Next-Generation Hedgehog/GLI Pathway Inhibitors for Cancer Therapy



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Cancer Stem Cells and Hedgehog/GLI Signal Transduction
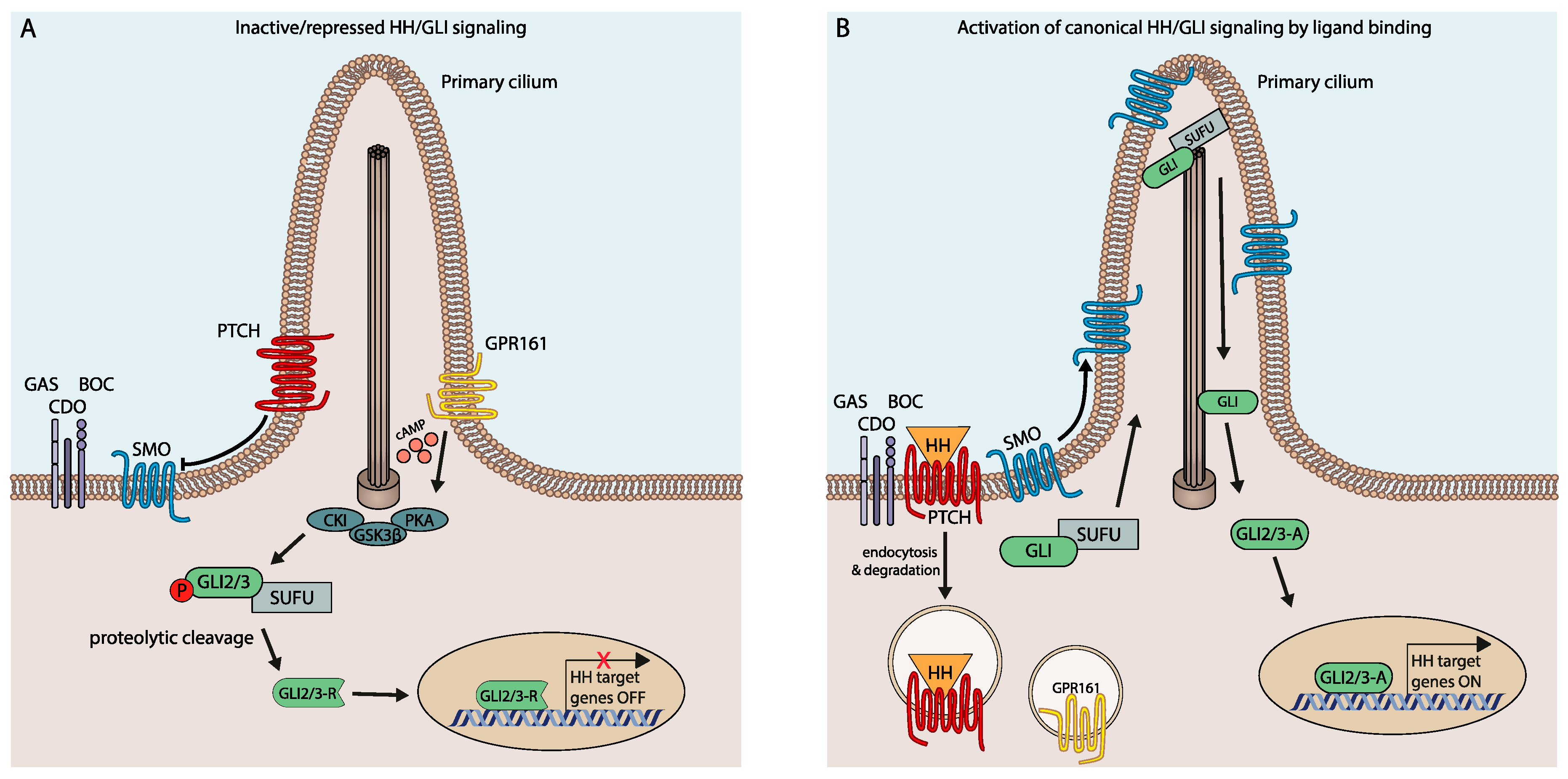
2. Canonical HH/GLI Signaling
3. Oncogenic HH/GLI Signaling
4. Therapeutic Targeting of Oncogenic HH/GLI Signaling
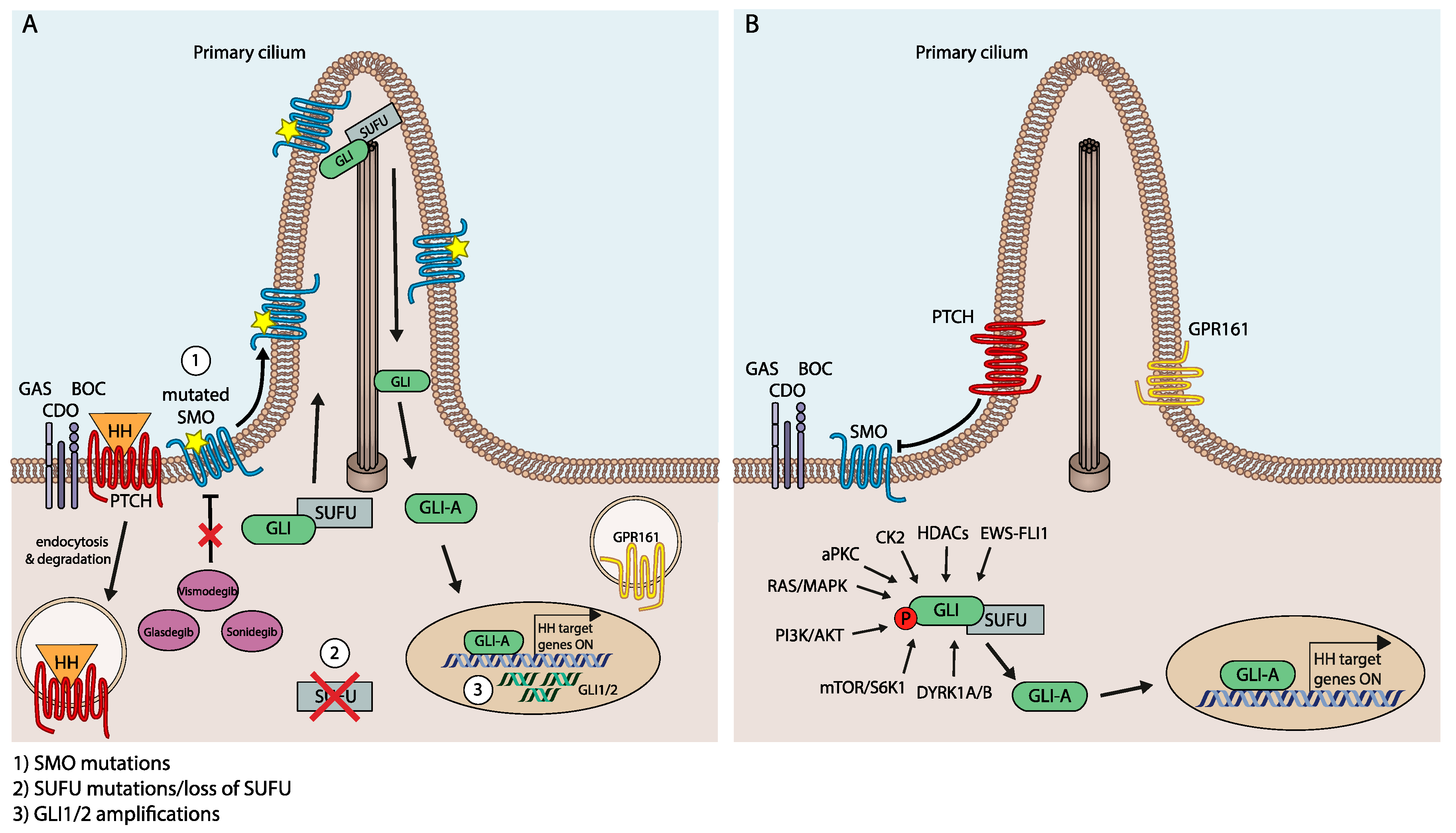
5. Mechanisms of Drug Resistance in HH/GLI Targeting
5.1. Resistance Mutations in HH/GLI Pathway Components
5.2. SMOi Resistance via SMO-Independent Induction of Oncogenic GLI Activity
5.3. Signal Crosstalk As Positive Modulator of Oncogenic HH/GLI Signaling and Mechanism of Drug Resistance
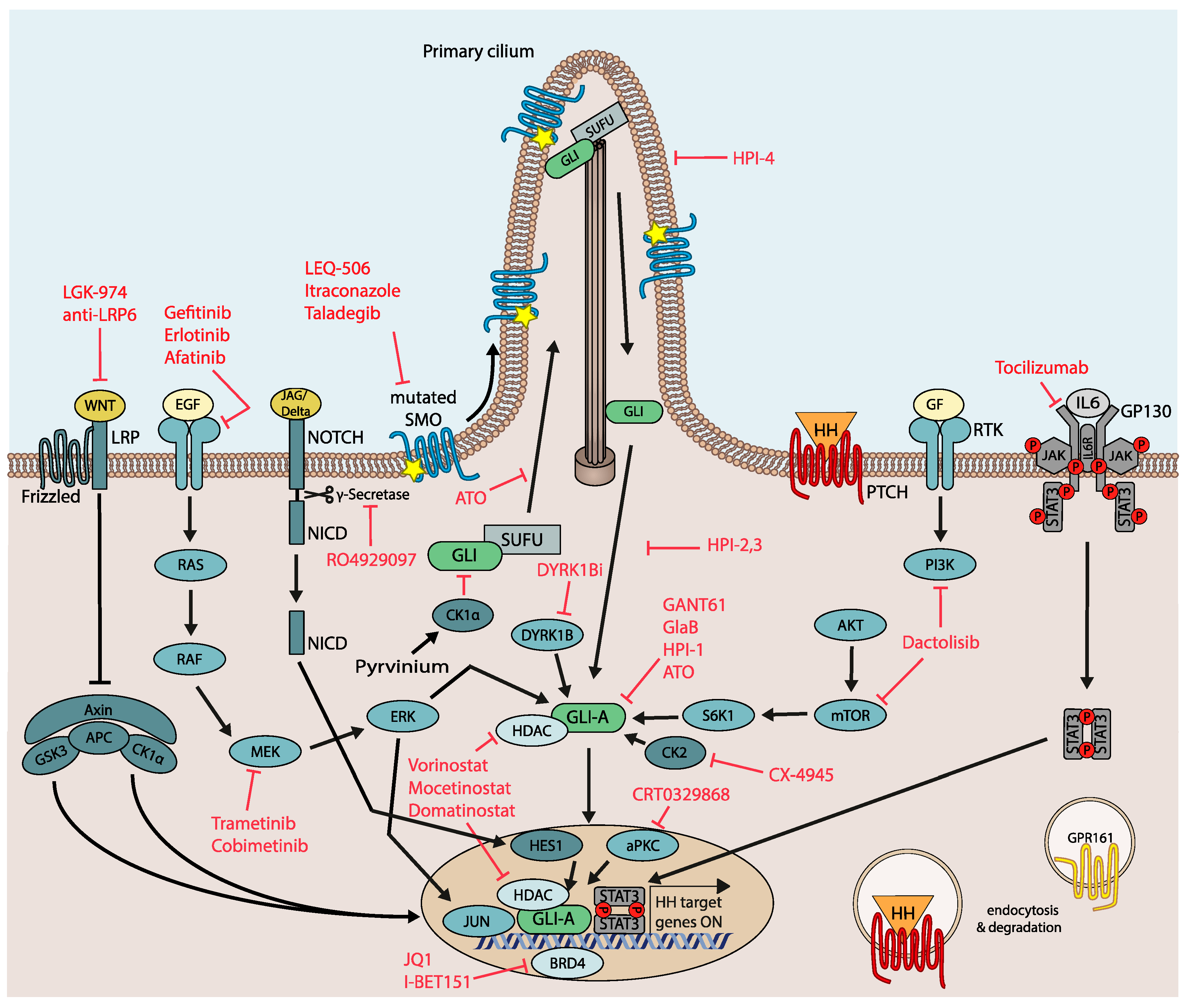
6. Overcoming SMO-Inhibitor Resistance by Next Generation HH/GLI Antagonists and Rational Combination Treatments
6.1. Targeting Drug Resistant SMO
6.2. Direct Targeting of Oncogenic GLI Transcription Factors
6.3. Targeting Modifiers of Oncogenic GLI Activity
6.4. Rational Combination Therapies Targeting Signal Integration and Cooperation—Future Applications of WNT, EGFR and IL6 Pathway Antagonists
7. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.; Blanpain, C. Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.D.; Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 2012, 488, 527–530. [Google Scholar] [CrossRef] [Green Version]
- Arwert, E.N.; Hoste, E.; Watt, F.M. Epithelial stem cells, wound healing and cancer. Nat. Rev. Cancer 2012, 12, 170–180. [Google Scholar] [CrossRef]
- Nawaz, M. Extracellular vesicle-mediated transport of non-coding RNAs between stem cells and cancer cells: Implications in tumor progression and therapeutic resistance. Stem Cell Investig. 2017, 4, 83. [Google Scholar] [CrossRef]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Rohatgi, R.; Scott, M.P. Patching the gaps in Hedgehog signalling. Nat. Cell Biol. 2007, 9, 1005–1009. [Google Scholar] [CrossRef]
- Hooper, J.E.; Scott, M.P. Communicating with Hedgehogs. Nat. Rev. Mol. Cell Biol. 2005, 6, 306–317. [Google Scholar] [CrossRef]
- Aberger, F.; Ruiz i Altaba, A. Context-dependent signal integration by the GLI code: The oncogenic load, pathways, modifiers and implications for cancer therapy. Semin. Cell Dev. Biol. 2014, 33, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Ruiz i Altaba, A.; Mas, C.; Stecca, B. The Gli code: An information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007, 17, 438–447. [Google Scholar] [CrossRef]
- Stecca, B.; Ruiz, I.A.A. Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. J. Mol. Cell Biol. 2010, 2, 84–95. [Google Scholar] [CrossRef]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Kasper, M.; Regl, G.; Frischauf, A.M.; Aberger, F. GLI transcription factors: Mediators of oncogenic Hedgehog signalling. Eur. J. Cancer 2006, 42, 437–445. [Google Scholar] [CrossRef]
- Di Magno, L.; Coni, S.; Di Marcotullio, L.; Canettieri, G. Digging a hole under Hedgehog: Downstream inhibition as an emerging anticancer strategy. Biochim. Biophys. Acta 2015, 1856, 62–72. [Google Scholar] [CrossRef]
- Van den Heuvel, M.; Ingham, P.W. Smoothened encodes a receptor-like serpentine protein required for hedgehog signalling. Nature 1996, 382, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bulkley, D.P.; Xin, Y.; Roberts, K.J.; Asarnow, D.E.; Sharma, A.; Myers, B.R.; Cho, W.; Cheng, Y.; Beachy, P.A. Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 2018, 175, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Price, M.A.; Kalderon, D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 2002, 108, 823–835. [Google Scholar] [CrossRef]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Unden, A.B.; Sandstedt, B.; Toftgard, R.; Zaphiropoulos, P.G. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat. Cell Biol. 1999, 1, 312–319. [Google Scholar] [CrossRef]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef]
