Orthotopic Patient-Derived Xenografts of Gastric Cancer to Decipher Drugs Effects on Cancer Stem Cells and Metastatic Dissemination

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
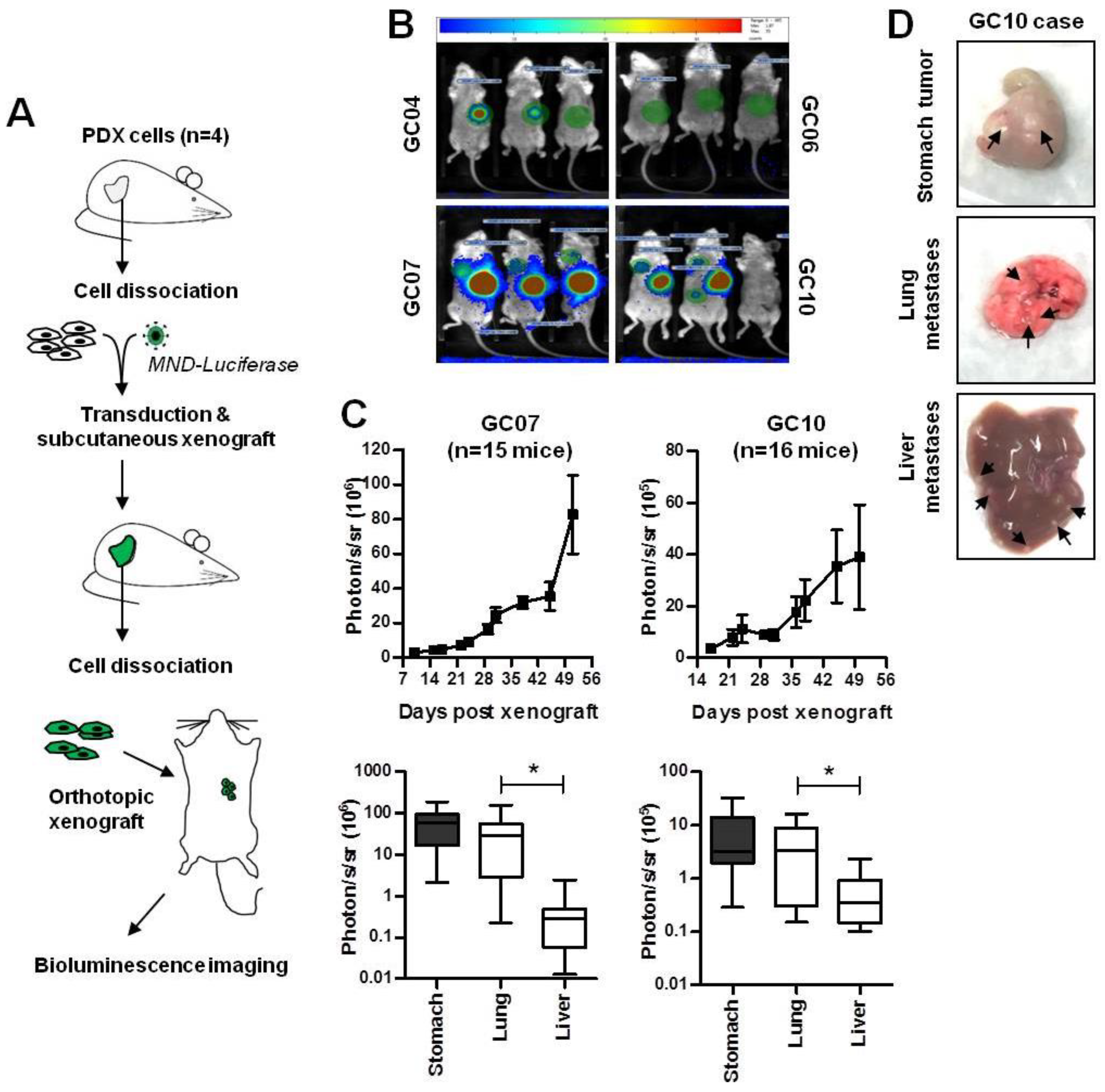
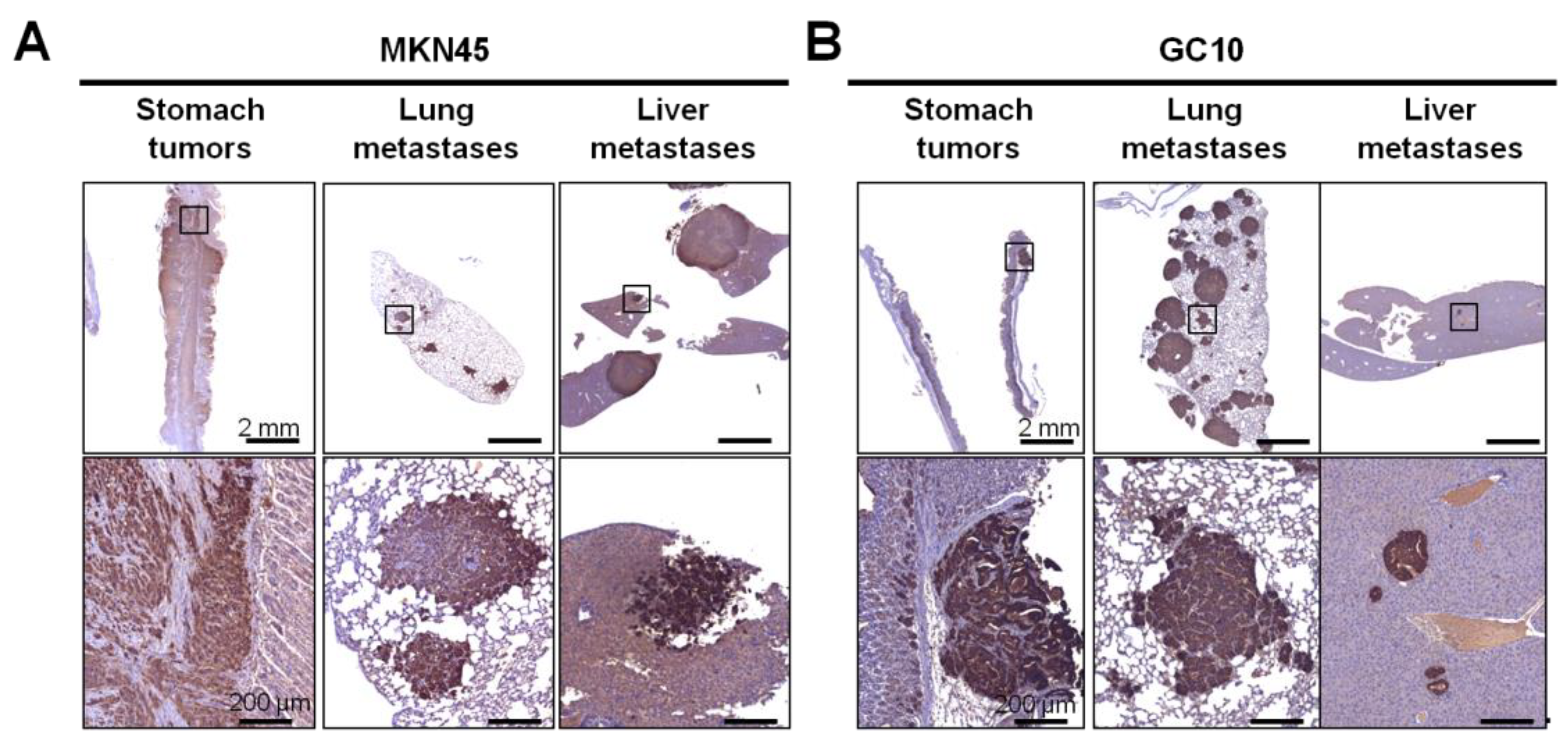
2.1. Development of Patient-Derived Orthotopic Xenograft Mouse Models Developing Distant Metastases
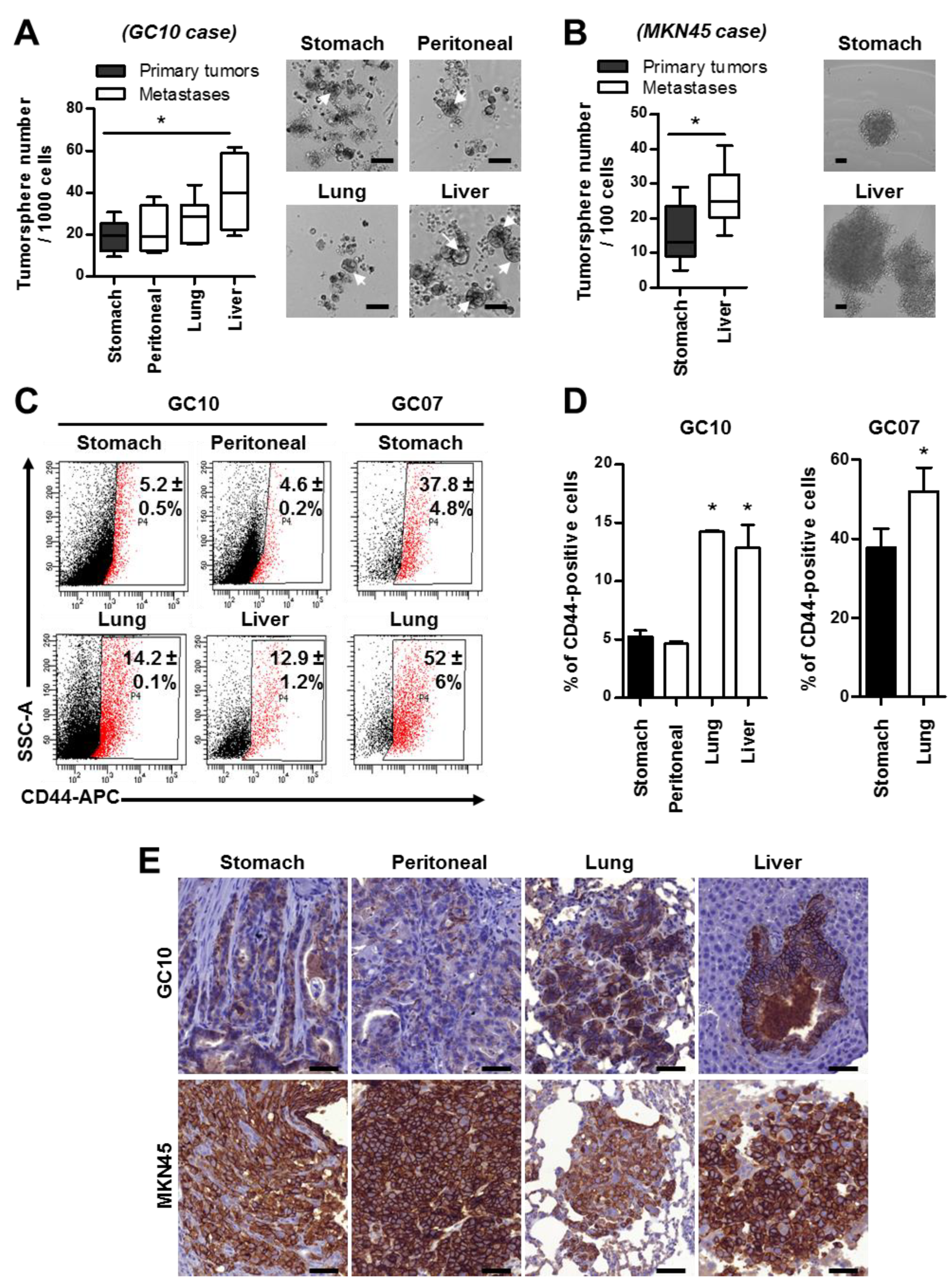
2.2. Metastases Are Enriched in Cancer Stem Cells
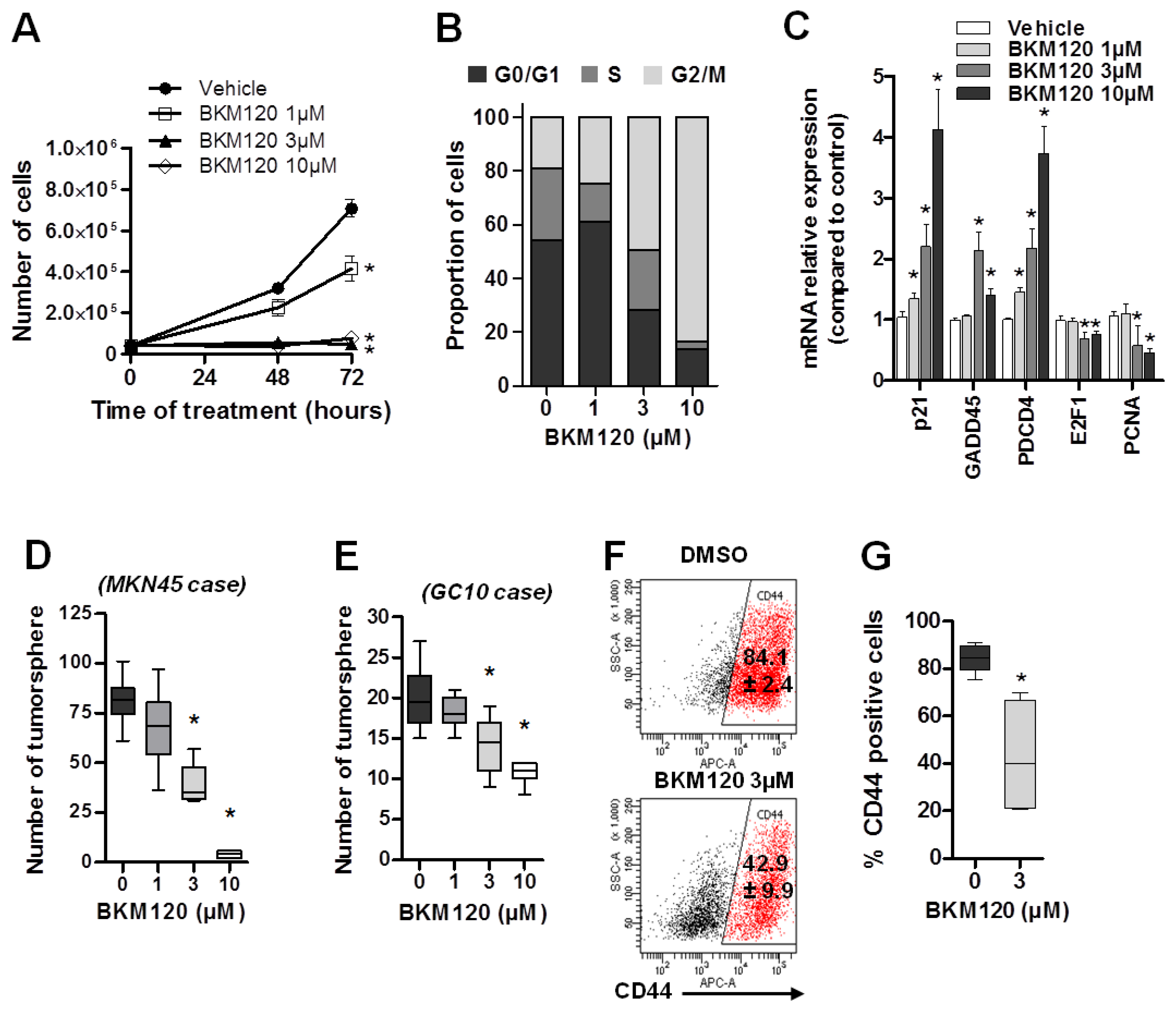
2.3. BKM120 Inhibits GC Proliferation and Decreases the Cancer Stem Cell Population and Properties
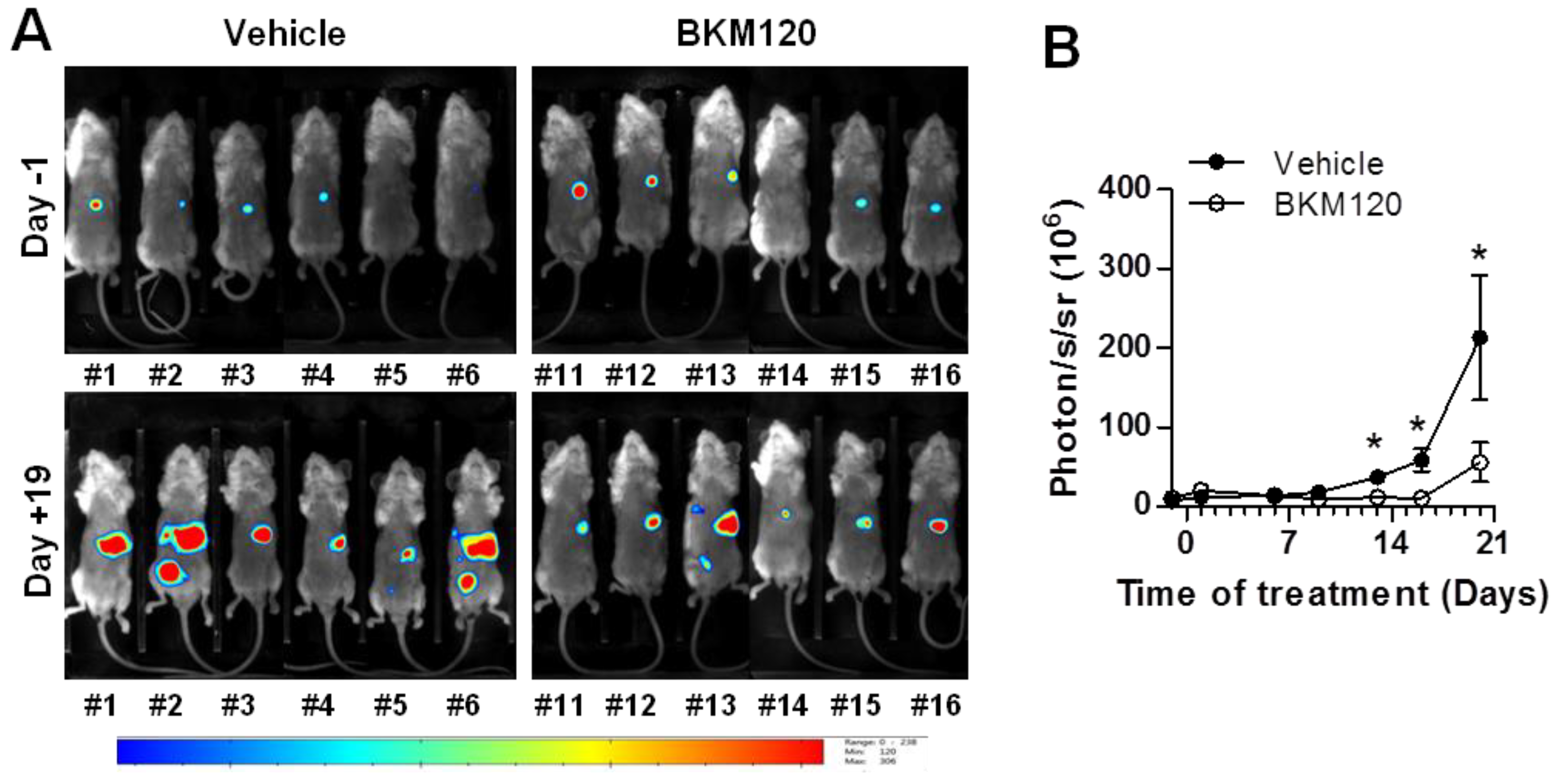
2.4. BKM120 Limits Tumor Growth and Dissemination In Vivo
3. Discussion
4. Materials and Methods
4.1. Cancer Cell Lines and Patient-Derived Xenograft Cell Culture
4.2. Production of Lentiviral Particles and Transduction of Cells
4.3. Orthotopic Xenograft and Resection Experiments
4.4. Bioluminescence Imaging
4.5. Immunohistochemistry
4.6. PI3K Inhibitor Preparation and Treatments
4.7. Tumorsphere Culture and Quantification
4.8. Flow Cytometry and Cell Cycle Assay
4.9. RNA Extraction and RT-qPCR
4.10. Protein Isolation and Western Blotting
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Mégraud, F.; Bessède, E.; Varon, C. Helicobacter pylori infection and gastric carcinoma. Clin. Microbiol. Infect. 2015, 21, 984–990. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Lei, Z.; Tan, I.B.; Das, K.; Deng, N.; Zouridis, H.; Pattison, S.; Chua, C.; Feng, Z.; Guan, Y.K.; Ooi, C.H.; et al. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology 2013, 145, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Association, J.G.C. Japanese gastric cancer treatment guidelines 2014 (ver. 4). Gastric Cancer 2017, 20, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Verheij, M.; Allum, W.; Cunningham, D.; Cervantes, A.; Arnold, D.; Committee, E.G. Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v38–v49. [Google Scholar] [CrossRef] [PubMed]
