechanisms, such as bromodomain and extra terminal (BET) inhibitors [
29], DNA hypomethylating agents [
18], and STAT inhibitors [
19] which have been shown to eliminate ALDH+ cells residual after platinum treatment. Interestingly, ALDH1A expression was shown to be inducible by BRD4 and by STAT3 at transcriptional level, however, agents targeting these circuitries may have other non-specific effects and could affect stemness phenotype through non-ALDH related mechanisms.
Here, we tested CM37, a first in class small molecule, discovered as the lead compound from a high-throughput screen of the ChemDiv library. While the compound was found to be potent at nanomolar concentrations in vitro and highly selective for ALDH1A1, its limited bioavailability and short half-life in vivo (~2 minutes) lessened its activity in vivo. Importantly, the inhibitor was tolerated in vivo at concentrations up to 20 mg/kg, without significant systemic toxicity. Efforts to improve its properties are ongoing by using rational chemistry design strategies specifically targeting the “arms” that extend from the central scaffold in order to improve the metabolic stability and modify the lipophilicity of the compound. The observed spectrum of activity of CM37 is in line with reports testing other inhibitors in this class. For example, disulfiram, a broad ALDH inhibitor, was shown to eradicate paclitaxel-resistant triple negative breast cancer cells [
24] as well as kinase inhibitor-tolerant lung cancer cells addicted to specific mutant oncogene drivers [
30]. However, disulfiram is non-specific and has high systemic toxicity, limiting its potential clinical applicability. Therefore, development of more selective inhibitors has garnered interest. A quinoline-based series of analogues which block ALDH1A1 has been recently described. The lead compound had activity similar to CM37 in OC spheroids and promoted synergy with paclitaxel [
31]. Optimization of other novel chemical structures inhibiting ALDH1A1, A2, and A3 has been reported [
32]. The lead compound was shown to deplete CSCs, synergize with platinum, and have in vivo activity when injected intra-tumorally [
32]. These emerging results point to growing interest exploring this pathway, which is clearly significant to the survival of drug-resistant cancer cells.
The mechanism by which ALDH inhibitors target CSCs remains not fully understood. Here we show that CM37 induces accumulation of ROS, which in turn, causes DNA damage. A disturbance in the intracellular redox balance was also shown to be induced by disulfiram in drug-tolerant lung cancer cells [
30] and is consistent with the known functions of the target enzyme. Knockdown of
ALDH1A1 by siRNA was shown to induce DNA damage in another report, consistent with our findings [
22]. Thus, we predict that sensitivity to ALDH inhibitors is likely be influenced by the selective expression of ALDH isoforms active in specific contexts and/or by the presence of other mechanisms that regulate oxidative stress in parallel. We cannot exclude that CM37 and other ALDH inhibitors may block stemness through other pathways (e.g., differentiation pathways). Collectively, our results consolidate the concept that ALDH1A1 plays an important role regulating stemness in ovarian cancer, describe the activity of a novel inhibitor targeting this rare and resistant cell population, and support continued efforts to optimize this new class of anti-cancer agents.
4. Materials and Methods
4.1. Chemicals and Reagents
CM37 was synthesized in the Chemical Genomics Medicinal Chemistry Core at Indiana University School of Medicine, Indianapolis. Trolox was purchased from Sigma-Aldrich (St. Louis, MO, USA). The ALDEFLUOR ALDH activity assay kit was purchased from StemCell Technologies (Vancouver, BC, Canada). Antibodies against phospho-histone H2AX (mAB #9718) and histone H2AX (2595S) were purchased from Cell Signaling Technology (Danvers, MA, USA), and against GAPDH from Biodesign International (Saco, ME, USA). The secondary HRP-conjugated antibodies were purchased from Amersham Biosciences (San Francisco, CA, USA) and Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). siRNAs targeting the ALDH1A1 isoform and non-targeting control siRNA were purchased from Dharmacon (Lafayette, CO, USA). Lentiviral particles encoding shALDH1A1 or scrambled, non-silencing control (Sh-control) siRNA were obtained from Sigma-Aldrich.