- Gulino, A.; Di Marcotullio, L.; Canettieri, G.; De Smaele, E.; Screpanti, I. Hedgehog/Gli control by ubiquitination/acetylation interplay. Vitam. Horm. 2012, 88, 211–227. [Google Scholar] [CrossRef]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Whalen, E.J.; Liu, R.; Xiao, K.; Kim, J.; Chen, M.; Wang, J.; Chen, W.; Lefkowitz, R.J. Beta-arrestin-mediated localization of smoothened to the primary cilium. Science 2008, 320, 1777–1781. [Google Scholar] [CrossRef]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Corcoran, R.B.; Scott, M.P. Hedgehog signal transduction by Smoothened: Pharmacologic evidence for a 2-step activation process. Proc. Natl. Acad. Sci. USA 2009, 106, 3196–3201. [Google Scholar] [CrossRef] [Green Version]
- Raleigh, D.R.; Sever, N.; Choksi, P.K.; Sigg, M.A.; Hines, K.M.; Thompson, B.M.; Elnatan, D.; Jaishankar, P.; Bisignano, P.; Garcia-Gonzalo, F.R.; et al. Cilia-Associated Oxysterols Activate Smoothened. Mol. Cell 2018, 72, 316–327. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Rohatgi, R. G-protein-coupled receptors, Hedgehog signaling and primary cilia. Semin. Cell Dev. Biol. 2014, 33, 63–72. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef]
- Sasaki, H.; Nishizaki, Y.; Hui, C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar]
- Huntzicker, E.G.; Estay, I.S.; Zhen, H.; Lokteva, L.A.; Jackson, P.K.; Oro, A.E. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006, 20, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Chuang, P.T.; Kawcak, T.; McMahon, A.P. Feedback control of mammalian Hedgehog signaling by the Hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev. 2003, 17, 342–347. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Struhl, G. Dual roles for patched in sequestering and transducing Hedgehog. Cell 1996, 87, 553–563. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Teperino, R.; Aberger, F.; Esterbauer, H.; Riobo, N.; Pospisilik, J.A. Canonical and non-canonical Hedgehog signalling and the control of metabolism. Semin. Cell Dev. Biol. 2014, 33, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Teperino, R.; Amann, S.; Bayer, M.; McGee, S.L.; Loipetzberger, A.; Connor, T.; Jaeger, C.; Kammerer, B.; Winter, L.; Wiche, G.; et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell 2012, 151, 414–426. [Google Scholar] [CrossRef]
- Hanna, A.; Shevde, L.A. Hedgehog signaling: Modulation of cancer properies and tumor mircroenvironment. Mol. Cancer 2016, 15, 24. [Google Scholar] [CrossRef]
- Yang, L.; Xie, G.; Fan, Q.; Xie, J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 2010, 29, 469–481. [Google Scholar] [CrossRef]
- Kasper, M.; Jaks, V.; Fiaschi, M.; Toftgard, R. Hedgehog signalling in breast cancer. Carcinogenesis 2009, 30, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, P.; Hernandez, A.M.; Stecca, B.; Kahler, A.J.; DeGueme, A.M.; Barrett, A.; Beyna, M.; Datta, M.W.; Datta, S.; Ruiz i Altaba, A. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12561–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, M.; Jaks, V.; Hohl, D.; Toftgard, R. Basal cell carcinoma—Molecular biology and potential new therapies. J. Clin. Investig. 2012, 122, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Aberger, F.; Kern, D.; Greil, R.; Hartmann, T.N. Canonical and noncanonical hedgehog/gli signaling in hematological malignancies. Vitam. Horm. Hedgehog Signal. 2012, 88, 25–54. [Google Scholar]