- Van Hagen, P.; Hulshof, M.C.; van Lanschot, J.J.; Steyerberg, E.W.; van Berge Henegouwen, M.I.; Wijnhoven, B.P.; Richel, D.J.; Nieuwenhuijzen, G.A.; Hospers, G.A.; Bonenkamp, J.J.; et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N. Engl. J. Med. 2012, 366, 2074–2084. [Google Scholar] [CrossRef]
- Carrasco-Garcia, E.; García-Puga, M.; Arevalo, S.; Matheu, A. Towards precision medicine: Linking genetic and cellular heterogeneity in gastric cancer. Ther. Adv. Med. Oncol. 2018, 10, 1758835918794628. [Google Scholar] [CrossRef]
- Hartgrink, H.H.; Jansen, E.P.; van Grieken, N.C.; van de Velde, C.J. Gastric cancer. Lancet 2009, 374, 477–490. [Google Scholar] [CrossRef] [Green Version]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Millis, S.Z.; Ikeda, S.; Reddy, S.; Gatalica, Z.; Kurzrock, R. Landscape of Phosphatidylinositol-3-Kinase Pathway Alterations Across 19 784 Diverse Solid Tumors. JAMA Oncol. 2016, 2, 1565–1573. [Google Scholar] [CrossRef]
- Kang, M.H.; Kim, J.S.; Seo, J.E.; Oh, S.C.; Yoo, Y.A. BMP2 accelerates the motility and invasiveness of gastric cancer cells via activation of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway. Exp. Cell Res. 2010, 316, 24–37. [Google Scholar] [CrossRef]
- Zang, M.; Zhang, B.; Zhang, Y.; Li, J.; Su, L.; Zhu, Z.; Gu, Q.; Liu, B.; Yan, M. CEACAM6 promotes gastric cancer invasion and metastasis by inducing epithelial-mesenchymal transition via PI3K/AKT signaling pathway. PLoS ONE 2014, 9, e112908. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Huang, J.; Xu, J.; Huang, Q.; Wang, J.; Zhong, W.; Lin, X.; Li, Y. ERp29 controls invasion and metastasis of gastric carcinoma by inhibition of epithelial-mesenchymal transition via PI3K/Aktsignaling pathway. BMC Cancer 2017, 17, 626. [Google Scholar] [CrossRef]
