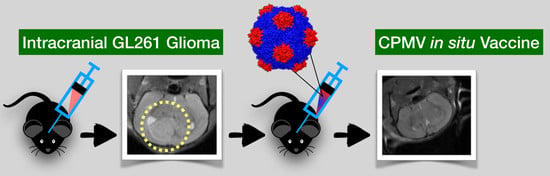
Plant Virus-Like Particle In Situ Vaccine for Intracranial Glioma Immunotherapy

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
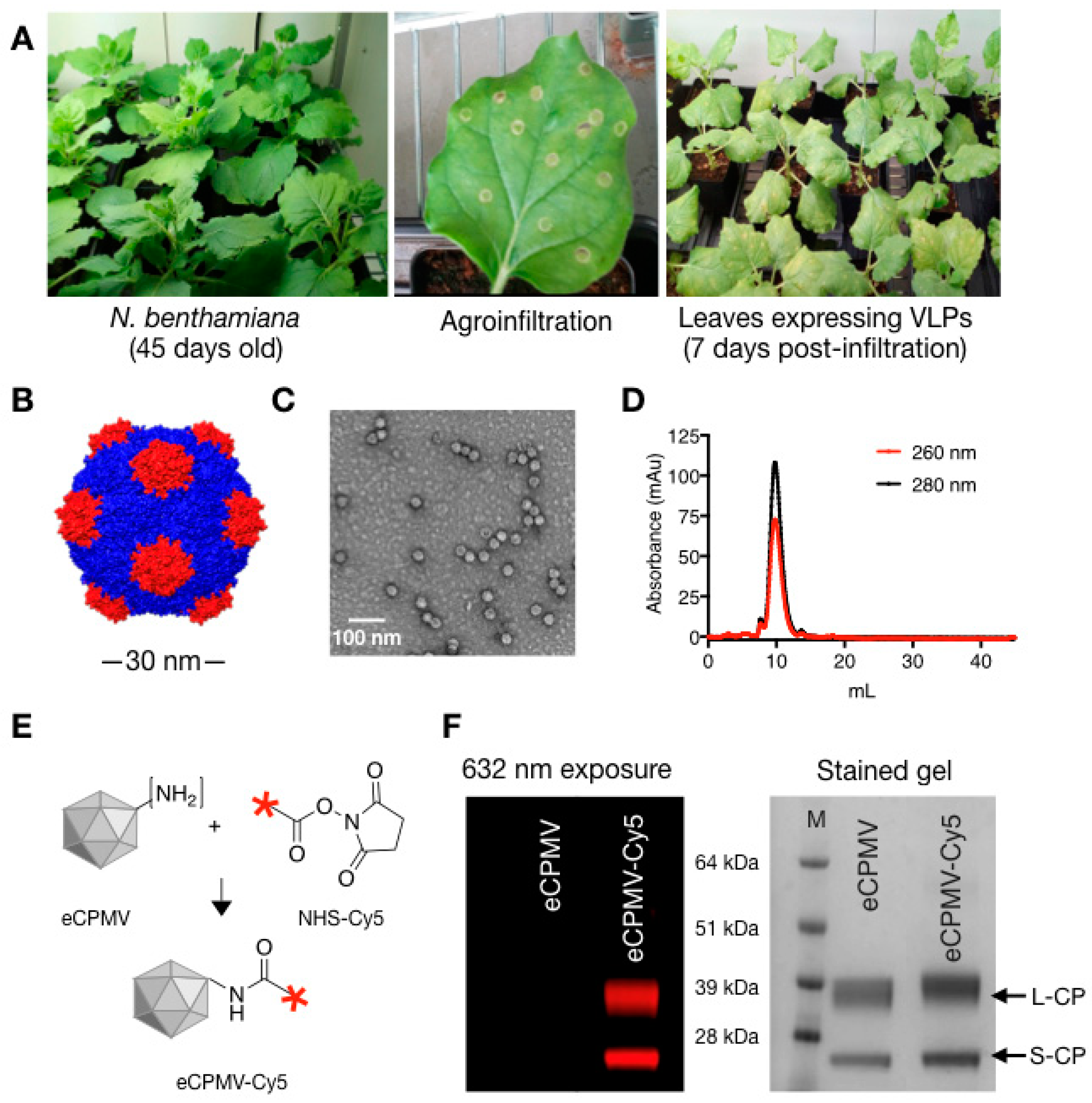
2. Results
3. Discussion
4. Materials and Methods
4.1. Production of eCPMV VLPs
4.2. Synthesis and Characterization of eCPMV-Cy5 Particles
4.3. Cell Line
4.4. Tumor Inoculation
4.5. Small Animal MRI
4.6. Flow Cytometry
4.7. Immunohistochemistry and H&E-Staining
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro Oncol. 2018, 20 (Suppl. 4), iv1–iv86. [Google Scholar] [CrossRef]
- Schima, W.; Ba-Ssalamah, A.; Kolblinger, C.; Kulinna-Cosentini, C.; Puespoek, A.; Gotzinger, P. Pancreatic adenocarcinoma. Eur. Radiol. 2007, 17, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Ningaraj, N.S. Drug delivery to brain tumours: Challenges and progress. Expert Opin. Drug Deliv. 2006, 3, 499–509. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells in radiation resistance. Cancer Res. 2007, 67, 8980–8984. [Google Scholar] [CrossRef]
- Boussiotis, V.A.; Charest, A. Immunotherapies for malignant glioma. Oncogene 2018, 37, 1121–1141. [Google Scholar] [CrossRef] [PubMed]
- Hoang-Minh, L.B.; Mitchell, D.A. Immunotherapy for Brain Tumors. Curr. Treat. Options Oncol. 2018, 19, 60. [Google Scholar] [CrossRef] [PubMed]
- Lyon, J.G.; Mokarram, N.; Saxena, T.; Carroll, S.L.; Bellamkonda, R.V. Engineering challenges for brain tumor immunotherapy. Adv. Drug Deliv. Rev. 2017, 114, 19–32. [Google Scholar] [CrossRef]
- Ratnam, N.M.; Gilbert, M.R.; Giles, A.J. Immunotherapy in CNS Cancers: The Role of Immune Cell Trafficking. Neuro Oncol. 2018, 21, 37–46. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.F.; Idema, A.J.; Bol, K.F.; Grotenhuis, J.A.; de Vries, I.J.; Wesseling, P.; Adema, G.J. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J. Neuroimmunol. 2010, 225, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.J.; Turaga, S.M.; Jarrar, A.; Reizes, O.; Longworth, M.S.; et al. Glioblastoma Cancer Stem Cells Evade Innate Immune Suppression of Self-Renewal through Reduced TLR4 Expression. Cell Stem Cell 2017, 20, 450–461.e4. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Bregy, A.; Wong, T.M.; Shah, A.H.; Goldberg, J.M.; Komotar, R.J. Active immunotherapy using dendritic cells in the treatment of glioblastoma multiforme. Cancer Treat. Rev. 2013, 39, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Kuramitsu, S.; Yamamichi, A.; Ohka, F.; Motomura, K.; Hara, M.; Natsume, A. Adoptive immunotherapy for the treatment of glioblastoma: Progress and possibilities. Immunotherapy 2016, 8, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Lassman, A.B.; van den Bent, M.; Kumthekar, P.; Merrell, R.; Scott, A.M.; Fichtel, L.; Sulman, E.P.; Gomez, E.; Fischer, J.; et al. Efficacy and safety results of ABT-414 in combination with radiation and temozolomide in newly diagnosed glioblastoma. Neuro Oncol. 