- Aberger, F.; Hutterer, E.; Sternberg, C.; del Burgo, P.J.; Hartmann, T.N. Acute myeloid leukemia—Strategies and challenges for targeting oncogenic Hedgehog/GLI signaling. Cell Commun. Signal. 2017, 15. [Google Scholar] [CrossRef]
- Lau, B.W.; Huh, K.; Madero-Marroquin, R.; De Marchi, F.; Lim, Y.; Wang, Q.; Lobo, F.; Marchionni, L.; Smith, D.B.; DeZern, A.; et al. Hedgehog/GLI1 activation leads to leukemic transformation of myelodysplastic syndrome in vivo and GLI1 inhibition results in antitumor activity. Oncogene 2019, 38, 687–698. [Google Scholar] [CrossRef]
- Lim, Y.; Gondek, L.; Li, L.; Wang, Q.; Ma, H.; Chang, E.; Huso, D.L.; Foerster, S.; Marchionni, L.; McGovern, K.; et al. Integration of Hedgehog and mutant FLT3 signaling in myeloid leukemia. Sci. Transl. Med. 2015, 7, 291ra96. [Google Scholar] [CrossRef]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog—A cancer stem cell pathway. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef]
- Lim, Y.; Matsui, W. Hedgehog signaling in hematopoiesis. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef] [Green Version]
- Teglund, S.; Toftgard, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Szczepny, A.; Rogers, S.; Jayasekara, W.S.N.; Park, K.; McCloy, R.A.; Cochrane, C.R.; Ganju, V.; Cooper, W.A.; Sage, J.; Peacock, C.D.; et al. The role of canonical and non-canonical Hedgehog signaling in tumor progression in a mouse model of small cell lung cancer. Oncogene 2017, 36, 5544–5550. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Martelotto, L.G.; Peifer, M.; Sos, M.L.; Karnezis, A.N.; Mahjoub, M.R.; Bernard, K.; Conklin, J.F.; Szczepny, A.; Yuan, J.; et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat. Med. 2011, 17, 1504–1508. [Google Scholar] [CrossRef] [Green Version]
- Syu, L.J.; Zhao, X.; Zhang, Y.; Grachtchouk, M.; Demitrack, E.; Ermilov, A.; Wilbert, D.M.; Zheng, X.; Kaatz, A.; Greenson, J.K.; et al. Invasive mouse gastric adenocarcinomas arising from Lgr5+ stem cells are dependent on crosstalk between the Hedgehog/GLI2 and mTOR pathways. Oncotarget 2016, 7, 10255–10270. [Google Scholar] [CrossRef] [Green Version]
- Merchant, J.L.; Saqui-Salces, M. Inhibition of Hedgehog signaling in the gastrointestinal tract: Targeting the cancer microenvironment. Cancer Treat. Rev. 2014, 40, 12–21. [Google Scholar] [CrossRef]
- Goodrich, L.V.; Scott, M.P. Hedgehog and patched in neural development and disease. Neuron 1998, 21, 1243–1257. [Google Scholar] [CrossRef]
- Goodrich, L.V.; Milenkovic, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Whitson, R.J.; Oro, A.E. Advanced treatment for basal cell carcinomas. Cold Spring Harb. Perspect. Med. 2014, 4, a013581. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef]
- Rusert, J.M.; Wu, X.; Eberhart, C.G.; Taylor, M.D.; Wechsler-Reya, R.J. SnapShot: Medulloblastoma. Cancer Cell 2014, 26, 940. [Google Scholar] [CrossRef]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. Embo Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef] [Green Version]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Mueller, M.T.; Hermann, P.C.; Witthauer, J.; Rubio-Viqueira, B.; Leicht, S.F.; Huber, S.; Ellwart, J.W.; Mustafa, M.; Bartenstein, P.; D’Haese, J.G.; et al. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology 2009, 137, 1102–1113. [Google Scholar] [CrossRef]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Penachioni, J.Y.; Montagnani, V.; Olivito, B.; Gattai, R.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L.; et al. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells 2012, 30, 1808–1818. [Google Scholar] [CrossRef]