- Sun, C.; Tao, Y.; Gao, Y.; Xia, Y.; Liu, Y.; Wang, G.; Gu, Y. F-box protein 11 promotes the growth and metastasis of gastric cancer via PI3K/AKT pathway-mediated EMT. Biomed. Pharmacother. 2018, 98, 416–423. [Google Scholar] [CrossRef]
- Wei, S.; Wang, L.; Zhang, L.; Li, B.; Li, Z.; Zhang, Q.; Wang, J.; Chen, L.; Sun, G.; Li, Q.; et al. ZNF143 enhances metastasis of gastric cancer by promoting the process of EMT through PI3K/AKT signaling pathway. Tumour Biol. 2016, 37, 12813–12821. [Google Scholar] [CrossRef]
- Lu, W.; Xu, Z.; Zhang, M.; Zuo, Y. MiR-19a promotes epithelial-mesenchymal transition through PI3K/AKT pathway in gastric cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 7286–7296. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Giraud, J.; Chambonnier, L.; Dubus, P.; Wittkop, L.; Belleannée, G.; Collet, D.; Soubeyran, I.; Evrard, S.; Rousseau, B.; et al. Characterization of Biomarkers of Tumorigenic and Chemoresistant Cancer Stem Cells in Human Gastric Carcinoma. Clin. Cancer Res. 2017, 23, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Bessède, E.; Staedel, C.; Acuña Amador, L.A.; Nguyen, P.H.; Chambonnier, L.; Hatakeyama, M.; Belleannée, G.; Mégraud, F.; Varon, C. Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene 2014, 33, 4123–4131. [Google Scholar] [CrossRef]
- Bessède, E.; Dubus, P.; Mégraud, F.; Varon, C. Helicobacter pylori infection and stem cells at the origin of gastric cancer. Oncogene 2015, 34, 2547–2555. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Giraud, J.; Staedel, C.; Chambonnier, L.; Dubus, P.; Chevret, E.; Bœuf, H.; Gauthereau, X.; Rousseau, B.; Fevre, M.; et al. All-trans retinoic acid targets gastric cancer stem cells and inhibits patient-derived gastric carcinoma tumor growth. Oncogene 2016, 35, 5619–5628. [Google Scholar] [CrossRef]