4.2. Cell Lines
The human OC cell lines COV362 and OVCAR5 were provided by Dr. Kenneth Nephew, (Indiana University). SKOV3, OVCAR3, and OV90 OC cell lines were purchased from American Type Culture Collection (Rockville, MD, USA). Cell lines were authenticated using Short Tandem Repeat (STR) DNA profiling analysis and tested mycoplasma negative (IDEXX Bioresearch, Columbia, MO, USA). SKOV3 and primary cells recovered from tumors or ascites were cultured in 1:1 MCDB 105 and M199 (Cellgro, Manassas, VA, USA) supplemented with 10% FBS (Cellgro) and 100 units/mL penicillin and 100 µg/mL streptomycin. OVCAR3 were cultured in DMEM high glucose medium supplemented with 10% FBS and 100 units/mL penicillin and 100 μg/mL streptomycin. OVCAR5 cells were grown in DMEM (high glucose), 10% (FBS), 0.1 mM non-essential amino acids (NEAA), 2 mM L-glutamine, and antibiotics. Spheroid cultures were maintained in Mammocult Complete medium (StemCell Technologies) and ultra-low attachment plates. Cells were cultured at 37 °C in a humidified incubator with 5% CO2 supply.
4.3. Patient-Derived Primary Human OC Cells
De-identified malignant ascites fluid was obtained from subjects with high-grade serous OC or with primary peritoneal carcinomatosis. The samples were obtained at the Indiana University Simon Cancer Center (IUSCC) and Northwestern University Robert H. Lurie Cancer Center under IRB approved protocols (Indiana University CRO#505; Northwestern University #STU00202468, respectively). For primary cell isolation from human specimens, we used previously described methods [
21]. In brief, freshly collected ascites specimens were centrifuged at low speed for 20 minutes. After removal of the fluid, cell pellets were dispersed mechanically and dissociated by using Cellstripper (Corning, Corning, NY, USA; Cat# 25-056-CI). Red blood cell (RBC) (BioLegend, San Diego, CA; Cat#420301) lysis buffer was used to remove RBC and DNase (Qiagen, Valencia, CA, USA; Cat# 79254) was used to digest free DNA in cell suspensions. The single cell suspensions were filtered through a 40 μm cell strainer (ThermoFisher Scientific, Waltham, MA, USA; Cat#NC0147038) to remove other debris before plating in non-adherent plates.
4.4. In Vivo Xenograft Studies
Foxn1nu nude mice were purchased from Harlan (Indianapolis, IN, USA), and were maintained at the Northwestern University Center for Comparative Medicine in accordance with the National Institutes of Health guidelines and following protocols approved by the Northwestern University Animal Use and Care Committee (Protocol #IS00003060). In brief, 20,000 OVCAR3 OC cells stably transfected with sh-control or sh-ALDH1A1 lentivirus were subcutaneously injected into the flanks of 6–8-week-old female nude mice. Tumors were assessed and measured twice weekly to assess tumor initiation. Time to tumor initiation was determined as the time from inoculation to the time when tumors were detectable by palpation (measuring at least 2 mm in greatest dimension). Tumors were allowed to form and grow until they reached 1500 mm3. At that point, mice were euthanized and tumors were harvested, measured, and weighed.
4.5. Sphere Formation Assay
The sphere formation assay was performed as previously described, with some modifications [
21,
25]. Briefly, OC or primary cells were seeded at a density of 10
4/100 μL in complete MammoCult media supplemented with 1.5% penicillin/streptomycin (Stemcell Technologies, Cambridge, MA, USA) in ultra-low attachment plates (Corning, Corning, NY, USA) and allowed to form spheroids for 7 days. Images were captured, and the numbers of spheres/well were quantified. The data are presented as means ± SEM of triplicate, independent experiments.