- Dierks, C.; Beigi, R.; Guo, G.R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog Signaling and Bmi-1 Regulate Self-renewal of Normal and Malignant Human Mammary Stem Cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [Green Version]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef]
- Tang, J.Y.; Ally, M.S.; Chanana, A.M.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Lindgren, J.A.; Ulerio, G.; Rezaee, M.R.; Gildengorin, G.; Marji, J.; et al. Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: Final results from the multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 2016, 17, 1720–1731. [Google Scholar] [CrossRef]
- Dlugosz, A.; Agrawal, S.; Kirkpatrick, P. Vismodegib. Nat. Rev. Drug Discov. 2012, 11, 437–438. [Google Scholar] [CrossRef] [Green Version]
- Hoy, S.M. Glasdegib: First Global Approval. Drugs 2019, 79, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Heidel, F.H.; Hellmann, A.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Pollyea, D.A.; DesJardins, P.; Ottmann, O.; et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 2019, 33, 379–389. [Google Scholar] [CrossRef]
- Savona, M.R.; Pollyea, D.A.; Stock, W.; Oehler, V.G.; Schroeder, M.A.; Lancet, J.; McCloskey, J.; Kantarjian, H.M.; Ma, W.W.; Shaik, M.N.; et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2294–2303. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N. Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.; et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Kuonen, F.; Huskey, N.E.; Shankar, G.; Jaju, P.; Whitson, R.J.; Rieger, K.E.; Atwood, S.X.; Sarin, K.Y.; Oro, A.E. Loss of primary cilia drives switching from Hedgehog to Ras/MAPK pathway in resistant basal cell carcinoma. J. Investig. Dermatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ponomaryov, T.; Ornell, K.J.; Zhou, P.; Dabral, S.K.; Pak, E.; Li, W.; Atwood, S.X.; Whitson, R.J.; Chang, A.L.; et al. RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway-Dependent Tumors. Cancer Res. 2015, 75, 3623–3635. [Google Scholar] [CrossRef]
- Gruber, W.; Hutzinger, M.; Elmer, D.P.; Parigger, T.; Sternberg, C.; Cegielkowski, L.; Zaja, M.; Leban, J.; Michel, S.; Hamm, S.; et al. DYRK1B as therapeutic target in Hedgehog/GLI-dependent cancer cells with Smoothened inhibitor resistance. Oncotarget 2016, 7, 7134–7148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.A.A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernandez-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Riobo, N.A.; Haines, G.M.; Emerson, C.P., Jr. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006, 66, 839–845. [Google Scholar] [CrossRef]
- Whisenant, T.C.; Ho, D.T.; Benz, R.W.; Rogers, J.S.; Kaake, R.M.; Gordon, E.A.; Huang, L.; Baldi, P.; Bardwell, L. Computational prediction and experimental verification of new MAP kinase docking sites and substrates including Gli transcription factors. PLoS Comput. Biol. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Po, A.; Silvano, M.; Miele, E.; Capalbo, C.; Eramo, A.; Salvati, V.; Todaro, M.; Besharat, Z.M.; Catanzaro, G.; Cucchi, D.; et al. Noncanonical GLI1 signaling promotes stemness features and in vivo growth in lung adenocarcinoma. Oncogene 2017, 36, 4641–4652. [Google Scholar] [CrossRef] [Green Version]