2017, 19, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Sirachainan, N.; Boongird, A.; Swangsilpa, T.; Klaisuban, W.; Lusawat, A.; Hongeng, S. Reported outcomes of children with newly diagnosed high-grade gliomas treated with nimotuzumab and irinotecan. Childs Nerv. Syst. 2017, 33, 893–897. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Dobrikova, E.Y.; Broadt, T.; Poiley-Nelson, J.; Yang, X.; Soman, G.; Giardina, S.; Harris, R.; Gromeier, M. Recombinant oncolytic poliovirus eliminates glioma in vivo without genetic adaptation to a pathogenic phenotype. Mol. Ther. 2008, 16, 1865–1872. [Google Scholar] [CrossRef]
- Delwar, Z.M.; Liu, G.; Kuo, Y.; Lee, C.; Bu, L.; Rennie, P.S.; Jia, W.W. Tumour-specific triple-regulated oncolytic herpes virus to target glioma. Oncotarget 2016, 7, 28658–28669. [Google Scholar] [CrossRef]
- Hardcastle, J.; Mills, L.; Malo, C.S.; Jin, F.; Kurokawa, C.; Geekiyanage, H.; Schroeder, M.; Sarkaria, J.; Johnson, A.J.; Galanis, E. Immunovirotherapy with measles virus strains in combination with anti-PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro Oncol. 2017, 19, 493–502. [Google Scholar] [CrossRef]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.D.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.M.; Zhang, R.; Shan, C.; et al. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 2017, 214, 2843–2857. [Google Scholar] [CrossRef]
- Lizotte, P.H.; Wen, A.M.; Sheen, M.R.; Fields, J.; Rojanasopondist, P.; Steinmetz, N.F.; Fiering, S. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat. Nanotechnol. 2016, 11, 295–303. [Google Scholar] [CrossRef]
- Hoopes, P.J.; Wagner, R.J.; Duval, K.; Kang, K.; Gladstone, D.J.; Moodie, K.L.; Crary-Burney, M.; Ariaspulido, H.; Veliz, F.A.; Steinmetz, N.F.; et al. Treatment of Canine Oral Melanoma with Nanotechnology-Based Immunotherapy and Radiation. Mol. Pharm. 2018, 15, 3717–3722. [Google Scholar] [CrossRef]
- Wen, A.M.; Shukla, S.; Saxena, P.; Aljabali, A.A.; Yildiz, I.; Dey, S.; Mealy, J.E.; Yang, A.C.; Evans, D.J.; Lomonossoff, G.P.; et al. Interior engineering of a viral nanoparticle and its tumor homing properties. Biomacromolecules 2012, 13, 3990–4001. [Google Scholar] [CrossRef]
- Lin, T.; Chen, Z.; Usha, R.; Stauffacher, C.V.; Dai, J.B.; Schmidt, T.; Johnson, J.E. The refined crystal structure of cowpea mosaic virus at 2.8 A resolution. Virology 1999, 265, 20–34. [Google Scholar] [CrossRef]
- Van Wezenbeek, P.; Verver, J.; Harmsen, J.; Vos, P.; van Kammen, A. Primary structure and gene organization of the middle-component RNA of cowpea mosaic virus. EMBO J. 1983, 2, 941–946. [Google Scholar] [CrossRef]