- Courtois, S.; Durán, R.V.; Giraud, J.; Sifré, E.; Izotte, J.; Mégraud, F.; Lehours, P.; Varon, C.; Bessède, E. Metformin targets gastric cancer stem cells. Eur. J. Cancer 2017, 84, 193–201. [Google Scholar] [CrossRef]
- Kim, M.Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.; Norton, L.; Massagué, J. Tumor self-seeding by circulating cancer cells. Cell 2009, 139, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Yang, G.; Ding, Y.; Huang, Y.; Liu, S.; Zhou, L.; Wei, W.; Wang, J.; Hu, G. Combined treatment with PI3K inhibitor BKM120 and PARP inhibitor olaparib is effective in inhibiting the gastric cancer cells with ARID1A deficiency. Oncol. Rep. 2018, 40, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Park, J.; Han, S.W.; Im, S.A.; Kim, T.Y.; Oh, D.Y.; Bang, Y.J. NVP-BKM120, a novel PI3K inhibitor, shows synergism with a STAT3 inhibitor in human gastric cancer cells harboring KRAS mutations. Int. J. Oncol. 2012, 40, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.; Bachmann, E.; Linnig, M.; Khillimberger, K.; Schimanski, C.C.; Galle, P.R.; Moehler, M. Selective PI3K inhibition by BKM120 and BEZ235 alone or in combination with chemotherapy in wild-type and mutated human gastrointestinal cancer cell lines. Cancer Chemother. Pharmacol. 2012, 69, 1601–1615. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M. Patient-derived orthotopic xenografts: Better mimic of metastasis than subcutaneous xenografts. Nat. Rev. Cancer 2015, 15, 451–452. [Google Scholar] [CrossRef]
- Furukawa, T.; Fu, X.; Kubota, T.; Watanabe, M.; Kitajima, M.; Hoffman, R.M. Nude mouse metastatic models of human stomach cancer constructed using orthotopic implantation of histologically intact tissue. Cancer Res. 1993, 53, 1204–1208. [Google Scholar]