- Riobo, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P., Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Bjorklund, M.; Cheng, F.; Syvanen, H.; Kivioja, T.; Kilpinen, S.; Sun, Z.; Kallioniemi, O.; Stunnenberg, H.G.; He, W.W.; et al. Application of active and kinase-deficient kinome collection for identification of kinases regulating hedgehog signaling. Cell 2008, 133, 537–548. [Google Scholar] [CrossRef]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Tostar, U.; Jin, Q.; Fendrich, V.; Guerra, C.; Barbacid, M.; Toftgard, R. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat. Struct. Mol. Biol. 2010, 17, 718–725. [Google Scholar] [CrossRef]
- Li, B.; Fei, D.L.; Flaveny, C.A.; Dahmane, N.; Baubet, V.; Wang, Z.; Bai, F.; Pei, X.H.; Rodriguez-Blanco, J.; Hang, B.; et al. Pyrvinium attenuates Hedgehog signaling downstream of smoothened. Cancer Res. 2014, 74, 4811–4821. [Google Scholar] [CrossRef]
- Purzner, T.; Purzner, J.; Buckstaff, T.; Cozza, G.; Gholamin, S.; Rusert, J.M.; Hartl, T.A.; Sanders, J.; Conley, N.; Ge, X.; et al. Developmental phosphoproteomics identifies the kinase CK2 as a driver of Hedgehog signaling and a therapeutic target in medulloblastoma. Sci Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Aikin, R.A.; Ayers, K.L.; Therond, P.P. The role of kinases in the Hedgehog signalling pathway. Embo Rep. 2008, 9, 330–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef]
- Coni, S.; Antonucci, L.; D’Amico, D.; Di Magno, L.; Infante, P.; De Smaele, E.; Giannini, G.; Di Marcotullio, L.; Screpanti, I.; Gulino, A.; et al. Gli2 acetylation at lysine 757 regulates hedgehog-dependent transcriptional output by preventing its promoter occupancy. PLoS ONE 2013, 8, e65718. [Google Scholar] [CrossRef]
- Long, J.; Li, B.; Rodriguez-Blanco, J.; Pastori, C.; Volmar, C.H.; Wahlestedt, C.; Capobianco, A.; Bai, F.; Pei, X.H.; Ayad, N.G.; et al. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J. Biol. Chem. 2014, 289, 35494–35502. [Google Scholar] [CrossRef]
- Whitson, R.J.; Lee, A.; Urman, N.M.; Mirza, A.; Yao, C.Y.; Brown, A.S.; Li, J.R.; Shankar, G.; Fry, M.A.; Atwood, S.X.; et al. Noncanonical hedgehog pathway activation through SRF-MKL1 promotes drug resistance in basal cell carcinomas. Nat. Med. 2018, 24, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef]
- Zwerner, J.P.; Joo, J.; Warner, K.L.; Christensen, L.; Hu-Lieskovan, S.; Triche, T.J.; May, W.A. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 2008, 27, 3282–3291. [Google Scholar] [CrossRef]
- Joo, J.; Christensen, L.; Warner, K.; States, L.; Kang, H.G.; Vo, K.; Lawlor, E.R.; May, W.A. GLI1 is a central mediator of EWS/FLI1 signaling in Ewing tumors. PLoS ONE 2009, 4, e7608. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Schnidar, H.; Neill, G.W.; Hanneder, M.; Klingler, S.; Blaas, L.; Schmid, C.; Hauser-Kronberger, C.; Regl, G.; Philpott, M.P.; et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol. Cell Biol 2006, 26, 6283–6298. [Google Scholar] [CrossRef]
- Eberl, M.; Klingler, S.; Mangelberger, D.; Loipetzberger, A.; Damhofer, H.; Zoidl, K.; Schnidar, H.; Hache, H.; Bauer, H.C.; Solca, F.; et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. Embo Mol. Med. 2012, 4, 218–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, C.; Gruber, W.; Eberl, M.; Tesanovic, S.; Stadler, M.; Elmer, D.P.; Schlederer, M.; Grund, S.; Roos, S.; Wolff, F.; et al. Synergistic cross-talk of hedgehog and interleukin-6 signaling drives growth of basal cell carcinoma. Int. J. Cancer 2018, 143, 2943–2954. [Google Scholar] [CrossRef]