- Chatterji, A.; Ochoa, W.F.; Paine, M.; Ratna, B.R.; Johnson, J.E.; Lin, T. New addresses on an addressable virus nanoblock; uniquely reactive Lys residues on cowpea mosaic virus. Chem. Biol. 2004, 11, 855–863. [Google Scholar] [CrossRef]
- Singh, P.; Prasuhn, D.; Yeh, R.M.; Destito, G.; Rae, C.S.; Osborn, K.; Finn, M.G.; Manchester, M. Bio-distribution, toxicity and pathology of cowpea mosaic virus nanoparticles in vivo. J. Control. Release 2007, 120, 41–50. [Google Scholar] [CrossRef]
- Szatmari, T.; Lumniczky, K.; Desaknai, S.; Trajcevski, S.; Hidvegi, E.J.; Hamada, H.; Safrany, G. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci. 2006, 97, 546–553. [Google Scholar] [CrossRef]
- Murray, A.A.; Wang, C.; Fiering, S.; Steinmetz, N.F. In Situ Vaccination with Cowpea vs. Tobacco Mosaic Virus against Melanoma. Mol. Pharm. 2018, 15, 3700–3716. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Lebel, M.E.; Chartrand, K.; Tarrab, E.; Savard, P.; Leclerc, D.; Lamarre, A. Potentiating Cancer Immunotherapy Using Papaya Mosaic Virus-Derived Nanoparticles. Nano Lett. 2016, 16, 1826–1832. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Alizadeh, D.; Zhang, L.; Brown, C.E.; Farrukh, O.; Jensen, M.C.; Badie, B. Induction of anti-glioma natural killer cell response following multiple low-dose intracerebral CpG therapy. Clin. Cancer Res. 2010, 16, 3399–3408. [Google Scholar] [CrossRef]
- Chicoine, M.R.; Zahner, M.; Won, E.K.; Kalra, R.R.; Kitamura, T.; Perry, A.; Higashikubo, R. The in vivo antitumoral effects of lipopolysaccharide against glioblastoma multiforme are mediated in part by Toll-like receptor 4. Neurosurgery 2007, 60, 372–380, Discussion 81. [Google Scholar] [CrossRef]
- Brandsma, D.; Stalpers, L.; Taal, W.; Sminia, P.; van den Bent, M.J. Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas. Lancet Oncol. 2008, 9, 453–461. [Google Scholar] [CrossRef]
- Kleijn, A.; Chen, J.W.; Buhrman, J.S.; Wojtkiewicz, G.R.; Iwamoto, Y.; Lamfers, M.L.; Stemmer-Rachamimov, A.O.; Rabkin, S.D.; Weissleder, R.; Martuza, R.L.; et al. Distinguishing inflammation from tumor and peritumoral edema by myeloperoxidase magnetic resonance imaging. Clin. Cancer Res. 2011, 17, 4484–4493. [Google Scholar] [CrossRef]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Sun, B.; Qin, W.; Song, M.; Liu, L.; Yu, Y.; Qi, X.; Sun, H. Neutrophil Suppresses Tumor Cell Proliferation via Fas/Fas Ligand Pathway Mediated Cell Cycle Arrested. Int. J. Biol. Sci. 2018, 14, 2103–2113. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell. Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef]