- Furukawa, T.; Kubota, T.; Watanabe, M.; Kitajima, M.; Hoffman, R.M. Orthotopic transplantation of histologically intact clinical specimens of stomach cancer to nude mice: Correlation of metastatic sites in mouse and individual patient donors. Int. J. Cancer 1993, 53, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, S.; Hotz, B.; Buhr, H.J.; Hotz, H.G. An orthotopic nude mouse model for preclinical research of gastric cardia cancer. Int. J. Colorectal Dis. 2009, 24, 31–39. [Google Scholar] [CrossRef]
- Busuttil, R.A.; Liu, D.S.; Di Costanzo, N.; Schröder, J.; Mitchell, C.; Boussioutas, A. An orthotopic mouse model of gastric cancer invasion and metastasis. Sci. Rep. 2018, 8, 825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Liu, J.; Yao, L.; Yin, J.; Su, N.; Liu, X.; Cao, F.; Liang, J.; Nie, Y.; Wu, K. Real-time bioluminescence and tomographic imaging of gastric cancer in a novel orthotopic mouse model. Oncol. Rep. 2012, 27, 1937–1943. [Google Scholar] [CrossRef]
- Feng, H.Y.; Zhang, Y.; Liu, H.J.; Dong, X.; Yang, S.C.; Lu, Q.; Meng, F.; Chen, H.Z.; Sun, P.; Fang, C. Characterization of an orthotopic gastric cancer mouse model with lymph node and organ metastases using bioluminescence imaging. Oncol. Lett. 2018, 16, 5179–5185. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells—An integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef]
- Morel, A.P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef]
- Bessède, E.; Molina, S.; Acuña-Amador, L.; Dubus, P.; Staedel, C.; Chambonnier, L.; Buissonnière, A.; Sifré, E.; Giese, A.; Bénéjat, L.; et al. Deletion of IQGAP1 promotes Helicobacter pylori-induced gastric dysplasia in mice and acquisition of cancer stem cell properties in vitro. Oncotarget 2016, 7, 80688–80699. [Google Scholar] [CrossRef] [Green Version]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef]