- Eberl, M.; Mangelberger, D.; Swanson, J.B.; Verhaegen, M.E.; Harms, P.W.; Frohm, M.L.; Dlugosz, A.A.; Wong, S.Y. Tumor Architecture and Notch Signaling Modulate Drug Response in Basal Cell Carcinoma. Cancer Cell 2018, 33, 229–243. [Google Scholar] [CrossRef]
- Sanchez-Danes, A.; Larsimont, J.C.; Liagre, M.; Munoz-Couselo, E.; Lapouge, G.; Brisebarre, A.; Dubois, C.; Suppa, M.; Sukumaran, V.; Del Marmol, V.; et al. A slow-cycling LGR5 tumour population mediates basal cell carcinoma relapse after therapy. Nature 2018, 562, 434–438. [Google Scholar] [CrossRef]
- Biehs, B.; Dijkgraaf, G.J.P.; Piskol, R.; Alicke, B.; Boumahdi, S.; Peale, F.; Gould, S.E.; de Sauvage, F.J. A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018, 562, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Jin, Q.; Koo, D.I.; Liao, X.; Englund, N.P.; Wang, Y.; Ramamurthy, A.; Schultz, P.G.; Dorsch, M.; Kelleher, J.; et al. Small molecule antagonists in distinct binding modes inhibit drug-resistant mutant of smoothened. Chem. Biol. 2011, 18, 432–437. [Google Scholar] [CrossRef]
- Tremblay, M.R.; Lescarbeau, A.; Grogan, M.J.; Tan, E.; Lin, G.; Austad, B.C.; Yu, L.C.; Behnke, M.L.; Nair, S.J.; Hagel, M.; et al. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J. Med. Chem. 2009, 52, 4400–4418. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Hatton, B.A.; Villavicencio, E.H.; Khanna, P.C.; Friedman, S.D.; Ditzler, S.; Pullar, B.; Robison, K.; White, K.F.; Tunkey, C.; et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc. Natl. Acad. Sci. USA 2012, 109, 7859–7864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimeno, A.; Weiss, G.J.; Miller, W.H., Jr.; Gettinger, S.; Eigl, B.J.; Chang, A.L.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2766–2774. [Google Scholar] [CrossRef]
- Peukert, S.; He, F.; Dai, M.; Zhang, R.; Sun, Y.; Miller-Moslin, K.; McEwan, M.; Lagu, B.; Wang, K.; Yusuff, N.; et al. Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened. ChemMedChem 2013, 8, 1261–1265. [Google Scholar] [CrossRef]
- Ohashi, T.; Oguro, Y.; Tanaka, T.; Shiokawa, Z.; Tanaka, Y.; Shibata, S.; Sato, Y.; Yamakawa, H.; Hattori, H.; Yamamoto, Y.; et al. Discovery of the investigational drug TAK-441, a pyrrolo[3,2-c]pyridine derivative, as a highly potent and orally active hedgehog signaling inhibitor: Modification of the core skeleton for improved solubility. Bioorg. Med. Chem. 2012, 20, 5507–5517. [Google Scholar] [CrossRef]
- Ibuki, N.; Ghaffari, M.; Pandey, M.; Iu, I.; Fazli, L.; Kashiwagi, M.; Tojo, H.; Nakanishi, O.; Gleave, M.E.; Cox, M.E. TAK-441, a novel investigational smoothened antagonist, delays castration-resistant progression in prostate cancer by disrupting paracrine hedgehog signaling. Int. J. Cancer 2013, 133, 1955–1966. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Shimizu, Y.; Nakashima, K.; Kondo, S.; Ogawa, K.; Sasaki, S.; Matsui, H. Inhibition mechanism exploration of investigational drug TAK-441 as inhibitor against Vismodegib-resistant Smoothened mutant. Eur. J. Pharmacol. 2014, 723, 305–313. [Google Scholar] [CrossRef]