4.6. ALDEFLUOR Assay and Fluorescence-Activated Cell Sorting
ALDH1 enzymatic activity was measured using the Aldefluor assay kit, per manufacturer recommendations (Stemcell Technologies). Briefly, OC monolayers were dissociated with trypsin and resuspended in Aldefluor assay buffer at a density of 2 × 106 cells/mL. The cell suspension was treated with 5 μL/mL bodipyaminoacetaldehyde (BAAA) and incubated in a 37 °C water bath for 60 minutes. The control sample (ALDH-negative cells), was derived from a 500 μL aliquot of BAAA-treated cell suspension incubated with a 15–30 μM of the ALDH inhibitor, diethylamino benzaldehyde (DEAB), and incubated as mentioned above. As such, the ALDH1A1-positive population was identified and gated using DEAB-treated cells as our control sample. The relative increase in FITC signal of the ALDH-positive cells was determined by a FACS Aria II flow cytometer (BD Biosciences, San Jose, CA, USA) and analyzed in at least two to three independent experiments.
4.7. CCK-8 Colorimetric Assay
The numbers of live cells were assessed by using the CCK-8 assay and following the manufacturer’s specifications (Dojindo Molecular Technologies, Rockville, MD, USA). Briefly, OC cells were seeded at a density of 104/100 μL in complete MammoCult media supplemented with penicillin/streptomycin (Stemcell Technologies, Cambridge, MA, USA) in 96-well ultra-low attachment plates (Corning, Corning, NY, USA). Numbers of live cells were assessed after 7 days by adding 10 μL of CCK-8 reagent to each well. Absorbance at 450 nm was quantified using a BioTek plate reader (Winooski, VT, USA). Assays were performed in at least four replicates. Data are presented as means ± SEM.
4.8. Cell Titer-Glo Cell Viability Assay
Spheroids were cultured as mentioned above. Numbers of live cells was measured by using Cell Titer-Glo kit (Promega, Madison, WI, USA) per the manufacturer’s recommendations with some modifications. Briefly, an equal volume of CellTiter- Glo reagent was added to each well and the plate was covered with aluminum foil and incubated on an orbital shaker for 30 min at RT. Lysates were transferred to opaque, low-binding polystyrene microplates (Corning, Corning, NY, USA) and allowed to equilibrate for 10 min. ATP abundance was quantified in a SpectraMax GeminiXS luminescence/fluorescence plate reader (Molecular Devices, Palo Alto, CA, USA). Experiments were performed in quadruplicate and repeated at least twice. Data are presented as means ± SEM.
4.9. Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted using RNA STAT-60 Reagent (Tel-Test Inc., Friendswood, TX, USA) and reverse-transcribed using an iScript cDNA kit (Bio-Rad, Hercules, CA, USA). Real-time PCR was used to measure
ALDH,
NANOG,
OCT4,
SOX2,
NIEL3,
RAD2,
RAD9A,
RAD9B,
RAD51, and
RAD23A expression, and human glyceraldehyde-3-phosphate dehydrogenase (
GAPDH), or beta-actin (
ACTB/
β-actin) were used as references. The relative expression of target genes was calculated using the ΔΔCt method. Results are presented as the means ±
SD of at least triplicate experiments. Primers are listed below (
Table 1).
4.10. DNA Damage Pathway PCR-Array
The human DNA Damage Pathway RT2 Profiler PCR Array and RT2 Real-Timer SyBR Green/ROX PCR Mix were purchased from Qiagen (Valencia, CA, USA). PCR was performed on an ABI Prism 7900 Sequence Detector (Applied Biosystems, Waltham, MA, USA). Data analysis was performed based on the ΔΔCt method with normalization of the raw data to the housekeeping genes, included in the array, using a Microsoft Excel algorithm provided by the manufacturer. For each gene, fold-changes were calculated as the difference in gene expression between control and CM37-treated spheroid cultures. An ontology classification assignment for each gene was performed, and fold-changes were calculated and expressed as percent of composition for each represented pathway in control versus treated spheres.