- Screpanti, V.; Wallin, R.P.; Ljunggren, H.G.; Grandien, A. A central role for death receptor-mediated apoptosis in the rejection of tumors by NK cells. J. Immunol. 2001, 167, 2068–2073. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef]
- Harris, M.G.; Hulseberg, P.; Ling, C.; Karman, J.; Clarkson, B.D.; Harding, J.S.; Zhang, M.; Sandor, A.; Christensen, K.; Nagy, A.; et al. Immune privilege of the CNS is not the consequence of limited antigen sampling. Sci. Rep. 2014, 4, 4422. [Google Scholar] [CrossRef]
- Cserr, H.F.; Harling-Berg, C.J.; Knopf, P.M. Drainage of brain extracellular fluid into blood and deep cervical lymph and its immunological significance. Brain Pathol. 1992, 2, 269–276. [Google Scholar] [CrossRef]
- Tian, G.; Courtney, A.N.; Jena, B.; Heczey, A.; Liu, D.; Marinova, E.; Guo, L.; Xu, X.; Torikai, H.; Mo, Q.; et al. CD62L+NKT cells have prolonged persistence and antitumor activity in vivo. J. Clin. Investig. 2016, 126, 2341–2355. [Google Scholar] [CrossRef]
- Ko, H.J.; Lee, J.M.; Kim, Y.J.; Kim, Y.S.; Lee, K.A.; Kang, C.Y. Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of activated NKT cells: An alternative cell-based antitumor vaccine. J. Immunol. 2009, 182, 1818–1828. [Google Scholar] [CrossRef]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehman, S.; Kalin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Yoo, B.C.; Jung, J.W.; Oh, E.S.; Hwang, J.S.; Shin, J.A.; Kim, S.Y.; Cha, S.H.; Han, I.O. Induction of glioma apoptosis by microglia-secreted molecules: The role of nitric oxide and cathepsin B. Biochim. Biophys. Acta 2009, 1793, 1656–1668. [Google Scholar] [CrossRef]
- Kees, T.; Lohr, J.; Noack, J.; Mora, R.; Gdynia, G.; Todt, G.; Ernst, A.; Radlwimmer, B.; Falk, C.S.; Herold-Mende, C.; et al. Microglia isolated from patients with glioma gain antitumor activities on poly (I:C) stimulation. Neuro Oncol. 2012, 14, 64–78. [Google Scholar] [CrossRef]
- Sarkar, S.; Doring, A.; Zemp, F.J.; Silva, C.; Lun, X.; Wang, X.; Kelly, J.; Hader, W.; Hamilton, M.; Mercier, P.; et al. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat. Neurosci. 2014, 17, 46–55. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Lisi, L.; Stigliano, E.; Lauriola, L.; Navarra, P.; Dello Russo, C. Proinflammatory-activated glioma cells induce a switch in microglial polarization and activation status, from a predominant M2b phenotype to a mixture of M1 and M2a/B polarized cells. ASN Neuro 2014, 6, 171–183. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Abou-Ghazal, M.; Reina-Ortiz, C.; Yang, D.S.; Sun, W.; Qiao, W.; Hiraoka, N.; Fuller, G.N. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin. Cancer Res. 2008, 14, 5166–5172. [Google Scholar] [CrossRef]
- DiDomenico, J.; Lamano, J.B.; Oyon, D.; Li, Y.; Veliceasa, D.; Kaur, G.; Ampie, L.; Choy, W.; Lamano, J.B.; Bloch, O. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology 2018, 7, e1448329. [Google Scholar] [CrossRef]