- Kim, J.; Tang, J.Y.; Gong, R.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; Reya, T.; et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Chen, B.; King, E.M.; Borodovsky, A.; Riggins, G.J.; et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Trang, V.; Lee, A.; Williams, N.S.; Wilson, A.N.; Epstein, E.H., Jr.; Tang, J.Y.; Kim, J. Posaconazole, a Second-Generation Triazole Antifungal Drug, Inhibits the Hedgehog Signaling Pathway and Progression of Basal Cell Carcinoma. Mol. Cancer Ther. 2016, 15, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Sivaraman, A.; Lee, K. Development of taladegib as a sonic hedgehog signaling pathway inhibitor. Arch. Pharmacal Res. 2017, 40, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, H.; Katritch, V.; Han, G.W.; Huang, X.P.; Liu, W.; Siu, F.Y.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Structure of the human smoothened receptor bound to an antitumour agent. Nature 2013, 497, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendell, J.; Andre, V.; Ho, A.; Kudchadkar, R.; Migden, M.; Infante, J.; Tiu, R.V.; Pitou, C.; Tucker, T.; Brail, L.; et al. Phase I Study of LY2940680, a Smo Antagonist, in Patients with Advanced Cancer Including Treatment-Naive and Previously Treated Basal Cell Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Chen, Z.; Lallemand-Breitenbach, V.; de The, H. How acute promyelocytic leukaemia revived arsenic. Nat. Rev. Cancer 2002, 2, 705–713. [Google Scholar] [CrossRef]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. Embo J. 2015, 34, 200–217. [Google Scholar] [CrossRef]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef] [Green Version]
- Chenna, V.; Hu, C.; Pramanik, D.; Aftab, B.T.; Karikari, C.; Campbell, N.R.; Hong, S.M.; Zhao, M.; Rudek, M.A.; Khan, S.R.; et al. A polymeric nanoparticle encapsulated small-molecule inhibitor of Hedgehog signaling (NanoHHI) bypasses secondary mutational resistance to Smoothened antagonists. Mol. Cancer Ther. 2012, 11, 165–173. [Google Scholar] [CrossRef]
- Infante, P.; Alfonsi, R.; Botta, B.; Mori, M.; Di Marcotullio, L. Targeting GLI factors to inhibit the Hedgehog pathway. Trends Pharmacol. Sci. 2015, 36, 547–558. [Google Scholar] [CrossRef]
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chene, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Rodon, J.; Perez-Fidalgo, A.; Krop, I.E.; Burris, H.; Guerrero-Zotano, A.; Britten, C.D.; Becerra, C.; Schellens, J.; Richards, D.A.; Schuler, M.; et al. Phase 1/1b dose escalation and expansion study of BEZ235, a dual PI3K/mTOR inhibitor, in patients with advanced solid tumors including patients with advanced breast cancer. Cancer Chemother. Pharmacol. 2018, 82, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.; Regl, G.; Hofbauer, S.W.; Altenhofer, P.; Achatz, G.; Dlugosz, A.; Schnidar, H.; Greil, R.; Hartmann, T.N.; Aberger, F. Hedgehog/GLI and PI3K signaling in the initiation and maintenance of chronic lymphocytic leukemia. Oncogene 2015, 34, 5341–5351. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, N.K.; Kling, M.J.; Coulter, D.W.; McGuire, T.R.; Ray, S.; Kesherwani, V.; Joshi, S.S.; Sharp, J.G. Improved therapy for medulloblastoma: Targeting hedgehog and PI3K-mTOR signaling pathways in combination with chemotherapy. Oncotarget 2018, 9, 16619–16633. [Google Scholar] [CrossRef] [PubMed]
- Coni, S.; Mancuso, A.B.; Di Magno, L.; Sdruscia, G.; Manni, S.; Serrao, S.M.; Rotili, D.; Spiombi, E.; Bufalieri, F.; Petroni, M.; et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci. Rep. 2017, 7, 44079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Tresckow, B.; Sayehli, C.; Aulitzky, W.E.; Goebeler, M.E.; Schwab, M.; Braz, E.; Krauss, B.; Krauss, R.; Hermann, F.; Bartz, R.; et al. Phase I Study of Domatinostat (4SC-202), a class I Histone Deacetylase Inhibitor in Patients with Advanced Hematological Malignancies. Eur. J. Haematol. 2018. [Google Scholar] [CrossRef]