4.11. ALDH Enzymatic Activity
Inhibitory activity of CM37 against ALDH orthologs was tested by using purified recombinant human ALDH1A1, ALDH1A2, ALDH1A3, ALDH2, and ALDH3A1. Dehydrogenase activity of ALDH1A1, ALDH1A2, ALDH1A3, and ALDH2 were measured in a solution containing 100–200 nM enzyme, 200 μM NAD+, 1% DMSO, and 100 μM propionaldehyde in 50 mM sodium BES, pH 7.5. ALDH3A1 activity was measured using 25 nM enzyme, 200 μM NAD+, 1% DMSO, and 1 mM benzaldehyde in 100 mM sodium phosphate buffer, pH 7.5. All assays were performed at 25 °C and were initiated by the addition of the aldehyde substrate following pre-incubation with compound and NAD+. The values represent the average of three independent experiments. Data are presented as percent inhibition.
4.12. Immunofluorescence (IF)
OVCAR5 and SKOV3 OC cells were seeded at a density of 5 × 104/mL in complete MammoCult media supplemented with penicillin/streptomycin (Stemcell Technologies, Cambridge, MA, USA) in 6-well ultra-low attachment plates (Corning, Corning, NY, USA) for 5 days and treated with CM37 (1 μM) or DSMO every other day. OC spheroids were fixed, permeabilized, and stained with γH2AX antibody (1:200, Cell Signaling Technology, Danvers, MA, USA). Spheroids were counterstained with Alexa fluor-488 anti-rabbit secondary antibody (1:1000; Molecular Probes, Eugene, OR, USA), and nuclei were visualized by Hoechst staining (Molecular Probes). Images were captured using a Nikon A1 Confocal Laser Microscope at the Northwestern University Center for Advanced Microscopy (Chicago, IL, USA).
4.13. Western Blot
Cells were lysed in buffer containing leupeptin (1 μg/mL), aprotinin (1 μg/mL), phenylmethylsulfonyl fluoride (PMSF; 400 μM), and sodium orthovanadate (Na3VO4; 1 mM). Lysates were briefly sonicated, then cleared by centrifugation (8000× g, 10 min, 4 °C), and total protein was measured using the Pierce BCA protein assay kit (ThermoFisher Scientific, Waltham, MA, USA). The lysates were resolved by SDS-PAGE (10% Tris-HCl polyacrylamide gels, Biorad, Hercules, CA, USA) and transferred to PVDF membranes (GE Healthcare Life Sciences, Pittsburg, PA, USA). The membranes were rinsed in Tris-buffered saline with 0.1% Tween 20 (TBS-T) and blocked for 30 min in TBS-T with 5% bovine serum albumin (BSA). Membranes were incubated with primary antibody overnight at 4 °C, and then with secondary antibody for 1 h. Antigen–antibody complexes were visualized using the enhanced chemiluminescence kit (Thermo Scientific, Waltham, MA, USA), and images were obtained using the ImageQuant LAS 4000 mini-imager (GE Healthcare, Pittsburgh, PA, USA). Images were quantified by densitometry using ImageJ32 software (National Institutes of Health, Bethesda, MD, USA). All values were corrected for the integrated optical density (iOD) and normalized to loading controls.
4.14. ROS Activity Assay
Mean reactive oxygen species (ROS) accumulation was measured by the DCFDA/H2DCFDA-Cellular Reactive Oxygen Species Detection Assay Kit (Abcam, Cambridge, MA, USA). Briefly, OC cells were resuspended at a concentration of 104 cells/mL in complete MammoCult media supplemented with penicillin/streptomycin (Stemcell Technologies, Cambridge, MA, USA) in 6-well ultra-low attachment plates (Corning, Corning, NY, USA) and allowed to form spheroids for 48 h. Spheroids were dissociated using trypsin, rinsed with complete media (DMEM + 10FBS, + Pen/Strep), then resuspended in complete MammoCult media supplemented with penicillin/streptomycin. Cell suspensions were treated with 20 µM DCFDA and incubated at 37 °C for 30 minutes, then treated with the indicated doses of CM37 or DSMO for 30 min–1 h, depending on the cell line. Positive control samples were treated with 100 µM TBHP. 1 µM propidium iodide was added for cell death exclusion immediately preceding mean ROS measurement by flow cytometry. Data are presented as means ± SEM of independent, triplicate experiments.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}