- Li, M.; Zhang, B.; Zhang, Z.; Liu, X.; Qi, X.; Zhao, J.; Jiang, Y.; Zhai, H.; Ji, Y.; Luo, D. Stem cell-like circulating tumor cells indicate poor prognosis in gastric cancer. Biomed. Res. Int. 2014, 2014, 981261. [Google Scholar] [CrossRef]
- Toyoshima, K.; Hayashi, A.; Kashiwagi, M.; Hayashi, N.; Iwatsuki, M.; Ishimoto, T.; Baba, Y.; Baba, H.; Ohta, Y. Analysis of circulating tumor cells derived from advanced gastric cancer. Int. J. Cancer 2015, 137, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Bedard, P.L.; Tabernero, J.; Janku, F.; Wainberg, Z.A.; Paz-Ares, L.; Vansteenkiste, J.; Van Cutsem, E.; Pérez-García, J.; Stathis, A.; Britten, C.D.; et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin. Cancer Res. 2015, 21, 730–738. [Google Scholar] [CrossRef] [PubMed]
- McRee, A.J.; Sanoff, H.K.; Carlson, C.; Ivanova, A.; O’Neil, B.H. A phase I trial of mFOLFOX6 combined with the oral PI3K inhibitor BKM120 in patients with advanced refractory solid tumors. Investig. New Drugs 2015, 33, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Bendell, J.C.; Rodon, J.; Burris, H.A.; de Jonge, M.; Verweij, J.; Birle, D.; Demanse, D.; De Buck, S.S.; Ru, Q.C.; Peters, M.; et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Ragon, B.K.; Kantarjian, H.; Jabbour, E.; Ravandi, F.; Cortes, J.; Borthakur, G.; DeBose, L.; Zeng, Z.; Schneider, H.; Pemmaraju, N.; et al. Buparlisib, a PI3K inhibitor, demonstrates acceptable tolerability and preliminary activity in a phase I trial of patients with advanced leukemias. Am. J. Hematol. 2017, 92, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann. Oncol. 