- Gruber, W.; Peer, E.; Elmer, D.P.; Sternberg, C.; Tesanovic, S.; Del Burgo, P.; Coni, S.; Canettieri, G.; Neureiter, D.; Bartz, R.; et al. Targeting class I histone deacetylases by the novel small molecule inhibitor 4SC-202 blocks oncogenic hedgehog-GLI signaling and overcomes smoothened inhibitor resistance. Int. J. Cancer 2018, 142, 968–975. [Google Scholar] [CrossRef]
- Mirza, A.N.; Fry, M.A.; Urman, N.M.; Atwood, S.X.; Roffey, J.; Ott, G.R.; Chen, B.; Lee, A.; Brown, A.S.; Aasi, S.Z.; et al. Combined inhibition of atypical PKC and histone deacetylase 1 is cooperative in basal cell carcinoma treatment. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [Green Version]
- Mullor, J.L.; Dahmane, N.; Sun, T.; Ruiz i Altaba, A. Wnt signals are targets and mediators of Gli function. Curr. Biol. 2001, 11, 769–773. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Andl, T.; Grachtchouk, V.; Wang, A.; Liu, J.; Syu, L.J.; Ferris, J.; Wang, T.S.; Glick, A.B.; Millar, S.E.; et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/beta3-catenin signaling. Nat. Genet. 2008, 40, 1130–1135. [Google Scholar] [CrossRef]
- Moreira, A.; Kirchberger, M.C.; Toussaint, F.; Erdmann, M.; Schuler, G.; Heinzerling, L. Effective anti-PD-1 therapy in a SUFU-mutated patient with Gorlin-Goltz syndrome. Br. J. Derm. 2018. [Google Scholar] [CrossRef]
- Fischer, S.; Ali, O.H.; Jochum, W.; Kluckert, T.; Flatz, L.; Siano, M. Anti-PD-1 Therapy Leads to Near-Complete Remission in a Patient with Metastatic Basal Cell Carcinoma. Oncol Res. Treat. 2018, 41, 391–394. [Google Scholar] [CrossRef]
- Chakrabarti, J.; Holokai, L.; Syu, L.; Steele, N.G.; Chang, J.; Wang, J.; Ahmed, S.; Dlugosz, A.; Zavros, Y. Hedgehog signaling induces PD-L1 expression and tumor cell proliferation in gastric cancer. Oncotarget 2018, 9, 37439–37457. [Google Scholar] [CrossRef]
- Lipson, E.J.; Lilo, M.T.; Ogurtsova, A.; Esandrio, J.; Xu, H.; Brothers, P.; Schollenberger, M.; Sharfman, W.H.; Taube, J.M. Basal cell carcinoma: PD-L1/PD-1 checkpoint expression and tumor regression after PD-1 blockade. J. Immunother Cancer 2017, 5, 23. [Google Scholar] [CrossRef]
- Holokai, L.; Chakrabarti, J.; Broda, T.; Chang, J.; Hawkins, J.A.; Sundaram, N.; Wroblewski, L.E.; Peek, R.M., Jr.; Wang, J.; Helmrath, M.; et al. Increased Programmed Death-Ligand 1 is an Early Epithelial Cell Response to Helicobacter pylori Infection. PLoS Pathog. 2019, 15, e1007468. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peer, E.; Tesanovic, S.; Aberger, F. Next-Generation Hedgehog/GLI Pathway Inhibitors for Cancer Therapy. Cancers 2019, 11, 538. https://doi.org/10.3390/cancers11040538
Peer E, Tesanovic S, Aberger F. Next-Generation Hedgehog/GLI Pathway Inhibitors for Cancer Therapy. Cancers. 2019; 11(4):538. https://doi.org/10.3390/cancers11040538
Chicago/Turabian StylePeer, Elisabeth, Suzana Tesanovic, and Fritz Aberger. 2019. "Next-Generation Hedgehog/GLI Pathway Inhibitors for Cancer Therapy" Cancers 11, no. 4: 538. https://doi.org/10.3390/cancers11040538
APA StylePeer, E., Tesanovic, S., & Aberger, F. (2019). Next-Generation Hedgehog/GLI Pathway Inhibitors for Cancer Therapy. Cancers, 11(4), 538. https://doi.org/10.3390/cancers11040538