- Hanihara, M.; Kawataki, T.; Oh-Oka, K.; Mitsuka, K.; Nakao, A.; Kinouchi, H. Synergistic antitumor effect with indoleamine 2,3-dioxygenase inhibition and temozolomide in a murine glioma model. J. Neurosurg. 2016, 124, 1594–1601. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2014, 20, 5290–5301. [Google Scholar] [CrossRef]
- Vom Berg, J.; Vrohlings, M.; Haller, S.; Haimovici, A.; Kulig, P.; Sledzinska, A.; Weller, M.; Bercher, B. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J. Exp. Med. 2013, 210, 2803–2811. [Google Scholar] [CrossRef]
- Kaurav, H.; Kapoor, D.N. Implantable systems for drug delivery to the brain. Ther. Deliv. 2017, 8, 1097–1107. [Google Scholar] [CrossRef]
- Pridgen, E.M.; Langer, R.; Farokhzad, O.C. Biodegradable, polymeric nanoparticle delivery systems for cancer therapy. Nanomedicine 2007, 2, 669–680. [Google Scholar] [CrossRef]
- Dagdeviren, C.; Ramadi, K.B.; Joe, P.; Spencer, K.; Schwerdt, H.N.; Shimazu, H.; Delcasso, S.; Amemori, K.; Nunez-Lopez, C.; Graybiel, M.; et al. Miniaturized neural system for chronic, local intracerebral drug delivery. Sci. Transl. Med. 2018, 10, eaan2742. [Google Scholar] [CrossRef]
- Lee, P.W.; Shukla, S.; Wallat, J.D.; Danda, C.; Steinmetz, N.F.; Maia, J.; Pokorski, J.K. Biodegradable Viral Nanoparticle/Polymer Implants Prepared via Melt-Processing. ACS Nano 2017, 11, 8777–8789. [Google Scholar] [CrossRef]
- Czapar, A.E.; Tiu, B.D.B.; Veliz, F.A.; Pokorski, J.K.; Steinmetz, N.F. Slow-Release Formulation of Cowpea Mosaic Virus for In Situ Vaccine Delivery to Treat Ovarian Cancer. Adv. Sci. 2018, 5, 1700991. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerstetter-Fogle, A.; Shukla, S.; Wang, C.; Beiss, V.; Harris, P.L.R.; Sloan, A.E.; Steinmetz, N.F. Plant Virus-Like Particle In Situ Vaccine for Intracranial Glioma Immunotherapy. Cancers 2019, 11, 515. https://doi.org/10.3390/cancers11040515
Kerstetter-Fogle A, Shukla S, Wang C, Beiss V, Harris PLR, Sloan AE, Steinmetz NF. Plant Virus-Like Particle In Situ Vaccine for Intracranial Glioma Immunotherapy. Cancers. 2019; 11(4):515. https://doi.org/10.3390/cancers11040515
Chicago/Turabian StyleKerstetter-Fogle, Amber, Sourabh Shukla, Chao Wang, Veronique Beiss, Peggy L. R. Harris, Andrew E. Sloan, and Nicole F. Steinmetz. 2019. "Plant Virus-Like Particle In Situ Vaccine for Intracranial Glioma Immunotherapy" Cancers 11, no. 4: 515. https://doi.org/10.3390/cancers11040515
APA StyleKerstetter-Fogle, A., Shukla, S., Wang, C., Beiss, V., Harris, P. L. R., Sloan, A. E., & Steinmetz, N. F. (2019). Plant Virus-Like Particle In Situ Vaccine for Intracranial Glioma Immunotherapy. Cancers, 11(4), 515. https://doi.org/10.3390/cancers11040515