2017, 28, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Heudel, P.E.; Fabbro, M.; Roemer-Becuwe, C.; Kaminsky, M.C.; Arnaud, A.; Joly, F.; Roche-Forestier, S.; Meunier, J.; Foa, C.; You, B.; et al. Phase II study of the PI3K inhibitor BKM120 in patients with advanced or recurrent endometrial carcinoma: A stratified type I-type II study from the GINECO group. Br. J. Cancer 2017, 116, 303–309. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Halabi, S.; Healy, P.; Alumkal, J.J.; Winters, C.; Kephart, J.; Bitting, R.L.; Hobbs, C.; Soleau, C.F.; Beer, T.M.; et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur. J. Cancer 2017, 81, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Soulières, D.; Faivre, S.; Mesía, R.; Remenár, É.; Li, S.H.; Karpenko, A.; Dechaphunkul, A.; Ochsenreither, S.; Kiss, L.A.; Lin, J.C.; et al. Buparlisib and paclitaxel in patients with platinum-pretreated recurrent or metastatic squamous cell carcinoma of the head and neck (BERIL-1): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol. 2017, 18, 323–335. [Google Scholar] [CrossRef]
- Baselga, J.; Im, S.A.; Iwata, H.; Cortés, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef]
- Kobayashi, I.; Semba, S.; Matsuda, Y.; Kuroda, Y.; Yokozaki, H. Significance of Akt phosphorylation on tumor growth and vascular endothelial growth factor expression in human gastric carcinoma. Pathobiology 2006, 73, 8–17. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Tumor-Bearing Mice 1/Number of Xenografted Mice | ||||||
|---|---|---|---|---|---|---|
| Experiment | Case | Stomach | Lung | Liver | Peritoneal Tumors | Weeks |
| a | GC04 | 3/4 | 2/4 | 0/4 | 1/4 | 13 |
b | GC06 GC07 GC10 GC07 GC10 | 0/4 4/4 4/4 12/12 14/15 | 0/4 4/4 3/4 12/12 11/15 | 0/4 0/4 2/4 3/12 13/15 | 0/4 0/4 0/4 1/12 5/15 | 25 8 10 8 8 |
| Number of Tumor-Bearing Mice 1/Number of Xenograft Mice | |||||
|---|---|---|---|---|---|
| Case | Stomach | Lung | Liver | Peritoneal Tumor | Weeks |
| MKN45 MKN74 NCI-N87 | 5/5 4/4 3/5 | 5/5 4/4 0/4 | 4/5 2/4 0/5 | 3/5 2/4 0/5 | 8 8 8–10 |
| Number of Tumor-Bearing Mice 1/Number of Xenograft Mice | ||||
|---|---|---|---|---|
| Case | Stomach | Lung | Liver | Spleen and Pancreas |
| Vehicle BKM120 | 9/9 8/8 | 9/9 7/8 | 7/9 4/8 | 6/9 2/8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giraud, J.; Bouriez, D.; Seeneevassen, L.; Rousseau, B.; Sifré, E.; Giese, A.; Mégraud, F.; Lehours, P.; Dubus, P.; Gronnier, C.; et al. Orthotopic Patient-Derived Xenografts of Gastric Cancer to Decipher Drugs Effects on Cancer Stem Cells and Metastatic Dissemination. Cancers 2019, 11, 560. https://doi.org/10.3390/cancers11040560
Giraud J, Bouriez D, Seeneevassen L, Rousseau B, Sifré E, Giese A, Mégraud F, Lehours P, Dubus P, Gronnier C, et al. Orthotopic Patient-Derived Xenografts of Gastric Cancer to Decipher Drugs Effects on Cancer Stem Cells and Metastatic Dissemination. Cancers. 2019; 11(4):560. https://doi.org/10.3390/cancers11040560
Chicago/Turabian StyleGiraud, Julie, Damien Bouriez, Lornella Seeneevassen, Benoit Rousseau, Elodie Sifré, Alban Giese, Francis Mégraud, Philippe Lehours, Pierre Dubus, Caroline Gronnier, and et al. 2019. "Orthotopic Patient-Derived Xenografts of Gastric Cancer to Decipher Drugs Effects on Cancer Stem Cells and Metastatic Dissemination" Cancers 11, no. 4: 560. https://doi.org/10.3390/cancers11040560
APA StyleGiraud, J., Bouriez, D., Seeneevassen, L., Rousseau, B., Sifré, E., Giese, A., Mégraud, F., Lehours, P., Dubus, P., Gronnier, C., & Varon, C. (2019). Orthotopic Patient-Derived Xenografts of Gastric Cancer to Decipher Drugs Effects on Cancer Stem Cells and Metastatic Dissemination. Cancers, 11(4), 560. https://doi.org/10.3390/cancers11040560